

Abstract
Biofilms are a major form of microbial life in which cells form dense surface associated communities that can persist for many generations. The long-life of biofilm communities means that they can be strongly shaped by evolutionary processes. Here, we review the experimental study of evolution in biofilm communities. We first provide an overview of the different experimental models used to study biofilm evolution and their associated advantages and disadvantages. We then illustrate the vast amount of diversification observed during biofilm evolution, and we discuss (i) potential ecological and evolutionary processes behind the observed diversification, (ii) recent insights into the genetics of adaptive diversification, (iii) the striking degree of parallelism between evolution experiments and real-life biofilms and (iv) potential consequences of diversification. In the second part, we discuss the insights provided by evolution experiments in how biofilm growth and structure can promote cooperative phenotypes. Overall, our analysis points to an important role of biofilm diversification and cooperation in bacterial survival and productivity. Deeper understanding of both processes is of key importance to design improved antimicrobial strategies and diagnostic techniques.
Keywords: biofilm, adaptive diversification, cooperation, experimental evoultion
This review paper provides an overview of (i) the different experimental models used to study biofilm evolution, (ii) the vast amount of diversification observed during biofilm evolution (including potential causes and consequences) and (iii) recent insights in how growth in biofilms can lead to the evolution of cooperative phenotypes.
INTRODUCTION
Biofilms are a major form of microbial life in which single or multiple species of bacteria form densely populated communities, typically enclosed in a matrix of secreted polymers (Costerton et al. 1995; Hall-Stoodley and Stoodley 2009; Steenackers et al. 2012; Hobley et al. 2015). Diffusion in the biofilm is limited, which allows gradients to arise and ultimately results in the formation of a spatially structured, heterogeneous environment (Stewart and Franklin 2008; Nadell, Xavier and Foster 2009). The ubiquity of biofilms is attributable to their ability to colonize many biotic and abiotic surfaces and suggests that living a collective life is critical for bacterial existence and evolution (Hall-Stoodley, Costerton and Stoodley 2004). Biofilm-dwelling bacteria are more tolerant to physical and chemical disruptions than planktonic cells and are a cause of major economic loss within industrial and medical sectors (Davies 2003; Ciofu et al. 2015).
Experimental evolution is the study of evolutionary changes that occur in an experimental population as a consequence of conditions imposed by the experimenter (Kawecki et al. 2012; Barrick and Lenski 2013). It offers the opportunity to study evolutionary processes experimentally in real time as they happen. Bacteria are very well suited to experimental evolution because of their short generation times and the viability of frozen organisms, which allow an experimenter to create a laboratory ‘fossil record’ for later study (Barrick and Lenski 2013). Numerous evolutionary studies have been performed on the planktonic form of bacteria and yeasts, under diverse environmental conditions (Lenski et al. 1991; Dunham et al. 2002; Nilsson et al. 2005; Hegreness et al. 2006; Ackermann et al. 2007; Lind and Andersson 2008; Cooper and Lenski 2010; Lang et al. 2013; Maddamsetti, Lenski and Barrick 2015) and several reviews discussing experimental evolution of planktonic bacteria (Elena and Lenski 2003; Behe 2010; Kawecki et al. 2012; Kassen 2014) are available. In contrast, and despite the predominance of biofilm growth in nature, surprisingly few evolution experiments have been performed with biofilm populations (for an overview; see Table 1).
Table 1.
Overview of experimental models used to study evolution in biofilms.
| Model | Description of model | Focus of studies using the model |
|---|---|---|
| Static microcosm of P. fluorescens | Non-shaking test tube, in which a biofilm mat forms at the broth–air interface | Effect of ecological opportunity on diversification (Rainey and Travisano 1998) and further evolutionary, ecological and genetic studies on this topic (Kassen and Rainey 2004; Fukami et al. 2007; Kassen 2009; Spiers 2014 and more*) |
| Bead transfer model of B. cenocepacia | In slowly rotating test tubes, biofilms are formed on plastic beads, which are regularly transferred to new test tubes. Cells must disperse and colonize a new bead in order to be transferred | Long-term evolution and diversification of B. cenocepacia and quantification of fitness of evolved variants (Poltak and Cooper 2011) with follow-up studies that unravel the underlying ecological and genetic mechanisms (Traverse et al. 2012; Cooper et al. 2014; Ellis et al. 2015)* |
| Spotting on solid agar plates | Agar plates with different compositions, in some cases complemented with disks* | Separate studies have used different agar compositions to investigate:
Additionally, growth media that mimic in vivo conditions have been used (Wong, Rodrigue and Kassen 2012; Gomez and Buckling 2013)* |
| Non-shaking microtiter plate | Biofilms are grown on the bottom or on discs on the bottom of the wells | Analysis of the evolved variants in E. coli (Kraigsley and Finkel 2009), S. aureus (Savage, Chopra and O'Neill 2013) and S. pneumonia (Allegrucci and Sauer 2007)* |
| Flow models | Biofilms experience flow conditions. Nutrients are continuously provided and dispersed, allowing unlimited growth | Various reactor devices* have been used in separate studies focusing on:
|
| In silico models | Mathematical modeling of biofilms | Evolution and stabilization of cooperation (Xavier and Foster 2007; Xavier, Martinez-Garcia and Foster 2009; Nadell, Foster and Xavier 2010; Mitri, Xavier and Foster 2011), polymer secretion by the QS system (Nadell et al. 2008) and the role of adhesion in biofilm evolution (Schluter et al. 2015)* |
| In vivo models | Biofilm isolates from CF patients | Evolution in patient isolates of P. aeruginosa (Smith et al. 2006; Kohler, Buckling and van Delden 2009; Hoboth et al. 2009 Huse et al. 2010; Cramer et al. 2011; Warren et al. 2011; Yang et al. 2011) and B. dolosa (Lieberman et al. 2011)* |
See Table S1(Supporting Information) for more detailed information about the referred studies.
Biofilm evolutionary studies provide important insights into biofilms, such as informing on the course of chronic infections and persistent contaminations, and aiding in the design of therapeutics and disinfectants (Traverse et al. 2012; McElroy et al. 2014). In addition, biofilm evolution has been used as a tool to study general evolutionary principles (Rainey and Travisano 1998; Kassen 2009; Spiers 2014). The study of biofilms has revealed striking analogies between life within biofilms and other biological systems. Gradients of oxygen in biofilms for example are analogous to light gradients in forests (Kim et al. 2014), while spatial heterogeneity within biofilms in general can be considered as the microscopic counterpart of landscape heterogeneity (Hallatschek et al. 2007). Moreover, biofilm evolution carries similarities to the development of malignant cancer tissue, where both are evolutionary and ecological systems where asexual cells divide in a structured environment (Martens et al. 2011).
In this review, we first discuss the advantages and disadvantages of a number of biofilm models which have been used for experimental evolution. We then organize our discussion according to two major themes that have emerged in the study of biofilm evolution. The first is the study of the rapid diversification often seen during biofilm evolution experiments and how this shapes both individual genotypes and phenotypes, and the biofilm as a whole. The second theme is how growth in biofilms can lead to the evolution of ‘cooperative’ phenotypes where cells, typically of the same genotype, work together as a collective in a manner that can promote both the growth and resilience of biofilms (Xavier and Foster 2007; Nadell, Xavier and Foster 2009; Drescher et al. 2014). We do not restrict ourselves to evolution experiments in the narrow sense of the word, but also discuss findings provided by fitness assays, competition experiments and mutational analyses.
MODELS TO STUDY EVOLUTION IN BIOFILMS
Biofilm formation is a particularly complex process that depends upon the many environmental factors that can influence the matrix composition and general structure of the biofilm (Goller and Romeo 2008). Importantly, as we will discuss, the evolutionary aspects of biofilms also strongly depend on environmental and experimental factors, such as nutrient levels, bacterial strains under study and their evolutionary history, inoculation sizes, flow rate, substrate area, physical disturbance, experiment duration, etc. For example, strains that have a higher level of preadaptation to the laboratory environment were found to undergo less phenotypic diversification in in vitro biofilm evolution experiments than clinical isolates without preadaptation (McElroy et al. 2014). Inoculation sizes and flow rates have been shown to influence the distribution of public goods within biofilms and as a consequence the evolution of cooperative traits (Drescher et al. 2014; van Gestel et al. 2014). And nutrient levels have been shown to affect the strength of diversifying selection and as such phenotypic diversification (Penterman et al. 2014).
The experimental model used to study biofilms then can strongly influence what one finds. Moreover, as for much of biology, the study of biofilms faces a strong tension between realism and complexity. While simplicity can be required to identify general evolutionary processes, resemblance to real-life situations is needed to mimic the course of chronic biofilm infections or persistent biofilm contaminations. This tension has led to a wide diversity of biofilm models used in evolutionary studies, and in this section we provide an overview of these models (Fig. 1 and Table 1), together with the associated advantages and disadvantages. A distinction is made between simple static in vitro models, in vitro flow models, in silico models and in vivo models. These are only a subset of all existing biofilm models, which have been extensively reviewed before (McBain 2009; Coenye and Nelis 2010; Rumbaugh and Carty 2011).
Figure 1.
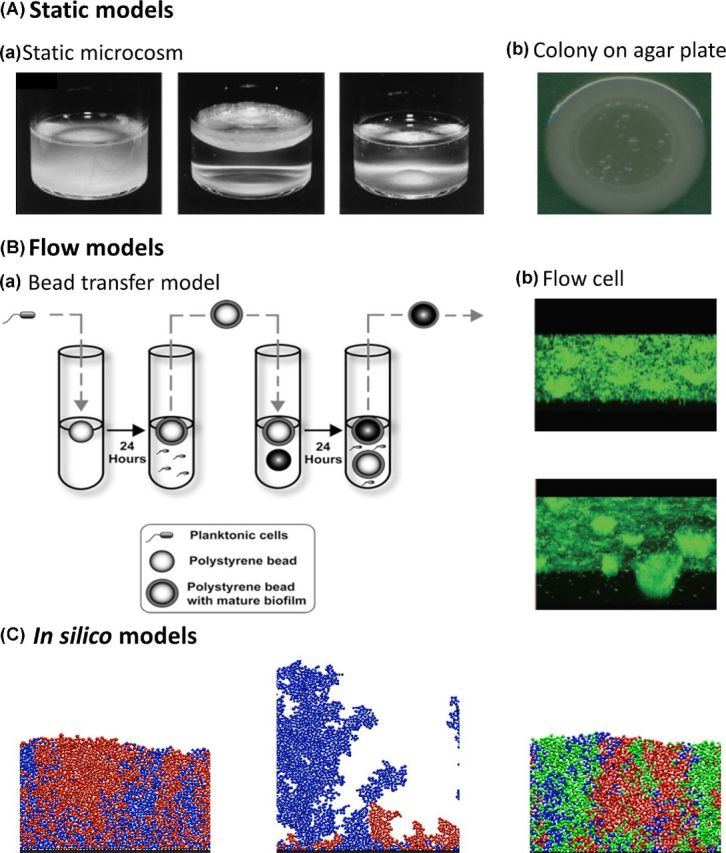
Illustration of biofilm models. (A) ‘Static models’: (a) The static microcosm model consists of a non-shaking test tube in which a biofilm mat like structure forms at the air–liquid interphase (Adapted from Rainey and Travisano 1998, also used by e.g. Kassen and Rainey 2004; Fukami et al. 2007; Kassen 2009; Spiers 2014). (b) Colonies on agar plates are considered to be suitable biofilm models due to the presence of gradients, an increased mutation rate and a structured environment (Adapted from Kim et al. 2014, also used by e.g. Korona et al. 1994; Perfeito et al. 2008; Koch et al. 2014; Saint-Ruf et al. 2014; van Gestel et al. 2014). (B) ‘Flow models’: (a) In the bead transfer model, plastic beads are put into slowly rotating test tubes. Biofilms grow on the beads and experience flow conditions due to the rotation of the test tubes. In every transfer cycle, the colonized beads are put in a new tube with new beads, without adding new bacteria (Adapted from Poltak and Cooper 2011, also used by e.g. Poltak and Cooper 2011; Traverse et al. 2012; Ellis et al. 2015; O'Rourke et al. 2015). (b) In flow cells, biofilms can grow in the presence of unlimited nutrients and dispersion (Adapted from Kirisits et al. 2005, also used by e.g. Boles, Thoendel and Singh 2004; Hansen et al. 2007; Koh et al. 2007; Yarwood et al. 2007; Lujan et al. 2011; Tyerman et al. 2013; McElroy et al. 2014; Penterman et al. 2014; Udall et al. 2015). (C) ‘In silico models’: agent-based in silico models consist of single dividing cells (the agents) that are programed to grow until a certain radius and then divide. Different parameters can be easily included and adapted (Adapted from Mitri, Xavier and Foster 2011, also used by e.g. Xavier and Foster 2007; Nadell et al. 2008; Nadell, Foster and Xavier 2010; Kim et al. 2014; van Gestel et al. 2014; Schluter et al. 2015).
Static in vitro biofilm models
The simplest in vitro model, used in one of the first evolution experiments in biofilms, consists of a glass test tube in which Pseudomonas fluorescens is grown under non-shaking conditions (Rainey and Travisano 1998). Due to the static conditions, spatial heterogeneity is created, and the bacteria are able to diversify and form a biofilm-like mat on the surface of the liquid culture. This model thus allows the study of factors driving the formation of biofilms as well as further diversification within biofilms. Other advantages of this model are its simplicity and ease of use, even though a non-shaking test tube is only a valid model for situations where no surface is present, which is for example the case for biofilm-like structures formed in granular sludge in wastewater treatments (Weber et al. 2007). Over the years, this P. fluorescens radiation in static broth-containing microcosms has become a paradigmatic experimental model for adaptive radiation, and has been applied for numerous evolutionary, ecological and genetic studies on this topic (Kassen and Rainey 2004; Fukami et al. 2007; Kassen 2009; Spiers 2014).
In other simple static models, a solid growth medium is used on which evolution in colonies is studied. The conditions in these colonies are considered similar to biofilm conditions. For example when gradients are present, the mutation rate is increased in older colonies, and a spatially structured environment is formed (Bjedov et al. 2003). Different growth media have been used including LB agar plates, (Saint-Ruf et al. 2014), minimal salt agar (Korona et al. 1994), minimal medium (Perfeito et al. 2008), modified Schaeffer's medium plates (van Gestel et al. 2014), Pseudomonas agar F-plates (Kim et al. 2014), TSB agar (Koch et al. 2014) and the cellulose static disk model (cellulose disks placed on Brain Heart Infusion agar) (Savage, Chopra and O'Neill 2013). In these models, the bacteria are fed from the surface they are growing on and this setup can be a model for biofilms associated with soft tissue infections, cystic fibrosis (CF) or food spoilage (McBain 2009).
Other models have incorporated a hard surface, and the bacteria obtain their nutrients from the liquid culture above the surface. For example, Kraigsley and Finkel (2009) and Boles, Thoendel and Singh (2004) studied evolution in a high-throughput biofilm model consisting of non-shaking polystyrene microtiter plates, in which biofilms grow on the bottom of the wells. This setup can be used for quick screenings, testing numerous different conditions or obtaining a high degree of replication. This model is representative of biofilms on plastic surfaces, such as catheters or food packaging (Hall-Stoodley, Costerton and Stoodley 2004). To study biofilms on additional types of surfaces, small disks composed of different materials may be incorporated into the wells of the microtiter plates, as done with hydroxyapatite plates for the study of oral biofilms (Guggenheim et al. 2001).
In vitro flow biofilm models
Many real-life biofilms are exposed to flow conditions and have constant supply of fresh nutrients. To mimic these conditions, flow reactor models can be used. Biofilms that grow under flow conditions are unlimited in their growth, in contrast to static models where nutrients are depleted in time. Also, depending on the flow, varying levels of nutrient dispersion can be achieved (Drescher et al. 2014). Different reactor devices have been used in evolution experiments, including drip flow reactors (Boles, Thoendel and Singh 2004; Yarwood et al. 2007; Penterman et al. 2014), drip-fed columns (Udall et al. 2015), tube reactors (Kirisits et al. 2005), rotating disk reactors (Boles and Singh 2008), flow cells (Hansen et al. 2007; Koh et al. 2007; Lujan et al. 2011; Tyerman et al. 2013; McElroy et al. 2014) and the Sorbarod biofilm model (Waite, Struthers and Dowson 2001; Savage, Chopra and O'Neill 2013). A remaining shortcoming is that many models use surfaces and growth media that do not reflect the in situ situation. However, flow models can be adapted to specific situations, such as dental plaque biofilms (Spencer et al. 2007) or chronic wound biofilms, by introducing other substrates like sliced pork, human tissue or portions of wound dressing (McBain 2009). On the other side, the use of simple surfaces and growth media might also be an advantage, as it allows the study of the effect of different parameters in a simple, defined and homogeneous environment.
Poltak and Cooper (2011) developed an elegant in vitro flow model, especially designed for biofilm evolution studies, that introduces the factor of biofilm dispersion. In this model, plastic beads are put into slowly rotating test tubes. Biofilms of Burkholderia cenocepacia grow on the beads and experience flow conditions due to the rotation of the test tubes. In every transfer cycle, the colonized beads are put in a new tube with new beads, without adding new bacteria. Thus, only bacteria that are able to attach to the beads in the first tube and disperse from the beads in the second tube are able to colonize the new beads and evolve further. As such, this setup takes into account dispersion in biofilms, colonization of new surfaces and biofilm maturation. Several parameters can be adjusted according to the needs of the experiment. For example, the ratio of colonized beads versus new beads, the time frame and the number of cycles can be changed. As described below, this B. cenocepacia bead transfer model has been used in a long-term (∼1500 generations) evolution experiment to unravel the underlying ecological and genetic mechanisms of phenotypic diversification (Poltak and Cooper 2011; Traverse et al. 2012; Ellis et al. 2015; O'Rourke et al. 2015).
In vitro models using growth media that resemble natural conditions
A further development in biofilm models is to use specific growth media that attempt to mimic natural conditions. A first evolution study with P. aeruginosa for example simulated the nutritional conditions in a CF lung, by using synthetic CF sputum (Wong, Rodrigue and Kassen 2012). A second study simulated a soil environment to grow P. fluorescens under natural conditions, including the presence of the resident microbial community in the soil (Gomez and Buckling 2013).
In silico models
Several in silico models have been used especially to study the evolution of cooperation in biofilms. Mainly agent-based models have been applied, in which a single dividing cell is the agent. These models have been developed over the last decade for applications in the field of biochemical engineering and employ mechanistic descriptions of solute diffusion and cell growth (Kreft et al. 2001; Picioreanu, Kreft and Van Loosdrecht 2004; Xavier, Picioreanu and van Loosdrecht 2005). Cells are programed to grow depending on the local substrate concentration, extracellular enzyme availability and the concentration of other extracellular products. When a certain radius is met, the cells divide and move until there is no overlap with other cells anymore. Several geometries, including growth on a 2D surface, radial expansions and 3D simulations are possible. The nutrient concentration in the local environment of dividing cells is a function of the nutrient concentration outside the biofilm, the diffusion rate and the consumption by growing cells. At a certain cost, cells may also secrete exopolymeric substances (EPS) or other extracellular products (public goods) such as nutrient scavenging enzymes, which become available to neighboring cells through diffusion and might offer them a benefit. These models have, for example, been applied to determine the outcome of competition (i) between an EPS producing and non-producing strain (Xavier and Foster 2007; Kim et al. 2014), (ii) between strains that differ in their EPS production and quorum sensing (QS) phenotype (Nadell et al. 2008), (iii) between a public good secretor and non-secretor under different levels of genotypic segregation (Nadell, Foster and Xavier 2010; van Gestel et al. 2014), between a public good secretor and a non-secretor in a multispecies biofilm (Mitri, Xavier and Foster 2011) and to study the role of adhesion in biofilm evolution (Schluter et al. 2015). An obvious advantage of in silico models is that all parameters can be easily adapted. The outcome of such experiments indicates that under certain experimental conditions (combination of parameter values) microorganisms have the ability to behave in a certain way. However, it might not be trivial to link these experimental conditions to real-life situations.
In vivo models
In vivo evolution models have mainly focused on P. aeruginosa biofilm isolates from CF patients. As CF is associated with an infection that can last for decades, samples from patients are easy to obtain and are invaluable to study bacterial evolution in biofilms in vivo (Folkesson et al. 2012). Many studies have characterized patient isolates, collected at one time point, which can be used to compare results of in vitro evolution experiments with isolated strains (Hogardt and Heesemann 2010; Akers et al. 2015). These studies however lack the ability to follow evolutionary dynamics. Nevertheless, a few designed evolution studies have been conducted in which patient isolates were collected over time. Examples include a study of P. aeruginosa isolates from the same patient after 6 or 96 months (Smith et al. 2006), the identification of mutations in P. aeruginosa strains that evolved in patients for 39 000 in vivo generations (Huse et al. 2010), the characterization of the evolutionary dynamics of P. aeruginosa in patients over 200 000 generations (Yang et al. 2011), a comparison between mutator and non-mutator strains that evolved in P. aeruginosa isolates, taken over 3 yr from 16 patients (Warren et al. 2011) and the evolution of QS and virulence in P. aeruginosa isolates from 31 patients, collected over 20 days (Kohler, Buckling and van Delden 2009). Whereas the in vivo approach provides the most realistic results (Bjarnsholt et al. 2013), drawbacks are that it might be difficult to repeat the experiment due to lack of sufficient sample material and to differentiate between adaptive mutations and ‘hitchhiking mutations’ (Wong, Rodrigue and Kassen 2012). Furthermore, many parameters in the in vivo experiments are difficult to control due to the heterogeneous conditions. As such, complementation with in vitro evolution experiments is very useful. Strikingly, as described below, the same mutations occurring in biofilm in vitro evolution experiments can often also be found in vivo.
Multispecies models
Only a few multispecies biofilm evolution experiments have been conducted so far. An in vitro two-species evolution experiment was performed with a flow cell biofilm model (Hansen et al. 2007). Additionally, evolution in two-species biofilms has been investigated by using the above described in silico agent-based model (Mitri, Xavier and Foster 2011).
DIVERSIFICATION DURING BIOFILM EVOLUTION
A key finding in many biofilm evolution experiments is that the initial microorganisms undergo diversification that is often not observed in planktonic experiments. These biofilm-specific variants can rapidly emerge and remain stable for many generations. In this section, we provide an overview of these diversifications, before discussing the potential ecological, evolutionary and genetic processes causing biofilm diversification. A focus on genetics reveals the remarkable degree of parallelism, both within evolution experiments themselves and between evolution experiments and real-life biofilms. Finally, we discuss the possible consequences of diversification, which include insurance effects, altered productivity, stabilization of cooperative traits and trade-offs between biofilm and free-living conditions.
Diversification in biofilms is widespread
A growing body of evidence supports the idea that even monospecies biofilms comprise a high level of morphotypic, phenotypic and genotypic heterogeneity. Indeed, in most evolution experiments several genetically stable variants are found to emerge within the first few days of biofilm formation, and these often show striking similarity to variants isolated from real-life biofilms (e.g. Kirisits et al. 2005; Smith et al. 2006; Cramer et al. 2011; Lieberman et al. 2011; Traverse et al. 2012; Savage, Chopra and O'Neill 2013; Penterman et al. 2014).
Morphotypic diversity
In the first evolution studies with biofilms, variants were classified based only on their morphotypic appearance. The observed colony morphologies include smooth, wrinkly, fuzzy, sticky, large, small, mucoid, colored and haemolytic. Morphotypic diversification in relation to biofilm evolution was first described by Korona et al. (1994) in Comomonas sp. and by Rainey and Travisano (1998) in the P. fluorescens radiation in static microcosms. In the latter study, three dominant morphotypic variants arose over the course of a few days: the ancestral broth-colonizing smooth morph, the wrinkly spreaders (WS) that form a biofilm mat at the broth–air interface and the fuzzy spreaders that seem to occupy the anoxic zone of the tubes. Later on, morphotypic diversity was also studied in other evolution experiments with amongst others P. fluorescens (Gomez and Buckling 2013; Kim et al. 2014), P. aeruginosa (Deziel, Comeau and Villemur 2001; Boles, Thoendel and Singh 2004; Kirisits et al. 2005) and Staphylococcus aureus (Yarwood et al. 2007).
A remarkable finding is that the timing and pattern of variant formation often follows a predictable sequence, where some variants can only be isolated after other variants emerge. The rise of such successive and dependent variants could either be an effect of epistasis, where the fitness effect of mutations in one gene is dependent upon mutations in other genes, or could—as explained below—be a consequence of modifications in the environment made by earlier variants (niche construction). The evolutionary dynamics of morphotypic diversification has first been explored by Rainey and Travisano (1998), who found that the three dominant morphs in the P. fluorescens radiation repeatedly occurred in the same order. Subsequently, Koh et al. (2007) showed that evolution of different morphotypes in Serratia marcescens biofilms occurred in a time-dependent manner. A defined order of morphotype evolution was also found in Streptococcus pneumoniae (Allegrucci and Sauer 2007) and S. aureus (Savage, Chopra and O'Neill 2013; Koch et al. 2014) biofilms, as well as in the B. cenocepacia bead transfer model (Poltak and Cooper 2011), where a smooth (S) variant was consistently identified first at ∼150 generations, followed by a ruffled spreader (R) and wrinkly (W) variant after, respectively, 300 and 300–450 generations.
Diversity in other phenotypes
Even though biofilm variants are often first classified by colony morphology, other properties of the evolved variants can also differ strongly, including biofilm formation, dispersion, capsule production, adhesion capacity, antibiotic resistance, antibiotic production, culturability, growth speed and swimming or swarming motility (Boles, Thoendel and Singh 2004; Kirisits et al. 2005; Allegrucci and Sauer 2007; Ponciano et al. 2009; Koch et al. 2014; Penterman et al. 2014). Moreover, specific colony characteristics can correlate with biofilm-related traits. For example, the different wrinkly variants isolated by Rainey and Travisano (1998); Boles et al. (2004); Poltak and Cooper (2011) in evolution experiments with P. fluorescens and B. cenocepacia all show increased attachment, increased biomass and increased cluster formation. Similarly, the small colony variants (SCV) isolated by Kirisits et al. (2005), Allegrucci and Sauer (2007) and McElroy et al. (2014) in P. aeruginosa and S. pneumoniae all show hyperattachment, increased hydrophobicity, increased biomass and more elaborate 3D biofilm structure. In contrast, the large mucoid (capsular) variants isolated by Allegrucci and Sauer (2007) form flat unstructured biofilms, fail to aggregate in liquid culture and adhere poorly to solid surfaces. However, colonies that are indistinguishable from one another on the basis of colony morphology, also often differ in certain phenotypic assays. For example, the SCVs isolated by Boles et al. (2004) showed hyperdetachment and a decreased biomass, in contrast to the earlier described phenotypes that were linked to the SCV morphotype, pointing to a higher underlying complexity and the need for genotypic analyses (Kirisits et al. 2005).
Genotypic diversity
Early studies on diversification occurred before low-cost sequencing and did not include a genomic analysis of the variants. In the first studies that did incorporate sequencing, the initial classification of the variants was still based on morphological differentiation, and the focus was only on one or two loci thought to be a potential cause of the morphotypic differentiation, based on literature data or suppressor analysis. As a first example, the Rainey group used suppressor analysis and sequencing to show that mutations in the wsp locus (which regulates the levels of c-di-GMP and as such production of acetylated cellulose polymer) are responsible for the emergence of mat forming wrinkly spreader (WS) variants in the P. fluorescens radiation in static microcosms (Spiers et al. 2002, 2003; Goymer et al. 2006; Bantinaki et al. 2007). To identify additional mutational routes to WS, this approach was repeated on WS strains evolved in an evolution experiment initiated by strains lacking the wsp operon, and later also by strains lacking both wsp and novel identified loci. This approach revealed two additional loci within which mutation generates the WS phenotype (McDonald et al. 2009). Other examples of genes identified by directed approaches include the cps3D gene which codes for capsulation in S. pneumoniae (Waite, Struthers and Dowson 2001; Allegrucci and Sauer 2007), the accessory gene regulator (agr) gene of the QS system in S. aureus (Yarwood et al. 2007; Koch et al. 2014) and the sigB gene, coding for the alternative sigma factor in S. aureus (Savage, Chopra and O'Neill 2013).
In addition to genotypic characterization of morphotypical variants, the changes in gene expression and the molecular mechanisms underlying phenotypic differentiation have also been studied. Transcriptome analysis was performed for example on P. aeruginosa variants and a change in expression of the psl and pel loci, which have been associated with the adhesion capacity to solid surfaces, was shown to be responsible for the observed phenotypes (Kirisits et al. 2005). In addition, the molecular mechanism underlying differences in the culturability of specific variants from P. aeruginosa biofilms was studied (Penterman et al. 2014).
Whole-genome sequencing and even metagenomic sequencing of evolving populations can now be performed at low cost. This not only allows to further investigate phenotypic variants but also to observe which mutations arise and remain stable without bias to visual variations (Brockhurst, Colegrave and Rozen 2011). A number of studies in this direction have been recently reported. As further discussed throughout the text, whole-genome sequencing was performed for example of phenotypic variants evolved in P. aeruginosa biofilms (Wong, Rodrigue and Kassen 2012) and P. fluorescens (Kim et al. 2014) and S. aureus (Koch et al. 2014) colonies. With the advent of deep-sequencing protocols, it has become possible to obtain a cross-section of within-population genetic diversity (Traverse et al. 2012; Pulido-Tamayo et al. 2015). Traverse et al. (2012) sequenced DNA from mixed communities, the complete genomes of representative clones and specific alleles of representative clones to reconstruct a nearly complete evolutionary history of long-term diversification in the B. cenocepacia bead transfer model. As explained below, striking findings were a recurrent evolution of biofilm specialist morphotypes from generalist types, strong interference competition between contending mutants and multiple adaptive alleles at relatively few loci. Finally, McElroy et al. (2014) performed deep sequencing of evolving biofilm populations of two strains of P. aeruginosa, 18A and PAO1, at two time points during short-term diversification (5–10 generations).
Causes of diversification
Several evolutionary and ecological processes have been proposed to explain diversification in biofilms (Fig. 2), although the relative role of each process remains to be determined.
Figure 2.

Overview of the causes of diversification. (A) ‘Ecological opportunity’: spatial structure and gradients present in a biofilm provide ecological opportunity in the form of vacant niches, which are unused or underutilized by the initially existing genotype(s) and are available to novel genotypes. (a) In a static microcosm, three morphological variants of P. fluorescens each occupy a preferred niche: the ancestral SM variant colonizes the broth, the WS variant forms a biofilm mat at the broth–air interphase and the FS seems to occupy the anoxic zone at the bottom of the microcosm (Rainey and Travisano 1998). (b) GFP-tagged MV emerge at the surface when introduced in a wild-type P. fluorescens colony. Consistently, MV's that spontaneously arise in P. fluorescens colonies push their way to the surface and dominate the colony (Kim et al. 2014). (B) ‘Ecological interaction’: genotypic variants may alter their environment and consequently create new niches. Ecological succession of the (Studded) S, (Ruffled) R and (Wrinkly) W variants of B. cenocepacia is enabled by this process of niche construction. A confocal image of the biofilm structure is shown, in which the entire biofilm is projected in blue, the W morphotype fluoresces red, and the R morphotype fluoresces green. The three endpoint morphotypes were found to partition biofilm space in such a way that the strong biofilm formers R (appearing in yellow) and W (appearing in purple) tightly associate with the beads in heterogeneous clumps and enhance the space available to S, which inhabits a unique layer at the outside of the biofilm on top of R and W (Poltak and Cooper 2011). This spatial segregation was less pronounced at earlier points in evolution (at 350 generations) at which S was found to constitute still a high proportion of the heterogeneous clumps near the bead surface. Thus, the S type appears to have evolved physical displacement from the other types in the biofilms. (C) ‘Population structure and drift’: population fragmentation enhances the influence of genetic drift. Drift can be further amplified because often only cells on the expanding edge of the biofilm can grow, which further reduces effective population size. This genetic drift at the expanding frontiers also drives strong population sectoring which can promote the maintenance of diversity. (a) A fluorescent image of a bacterial colony, grown from a mixture of CFP- and YFP-labeled cells reveals spatial segregation of these neutral genetic markers. Only a very thin active layer of growing cells at the boundary of the colony is able to pass on their genes to the next layer of outwardly growing cells, causing a continual bottleneck. The resulting reduction in effective population size promotes a quick segregation of mutants into monoclonal domains (Hallatschek et al. 2007). (b) A simulation of surface growth that started with a 1:1 mixture of red and blue cells under low growth substrate conditions. Cell color served as a neutral marker for the lineage segregation in space (Nadell, Foster and Xavier 2010). (D) ‘Gradients of stress factors (e.g. antibiotics)’: provide stepping stones that, along with speeding up evolution, also might increase diversification. Indeed, small increases in resistance against the stress factor might be caused by a diversity of mutations of small effect, possibly resulting in a variety of combinations of mutations. Gradients in this biofilm are visualized by imaging the diffusion of the red fluorescent dye rhodamine B into the interior of cell clusters (Rani, Pitts and Stewart 2005; Stewart and Franklin 2008). A movie of the entire sequence can be viewed at https://www.biofilm.montana.edu/resources/movies/2005/2005m01.html. (E) ‘Clonal interference’: competition between simultaneous beneficial mutations is expected to lead to longer fixation times and as a consequence to temporally higher diversity. Mutational dynamics within and among niches are shown over time for three evolving morphotypes. Each color transition represents a new haplotype and clonal interference is observed in the case of haplotypes cooccurring within the same niche, for example the haplotypes of the R morphotype (green) (Traverse et al. 2012). (F) ‘Mutation rate’: mutations are the source of genetic variation for diversifying selection and drift to act on and increased mutation rate enhances the potential for clonal interference, and thus diversity. Mutation frequency in P. aeruginosa biofilms is observed using fluorescence (GFP) inducing reversion mutations and clusters of GFP cells within microcolonies are shown (Conibear, Collins and Webb 2009).
Environmental heterogeneity within biofilms provides ecological opportunity
A biofilm consists of microbial cells that are embedded in a self-produced matrix of extracellular polymeric substances (EPS). The presence of EPS reduces microbial mobility and limits the transfer of chemicals, giving rise to gradients of amongst others nutrients and oxygen (Stewart and Franklin 2008; Nadell, Xavier and Foster 2009). A biofilm is thus a spatially structured and heterogeneous environment, which from an ecological perspective can be seen as a collection of different niches (Chase and Leibold 2003; Kassen and Rainey 2004). A longstanding notion in ecology is the competitive exclusion principle, which states that a single niche can support no more than one genotype (Hardin 1960; Macarthur and Levins 1964). The spatial structure and gradients present in a biofilm provide ecological opportunity in the form of vacant niches, which are unused or underutilized by the the initially existing genotype(s) and are available to novel genotypes (Simpson 1953; Kassen 2009; Yoder et al. 2010). Such vacant niches may also exist in free-living populations e.g. when multiple substitutable substrates are present (Barrett, MacLean and Bell 2005), but are expected to be more abundant within biofilms (Habets et al. 2006; Kassen 2009).
Theory predicts that diversifying selection, generated by resource competition, favors the emergence of ecological niche specialists, a process called niche partitioning or character displacement (Schluter 2000; Kassen 2009). Ecological specialists trade-off their enhanced competitive advantage in one niche against reduced competitive ability in another. The existence of these trade-offs precludes competitive exclusion by one genotype and makes stable coexistence of genotypes possible (Kassen 2002). One hallmark of this hypothesized mechanism of competitive diversification is negative frequency-dependent selection, which means that the direction of natural selection depends on the frequency of genotypes in such a way that rare types have a fitness advantage. The fitness of the different genotypes changes as a negative function of their frequency, such that variation is maintained (Rainey and Travisano 1998; Brockhurst et al. 2006). There is abundant evidence from evolution experiments that the ecological opportunity in biofilms and diversifying selection driven by resource competition play a crucial role in adaptive diversification in biofilms:
In the P. fluorescens radiation in static broth-containing microcosms, a striking relationship was observed between the colony morphotype of the emerging variants and their niche preference: the ancestral smooth morph (SM) variant colonizing the broth, the WS variant, being an EPS hyperproducer and forming a biofilm mat at the broth–air interphase and the fuzzy spreader (FS) seeming to occupy the anoxic zone at the bottom of the microcosm (Rainey and Travisano 1998; Kassen 2009). Later, it was found that the FS in fact forms cellular rafts at the meniscus of the microcosms, which then collapse to the vial bottom and repeatedly reform rafts only to again collapse (Ferguson, Bertels and Rainey 2013). These morphotypes are therefore refered to as ‘ecomorphs’. Moreover, the emergence and maintenance of diversity was shown to require spatially structured (static) microcosms. Indeed, when spatial structure was destroyed by shaking the microcosms, thereby eliminating the multiplicity of niches, diversity was lost from previously diversified populations and did not evolve from genetically uniform ones (Rainey and Travisano 1998; Buckling et al. 2000; Kassen, Llewellyn and Rainey 2004; Hall et al. 2012). Further, support for the need of ecological opportunity was provided by manipulating the ecological opportunity by inoculating the static microcosms with different combinations of distinct niche specialists prior to introduction of the ancestral smooth morph. The extent of diversification clearly decreased as the ecological opportunity decreased (Brockhurst et al. 2007; Gomez and Buckling 2013). An important consequence of this dependency of diversification on prior niche occupation is that immigration history will also influence diversification (Fukami et al. 2007). The role of competitive trade-offs was supported by the observation of negative frequency-dependent selection acting in pairwise competitions between the ancestor and derived morphs (Rainey and Travisano 1998). Competition for oxygen was shown to selectively favor the WS variants (Koza et al. 2011), which firmly attach to each other and form a self-supporting biofilm mat with superior access to oxygen (Rainey and Travisano 1998; Rainey and Rainey 2003). However, once the mat becomes too heavy it sinks, explaining the frequency-dependent nature of selection (Rainey and Travisano 1998). An important note is that a substantial level of morphological and metabolic variation occurs within each ecomorph (mainly within the WS biofilm mat), which is also associated with a variance in fitness (MacLean, Bell and Rainey 2004; Bantinaki et al. 2007; McDonald et al. 2009). This intraecomorph diversity rapidly increases over the first few days, after which it is slowly lost (Fukami et al. 2007; Meyer et al. 2011). This so-called overshooting dynamics was shown to be caused by persistent diversifying selection, which first drives diversification into ecomorphs and intraecomorph variants with distinct resource use, but afterwards leads to loss of ecologically intermediate intraecomorph variants with lower fitness than more extreme types (Meyer et al. 2011).
Where the previous study illustrates the role of spatial heterogeneity, ecological opportunity and diversifying selection in the origin of a biofilm in a static microcosm, similar processes are important as well for evolution inside biofilm communities. Xavier and Foster (2007) used an agent-based model to predict that gradients of oxygen in biofilms can provide a strong selection pressure for copious exopolymer production. This model includes a biochemical description of the carbon fluxes for growth and polymer production, and explicitly calculates diffusion–reaction effects and the resulting solute gradients in the biofilm. It was found that secretion of extracellular polymers by a cell allows it to push descendants up and out into better oxygen conditions. At the same time polymer production harms neighboring non-producers by suffocating them. This effect is analogous to the evolution of woody tissue in plants to grow tall and gain better access to light. Interestingly, under certain conditions, a rare polymer producer can invade a population of non-producers, while the reverse invasion of rare non-producers also happens. This negative frequency-dependent selection predicts a stable coexistence of producers and non-producers and can easily be explained by the suffocation effect, which causes decreasing opportunity for producers to overgrow non-producers when their frequency increases. Although phrased in terms of oxygen gradients, similar results can be expected whenever the biofilm forms a barrier to a limiting resource. Kim et al. (2014) provided empirical evidence for this concept, by showing that in colonies of P. fluorescens spontaneous mucoid variants (MV) repeatedly arise that push their way to the surface and dominate the colony. The MV do not gain their advantage from a faster intrinsic growth rate, but use secretions to collectively expand and push themselves to the surface of the wild-type colony. Consistently, the MV do not have an advantage in competition with wild-type cells when structure is removed by growing them in liquid cultures or by regularly mixing up the colony.
Kirisits et al. (2005) reported that the isolation of small, wrinkled, sticky colony variants (ST) of P. aeruginosa PAO1 from a tube biofilm reactor. Again these variants did not evolve in a shaken liquid culture, where spatial structure was absent. Despite the ST phenotype's increased adherence in short-term adhesion assays, its tendency to stay near the point of initial attachment in a biofilm, and its ability to form large cellular aggregates, it did not predominate the biofilm, but accounted for less than 20% of the population. Even if the biofilm reactor was inoculated with high ST-to-WT ratios, the biofilm always reached a steady-state value around 30%. This observation of negative frequency-dependent selection supports the hypothesis that ST only has a competitive advantage in a particular niche within the biofilm.
Finally, niche partitioning could also explain the evolution of certain genotypes that at first sight seem to have acquired deleterious mutations. Examples include the stable small non-mucoid variant in a S. pneumoniae biofilm, which lacks capsule production, important for the pathogenicity in terms of being able to evade the immune system (Allegrucci and Sauer 2007) and the culture-impaired P. aeruginosa variants, which are killed by a self-produced toxin under planktonic conditions (Penterman et al. 2014). A possible explanation for why these variants could persist is that they only have an advantage compared to the wild type in a specific niche in the biofilm. Consistent with this idea, these variants did not emerge in shaking liquid cultures.
Environmental modification promotes facilitation and ecological succession
Once separated in niche space, the emerging genotypic variants may alter their environment by changing the gradients in existing biotic and abiotic factors or by providing new resources (e.g. metabolic by-products on which new specialists can feed or increased available space), among other factors. This process of environmental modification by organisms has been called ‘niche construction’ (Day, Laland and Odling-Smee 2003) and may increase diversity by providing novel niches available to emerging genotypes (Kassen and Rainey 2004; Kassen 2009; Poltak and Cooper 2011). As such positive, facilitative interactions between genotypes may arise, in which one genotype depends on the presence of the other (Day and Young 2004). Furthermore, changes in the environment can lead to ecological succession (Poltak and Cooper 2011). This phenomenon can be defined using three parameters: (i) the process is orderly, reasonably directional and predictable, (ii) it results from modification of the environment by the community and (iii) it leads to increased productivity for the population (Odum 1975).
A first piece of data suggesting the importance of facilitation comes from the P. fluorescens radiation in static broth-containing microcosms (Rainey and Travisano 1998; Day and Young 2004). As described above, the ability of the three morphotypes to invade a population of a different morphotype from rare was evaluated. All pairwise combinations showed negative frequency-dependent selection, except for the FS type, which cannot invade a population of WS. As such, the ability of FS to emerge after WS in the radiation and their stable coexistence suggest a facilitative interaction of FS with the ancestral smooth type SM.
- A second example indicating the importance of ecological succession has been provided by Poltak and Cooper (2011). In their long-term experiment (1050 generations) of B. cenocepacia biofilm propagation using the bead transfer model, three morphotypes evolved in a common pattern of succession: the smooth or studded morphotype (S) at 150 generations, the ruffled spreader (R) at 300 generations and the wrinkly morphotype (W) between 300 and 450 generations. Again the emergence and maintenance of diversity was demonstrated to require spatial structure, since diversity did not evolve in planktonic control cultures and eliminating structure from previously diversified populations by removing the bead significantly reduced diversity. However, positive interactions between the evolving morphotypes were also shown to be important and at the basis of a higher cellular productivity than expected from the productivity of the constituent morphotypes in monoculture:
- Evaluation of variants isolated from the end of the experiment indicated that cross-feeding plays an important role in this system. Only S is self-sustaining, while neither R nor W can grow in their own supernatants and greatly depend on the metabolic by-products of the other, previously evolved, community members (Poltak and Cooper 2011).
-
The three endpoint morphotypes were found to partition biofilm space in such a way that the strong biofilm formers R and W tightly associate with the beads in heterogeneous clumps and enhance the space available to S, which inhabits a unique layer at the outside of the biofilm on top of R and W (Poltak and Cooper 2011). This spatial segregation was less pronounced at earlier points in evolution (at 350 generation) at which S was found to constitute still a high proportion of the heterogeneous clumps near the bead surface. Thus, the S type appears to have evolved physical displacement from the other types in the biofilms. As expected, this process of niche construction and character displacement was shown to coincide with a reduced competition and enhanced community productivity. Also, trade-offs were associated with niche adaptation since the biofilm output of the late morphotypes inversely correlated with their growth rate (Ellis et al. 2015).Overall, the observed dynamics may be interpreted as one of ecological succession enabled by niche construction. S likely arose first because it could better exploit the selective environment than its ancestors because of its higher growth rate and biofilm productivity. R and W may have evolved in response by consuming specific metabolites provided by S. Finally, S evolved to occupy a specific biofilm layer, with increased availability of space, provided by R and W.
Another example is the emergence of six genetically stable morphological variants in a predictable sequence throughout the development of microcolony-type biofilms of S. marcescens (Koh et al. 2007). The biofilm development progresses through a series of stages during 10 days (formation of microcolonies, emergence of hollow microcolonies, rapid biofilm expansion, cell death and biofilm detachment) and the diversification is strongly correlated with these stages. A sticky-smooth variant (SSV) consistently occurred during hollow microcolony formation, four morphotypes appeared during biofilm expansion, and a smooth-ultramucoid variant (SUMV) (which is derived from SSV) consistently emerged when cell death and biofilm detachment occurred. Moreover, the variants show specialized colonization traits (motility, attachment, biofilm morphology), which are associated with the biofilm stage at which they were isolated. These results again suggest a dynamic of ecological succession in which previous developed morphotypes alter the environment and set the stage for emergence of novel types (Koh et al. 2007).
Ecological competition causes environmental modification and diversification
Ecological competition between emerging variants can also cause environmental modification. As a response to this competitive environment, strains can further evolve to defend themselves. Koch et al. (2014) for instance showed how a single staphylococcal isolate spontaneously diversified by evolving new competitive phenotypes in a coevolutionary arms race. A first evolved strain produced a toxic bacteriocin active against the parent strain. The parent strain counter-adapted to this challenge by generating a second new strain resistant to the bacteriocin.
Population structure promotes fixation of diverse mutations of smaller effect
In a spatially structured environment such as a biofilm, the population may be subdivided into a number of more or less independently evolving subpopulations, a phenomenon called population fragmentation (Habets et al. 2006). From a theoretical point of view, population fragmentation has two key effects that can lead to enhanced diversity (Habets et al. 2006). The first is that small populations experience a higher influence of genetic drift (Wright 1931; Gomez and Buckling 2013; Habets et al. 2006). In biofilms, drift can be further amplified because often only cells on the expanding edge of the biofilm can grow, which further reduces effective population size (Golding, Cohen and Ben-Jacob 1999; Hallatschek et al. 2007). This genetic drift at the expanding frontiers also drives strong population sectoring which can promote the maintenance of diversity (Klopfstein, Currat and Excoffier 2006; Hallatschek et al. 2007; Hallatschek and Nelson 2008). Key to this process is nutrient limitation that ensures that only a subset of the cells in the biofilm can grow at the expanding edge (Nadell, Foster and Xavier 2010; Mitri, Clarke and Foster 2015). A potential second effect of population fragmentation in biofilms is that there can be less access to rare beneficial mutations of large effect. As such, the smaller sub-populations in biofilms may tend to fix beneficial mutations of smaller effect. And because these mutations are more abundant, spatially subdivided populations are likely to follow more diverse adaptive routes (Rozen, de Visser and Gerrish 2002; Rozen et al. 2008). Moreover, this fixation of mutations of smaller effect is predicted to slow down the rate of adaptation. Interestingly, the occurrence of slow adaptation and a diversity of adaptive routes may, in the longer term, lead to improved adaptation if it allows the population to discover a rare or complex strategy that would have been lost in a homogeneous fast adapting population (Miralles et al. 1999; Nahum et al. 2015).
To provide experimental evidence for the role of population fragmentation in biofilm diversification, Habets et al. (2006) propagated populations founded by a single genotype of Escherichia coli for 900 generations in either a homogeneous environment (shaken liquid culture), a heterogeneous environment with a population structure that was kept intact (colony on agar plate, transferred by a stamp) or a heterogeneous environment in which the population structure was destroyed each day (colony on agar plate, mixed every day before transfer to a new plate). In line with theory, population structure enhanced adaptive radiation, since significant diversity in catabolic activity among evolved clones was only observed in spatially structured environments with intact population structure. Moreover, negative frequency-dependent fitness interactions were found, suggesting that diversity is stably maintained. An interesting note is that the fitness compared to the ancestor was lower for populations evolved in the heterogeneous environment with intact population structure than those with destroyed population structure, as predicted in case of fixation of beneficial mutations of smaller effect. This study did not address whether this heterogeneity can ultimately improve the process of adaptation. However, another recent study using liquid culture did find that population subdivision can improve adaptation in E. coli experimental evolution (Nahum et al. 2015).
Presence of gradients provides stepping stones for diverse mutations of smaller effect
Often several mutations are required for a bacterium to obtain significant resistance to stresses like antibiotics (Lipsitch 2001). In a homogeneous environment, a bacterium has to rapidly acquire all these mutations to be able to survive. The presence of gradients in the stress factors however might provide stepping stones (also called ‘sanctuaries’ or ‘resistance-selective environments’) allowing these mutations to be selected one by one (Baquero et al. 1998; Baquero and Coque 2014). Ecologically, these functions by the sanctuary providing a source of immigrants that continually seed the toxic environment giving the population many opportunities to adapt (Perron, Gonzalez and Buckling 2007). Zhang et al. (2011) used an ingenious assay to demonstrate that concentration gradients can indeed strongly promote antibiotic resistance development. By means of a complex microfluidic device, a smoothly varying concentration gradient of ciprofloxacin was set up in a two-dimensional landscape of connected hexagonal wells. Remarkably, high resistance, due to four single-nucleotide substitutions in three genes, evolved in only 10 h after inoculation of the E. coli bacteria in the center of the device.
Hermsen, Deris and Hwa (2012) developed a mathematical model, called the ‘staircase model’ to explain how antibiotic gradients can promote adaptation. This suggests that the gradients allow resistant mutants to evade competition and circumvent the slow process of fixation by invading compartments with higher drug concentrations, where less resistant strains cannot subsist. Although the role of this mechanism within biofilms remains to be proven, the common presence of gradients within biofilms means it may be relevant in nature (Stewart and Franklin 2008). How could this process, in addition to increasing the rate of adaptation, also increase diversification? First, small increases in resistance might be caused by a diversity of mutations of small effect (Martinez and Baquero 2000), possibly resulting in a variety of combinations of mutations. Second, antibiotic resistance (or stress resistance in general) often requires adaptations that carry a fitness cost in the absence of the antibiotic (Andersson 2006). The staircase model predicts that this fitness trade-off between niches with high and low antibiotic concentration will prevent resistant strains from outcompeting less-resistant strains in regions of lower antibiotic concentration, which may stabilize diversity (Hermsen, Deris and Hwa 2012).
Clonal interference increases fixation times and enhances temporal diversity
The traditional view of evolution by natural selection is that adaptation occurs via rare beneficial ‘driver’ mutations, which will sequentially reach fixation via selective sweeps that purge all variation and preserve the clonal genotype (i.e. periodic selection). However, recent evolution experiments have shown that beneficial mutation rates are typically high enough so that multiple driver populations can cooccur. Competition between these alternative beneficial mutations, a phenomenon called clonal interference, is expected to lead to longer fixation times and as a consequence to temporally higher diversity (Greaves and Maley 2012; Barrick and Lenski 2013). Theory suggests that such competition can also produce a simultaneous sweep of multiple linked mutations, because more than one beneficial mutation may be required to prevail in competition (Park and Krug 2007; Sniegowski and Gerrish 2010). In silico simulations have predicted that clonal interference is more prevalent with spatial structure than without, due to the slow wave-like spread of beneficial mutations through space in structured populations, compared to the fast Malthusian sweeps in well-mixed populations (Martens and Hallatschek 2011; Martens et al. 2011). Consistently, Traverse et al. (2012) observed a large prevalence of clonal interference in the B. cenocepacia bead transfer model, since new mutants coexisted with each other for hundreds of generations.
Increased mutation rate and horizontal gene transfer
An increased mutation rate and horizontal gene transfer (HGT) may also promote diversification in biofilms. Indeed, bacteria in a biofilm can exhibit a 2- to 60-fold increase in mutation rate compared to planktonic cultures (Loewe, Textor and Scherer 2003; Conibear, Collins and Webb 2009; Ryder, Chopra and O'Neill 2012). Mutation rates are historically seen largely invariant and driven by inevitable errors in DNA replication (Drake et al. 1998). However, it is now appreciated that bacterial mutation rates and rates of horizontal DNA transfer can increase greatly under certain stresses, with the effect that the rate of adaptation will increase under conditions where a cell is not well adapted to its environment. Additionally, mutational hotspots can occur because stress-induced mutations are linked to local events such as DNA breaks (Galhardo, Hastings and Rosenberg 2007; Rosenberg and Queitsch 2014).
Oxygen stress can be a reason for increased mutation rate in biofilms, caused by deleterious effects on the DNA by reactive oxygen species (ROS) themselves, or by the induction of the ROS stress response. The presence of ROS has been shown to be a source of genotypic, morphotypic and/or phenotypic diversification in S. pneumoniae (Allegrucci and Sauer 2007), P. aeruginosa (Boles and Singh 2008; Driffield et al. 2008; Conibear, Collins and Webb 2009; Starkey et al. 2009), S. aureus (Ryder, Chopra and O'Neill 2012) and E. coli (Saint-Ruf et al. 2014) biofilms. Addition of antioxidants or the use of mutants affected in ROS production reduced diversification in these studies; addition of hydrogen peroxide or the use of oxidant hypersensitive mutants increased diversification. A possible explanation for increased oxygen stress in biofilms is the presence of steep gradients, which rapidly generate stress by accumulation of toxic metabolites (Stewart and Franklin 2008; Conibear, Collins and Webb 2009; Saint-Ruf et al. 2014). Consistently, genes involved in the oxidative stress response were found to be upregulated in E. coli colonies (Saint-Ruf et al. 2014) and S. aureus biofilms (Ryder, Chopra and O'Neill 2012). On the contrary, in P. aeruginosa biofilms it was found that several genes coding for oxidative DNA damage protection enzymes were downregulated (Driffield et al. 2008). This imbalance between oxidant burden and antioxidant defense might make the biofilm cells even more susceptible to oxygen stress.
The mechanisms by which ROS induce DNA damage in biofilms are not completely clear. In E. coli, ROS can induce the general stress RpoS response and the SOS response, which are required in stress-induced mutagenesis systems. However, Boles and Singh (2008) found none of these responses to be necessary for the generation of variation in their S. aureus biofilm model. Instead, it was shown that ROS can cause double-stranded breaks in the DNA, which give rise to genetic variants when they are repaired by a mutagenic mechanism involving combinatorial DNA repair genes (amongst others recA) (Rosenberg 2001; Boles, Thoendel and Singh 2004). An interesting study on the relative role of ROS-induced mutation rate and diversifying selection in the generation of morphotypic variation was conducted by Allegrucci and Sauer (2008). Addition of hydrogen peroxide to planktonic S. pneumomiae cultures was shown to generate a similar diversity as observed in biofilms: the same morphotypes arose, in the same order and in similar frequencies. These results suggest that at least in this system, morphotypic diversification is not due to selection for variants better equipped for adherence and biofilm growth but instead is likely the result of increased mutation rates induced by oxidative stress conditions. In contrast, Wrande, Roth and Hughes (2008) proved the accumulation of rifampicin-resistant (RifR) mutants, in Salmonella enterica and E. coli colonies to be caused by selection mechanisms rather than stress-induced mutagenesis, which was previously thought to be the cause of these observed mutations (Taddei, Matic and Radman 1995). In these colonies, the general mutation rate was not enhanced as only an increase in RifR mutants, which have a growth advantage, was observed (Wrande, Roth and Hughes 2008). Next, to the ROS-mediated increased mutation rate, nutrient limitation can also be a cause of stress-mediated mutations, as observed in E. coli biofilms (Ponciano et al. 2009). Finally, other stress responses that induce mutations in bacteria have been reviewed by Galhardo, Hastings and Rosenberg (2007).
Mutation rate itself can also evolve independently of stress responses. Indeed, hypermutator phenotypes with an increased mutation rate can emerge. Natural selection can favor these hypermutator strains when adapting to a new environment because the hypermutator genes can hitchhike with beneficial mutations they generate (Metzgar and Wills 2000; Denamur and Matic 2006). In addition, spatial heterogeneity can favour such hypermutators by allowing them to spread to favorable environments should conditions change again (Travis and Travis 2004). Consistent with these ideas, hypermutator phenotypes are found in many biofilms. For example, by studying 90 isolates that were obtained over time from 29 CF patients, it was found that 17% of these strains contained a mutation in the MutS-MutL mismatch repair system, causing a mutator phenotype. Other mutations that had an effect on the virulence and drug efflux pumps were found as well, possibly under influence of the mutator phenotype (Smith et al. 2006; Mena et al. 2008). In order to better understand the impact of mutator phenotypes, Lujan et al. (2011) competed artificially constructed mutS mutants of P. aeruginosa with the wild type in a flow cell biofilm model and in planktonic phase. The mutators showed an enhanced adaptability over the wild type when grown in biofilms but not as planktonic cells. This advantage was associated with enhanced micro-colony development and biofilm architecture by the mutators, as also seen by Conibear, Collins and Webb (2009), and by a faster and more extensive phenotypic diversification. Similarly, Saint-Ruf et al. (2014) observed a faster and more extensive diversification in E. coli colonies founded by mutS and mutT mutator strains compared to those founded by wild-type cells. However, mutator phenotypes are self-limiting due to the high risk of deleterious mutations (Denamur and Matic 2006).
HGT is yet another proposed cause of the increased mutation rate in biofilms, because the HGT rate is often higher in biofilms compared with planktonic cultures, due to a higher conjugation rate, a higher transformation rate, an increased plasmid stability, high cell density, stable environment for physical cell–cell contact and high amount of eDNA present in the matrix (Madsen et al. 2012; Burmolle et al. 2014). Furthermore, Antonova and Hammer (2011) showed that HGT can be induced by autoinducers in a mixed species biofilm containing Vibrio cholera. HGT is thought to be an important process in the acquisition of mutations that induce social phenotypes because genes of social traits can be acquired (Mitri and Foster 2013). Although in most studies the increased HGT is not mentioned as possible explanation, it is highly plausible that this effect is an important cause of diversification in many biofilm evolution experiments. In addition, other possible causes of an increased mutation rate include genomic rearrangements and mobility of insertion sequences (Saint-Ruf et al. 2014).
Genetic mechanisms behind adaptive diversification
The genetic mechanisms underlying adaptive diversification have been studied in most detail in the P. fluorescence radiation in static microcosms and the B. cenocepacia bead transfer model. These studies also revealed insight into the molecular basis of fitness trade-offs across niches and the competitive dynamics of genotypes within and across niches.
Antagonistic pleiotropy in the P. fluorescens radiation in static microcoms
The transition from smooth to WS in the P. fluorescens radiation in static broth-containing microcosms and the associated invasion of the air–broth interface is caused by the constitutive activation of a pathway for the production of a cellulose-like polymer, encoded by the wss operon. The activation of Wss occurs through a range of loss-of-function mutations in the methylesterase WspF (part of the Wsp signal transduction pathway), leading to increased methylation of the Wsp receiver complex and a constitutive activation (through phosphorylation) of the diguanylate cyclase response regulator WspR, resulting in enhanced levels of cyclic diguanosine monophosphate (c-di-GMP) (Spiers et al. 2002, 2003; Goymer et al. 2006; Bantinaki et al. 2007). C-di-GMP is an intracellular signalling molecule that plays a central role in the regulation of motility, virulence and biofilm formation in P. fluorescens and many other bacteria (Hengge 2009; Romling, Galperin and Gomelsky 2013). As such enhanced WspR activity results in the constitutive activation of Wss and an increased production of acetylated cellulose needed for forming the biofilm mat (Spiers et al. 2003). Interestingly, the genetically distinct mutations leading to the WS morphotype are associated with a diverse array of fitness values, which sets the stage for divergence by selection among independently arisen WS (Bantinaki et al. 2007).
The evolution of a diverse community in a heterogeneous environment requires that no single generalist type is able to outcompete all the others in all niches of the environment. The evolution of such generalists is thought to be unlikely, because trade-offs in fitness across environments are expected, either because of antagonistic pleiotropy (alleles beneficial in one environment are deleterious in others) or mutation accumulation (mutations neutral in the environment of selection that disrupt function in novel environments) (Kassen 2009). Antagonistic pleiotropy is clearly implicated in the emergence of the biofilm mat-forming WS in the P. fluorescens radiation in static microcosms. Biolog fitness assays and proteome analysis indicated that WS genotypes express catabolic defects, which could be attributed to the same mutations causing the overexpression of acetylated cellulose needed for mat-formation (MacLean, Bell and Rainey 2004; Knight et al. 2006). Interestingly, prolonged selection in the spatially structured microcosms caused the cost of adaptation to decline, without loss of the benefits associated with adaptation, an observation which could either be explained by compensatory adaption with specialist lineages or clonal competition among specialist lineages.
Competition occurs within and between niches in the B. cenocepacia bead transfer model
Metagenome sequencing of the diversifying population in the B. cenocepacia bead transfer model revealed that changes in c-di-GMP metabolism are also underlying the emergence of the S, R and W morphotypes in this system and are at the basis of their distinct biofilm attachment patterns (and thus niche preference) (Traverse et al. 2012). The initial S and R morphotypes originated from the ancestor by two different SNPs in the yciR gene, which encodes both a diguanylate cyclase (synthesizing c-di-GMP) and phosphodiesterase (degrading c-di-GMP) domain. Alternative yciR alleles thus define morphological and ecological differences in this system. Finally, the initial W morphotype acquired a mutation in wspA, part of the Wsp receiver complex described above. In these, mutations were associated with a strong increase in frequency of each morphotype, and the enhanced access to beneficial mutations resulted in further adaptation of each morphotype. The S lineage for example consequently accumulated (i) a SNP in a TCA cycle enzyme, (ii) a large deletion that removed the previously mutated yciR locus, (iv) a single bp deletion in manC which promoted biofilm production, (v) a change in the promoter of the iron storage gene bacterioferritin that increased its expression and (vi) a deletion of 49 diverse genes. An interesting finding was that different new mutants coexisted with each other for hundreds of generations (a dynamics called clonal interference), which contrasts with the sequential replacement that often occurs in unstructured environments (periodic selection). As described above this provides an additional source of genetic diversity.
An intriguing observation was that competition between genotypes (clonal interference) did not only occur within niches but also between niches, suggesting a more fluid community structure (Traverse et al. 2012). Indeed, the biofilm specialist types R and W did recurrently evolve anew from the single evolving S lineage. Remarkably, a W haplotype that gained a fitness advantage by a mutation in the promoter of bacterioferritin was not able to spread beyond this niche. Instead, new mutants of S with upregulated bacterioferritin evolved as a response and succeeded to invade other niches again. Only the S lineage, which balances fitness between planktonic and biofilm conditions (and can be considered as a generalist) was thus able to spawn new R and W types at least seven times. The S lineage may be more evolvable because of its larger population size, absence of negative epistasis, or larger ecological breadth. As a consequence, the endpoint S, R and W morphotypes share a common adaptive haplotype derived from the early S clone (with mutations in TCA cycle, yciR, manC and bacterioferritin) and have only a few mutations distinguishing them from each other. The endpoint R type evolved from S by four mutations, whereas the endpoint W types evolved by single mutations in wspA and wspE, similar to the original W lineage. Finally, the S lineage experienced a further 49-gene deletion.
Parallelism in diversification
The increasing amount of reports on parallel evolution in nature and in vitro show that evolution can be highly reproducible (Gerstein, Lo and Otto 2012; Meyer et al. 2012; Herron and Doebeli 2013; Stern 2013). Also biofilm evolution experiments have indicated a striking amount of parallelism at the morphotypic, phenotypic and genotypic level, both between replicate lineages within the same evolution experiment, between different evolution experiments (performed in different labs) and importantly, between in vitro evolution experiments and evolution in chronic infections. The rapidity and repeatability of biofilm evolution are indicative of strong selective pressures within biofilms. The exceptional parallelism found between laboratory-derived variants and in vivo isolates indicates that some of the same forces that drive biofilm adaptation in vitro also contribute to adaptation during chronic infections.
Replicate lineages within the same evolution experiment
In the P. fluorescens radiation in static broth-containing microcosms, the three dominant morphological classes (S, WS and FS) showed highly repeatable evolutionary dynamics across replicate populations. However, slight variation was always encountered within the morphological classes across microcosms. The mutational origins of 26 independent WS genotypes were unraveled by suppressor analysis and sequencing (Bantinaki et al. 2007; McDonald et al. 2009). All mutations reside exclusively in one of three pathways (Wsp, Aws and Mws), each harboring a diguanylate cyclase responsible for the production of c-di-GMP. The majority of mutations were found to cause loss-of-function changes in a few negative regulators of the diguanylate cyclases, as such enhancing the levels of c-di-GMP and acetylated cellulose required to form the biofilm mat. Eliminating the three pathways from the P. fluorescens genome and replaying evolution revealed 13 new mutational pathways that allow realization of the WS phenotype. Remarkably, all 13 pathways harbor diguanylate cyclases and overexpression of c-di-GMP and acetylated cellulose is thought to be the cause of the WS phenotype in each case. WS morphotypes with mutations in these new pathways however took longer to arise, which explains why they were not present among the 26 WS genotypes derived from the ancestral strain (Lind, Farr and Rainey 2015). Since these WS types arisen via the new pathways do not differ in fitness relative to the earlier identified WS types, their later appearance cannot be attributed to a selective disadvantage. Genetic constraints, mediated by genetic architecture and the mutational target size, have therefore been proposed to be of crucial importance as well to explain genetic parallel evolution (McDonald et al. 2009; Lind, Farr and Rainey 2015). Indeed, certain pathways have a greater capacity than others to translate mutation into phenotypic variation, and a number of principles have been derived from the data: evolution proceeds firstly via pathways subject to negative regulation, then via promoter mutations and gene fusions, and finally via activation by intragenic gain-of-function mutations (Lind, Farr and Rainey 2015).
In the B. cenocepacia bead transfer model, each of the six replicate biofilm populations underwent a common pattern of adaptive morphological diversification, in which the three ecologically distinct morphotypes (S, R and W) arose in the same order of succession. Although a detailed evolutionary model was only assembled for one lineage (see above), mutations associated with adaptation were identified by metagenome sequencing in early and late samples of all six lineages. An exceptional parallelism among adaptive targets was observed with at least 26 mutations involved in c-di-GMP metabolism (Seven mutations in yciR, 18 mutations in the wsp operon), 20 mutations in a gene cluster involved in LPS biosynthesis, and several mutations in the TCA cycle, RNA polymerase subunits rpoC and ropD and a galactose metabolism operon (Poltak and Cooper 2011; Traverse et al. 2012). This parallelism can likely be attributed to strong natural selection. Indeed, metagenome sequencing revealed several features that suggest a strong role for selection in this system as follows. (i) Specific mutations rapidly rose to high frequency (e.g. five mutations became detectable in the S lineage within 315 generations). (ii) Mutations in coding sequences were mostly nonsynonymous, intergenic mutations were associated with likely promoters, and deletions affected genes that were plausible targets of selection. (iii) The rapid rise of the different lineages combined with the low per-genome mutation rate should theoretically hinder hitchhiking of neutral mutations.
McElroy et al. (2014) sequenced the metagenome of populations of two P. aeruginosa strains (the lab strain PAO1 and the clinical isolate 18A) at two time points during short-term biofilm evolution (∼5.3 and 10.3 generations in total for PAO1 and 18A, respectively). Also, here a strong within-strain parallel evolution was observed, often involving identical nucleotides, which—in agreement with an enhanced mutation rate within biofilms–indicates that the mutation rate was not limiting. In contrast, there was an almost complete lack of non-coding and synonymous mutations, suggesting that the majority of the P. aeruginosa genome is constrained by negative selection, with strong positive selection acting on an accessory subset of genes facilitating adaptation to the in vitro biofilm lifestyle (McElroy et al. 2014). Parallel evolution at the nucleotide level was a striking finding, as this is considered to be a rare event for bacteria (Dettman et al. 2012).
Finally, Kim et al. (2014) showed that in colonies of P. fluorescens spontaneous MV repeatedly arise that produce secretions to push their way to the surface and gain better access to oxygen. Analysis of over 500 independent adaptation events revealed a striking level of parallelism because all of them occurred through mutation of a single repressor of secretions, RsmE. For many positions, the same mutation was even found multiple times, up to a maximum of 69 cases of a particular SNP. Since none of the mutations were synonymous and the mutation rate of rsmE does not exceed the genome average, this parallelism was attributed to strong natural selection. Consistently, all mutants were able to outcompete the wild type, although a strong variability in competitive ability among the mutants was observed. Interestingly, generation of a fine-scale map of the mutations provided an explanation for this variability in competitiveness in terms of molecular structure and function.
Comparison of different in vitro evolution experiments
Colony types with similar characteristics have been isolated in different evolution experiments, even across different bacterial species. SCV were for example isolated by Kirisits et al. (2005), Allegrucci and Sauer (2007) and McElroy et al. (2014) in P. aeruginosa and S. pneumoniae. As discussed above, in some cases these SCV also show similarity in other phenotypes, such as hyperattachment, increased biomass and more elaborate 3D biofilm structure. Another example is represented by the wrinkly variants of P. fluorescens and B. cenocepacia isolated in different evolution experiments by Rainey and Travisano (1998), Boles et al. (2004) and Poltak and Cooper (2011), which all show increased attachment, increased biomass and increased cluster formation.
Strong parallelism between experiments was also observed at the genomic and molecular level. Despite the limited number of genomic studies, changes in c-di-GMP metabolism, often mediated by mutations in the wsp operon, were found to play a major role in morphotypic and phenotypic differentiation in at least three independent evolution experiments (McDonald et al. 2009; Traverse et al. 2012; McElroy et al. 2014). Additionally, mutations that caused an effect on the LPS due to a mutation in glycosyltransferase genes were found both in P. fluorescens (fuzY) (Ferguson, Bertels and Rainey 2013), B. cenocepacia (manC) (Traverse et al. 2012) and P. aeruginosa (wbpJ) (Penterman et al. 2014).
Comparison with naturally occurring biofilms
For some of the in vitro studies, similar variants could be found in isolates from real-life biofilms. For example, the laboratory-derived SCV (sticky variants) of P. aeruginosa described by Kirisits et al. (2005) are similar in colony morphology to clinical isolates from a CF lung and also other phenotypes are consistent, including aggregation in liquid culture, hyperadherence and reduced motility. Also for the experimentally evolved agr and sigB variants in S. aureus, clinical isolates containing mutations in the same genes have been isolated (Karlsson-Kanth et al. 2006; Traber et al. 2008; Savage, Chopra and O'Neill 2013). Furthermore, variants with an altered outer-membrane lipopolysaccharide structure that emerged in an evolution experiment with P. aeruginosa have been proposed to also occur in isolates from CF patients (Penterman et al. 2014). Indeed, mutations in the same genes were detected both in the variants from the in vitro experiment and isolates from CF patients (Warren et al. 2011; Davis et al. 2013). A final example are the morphological variants that occurred in the B. cenocepacia bead transfer model by Poltak and Cooper (2011), which were also found in CF patients isolates (Haussler et al. 2003). The same four classes of mutations found in the replicate in vitro populations (c-di-GMP metabolism, LPS gene cluster, Transcription and TCA cycle) were also identified in studies of isolates of B. dolosa and P. aeruginosa that evolved during CF infections (Smith et al. 2006; Cramer et al. 2011; Lieberman et al. 2011; Traverse et al. 2012). In the isolates, many more mutations were observed; however, the amount of mutation overlap was more than expected by chance, which suggests convergent evolution. This parallelism between variation in in vitro biofilm evolution experiments and isolates from chronic infections, suggests that adaptation during chronic infections may be at least partly driven by selection in biofilms.
Parallelism has also been observed between mutations in different in vivo P. aeruginosa (Smith et al. 2006; Cramer et al. 2011; Warren et al. 2011; Yang et al. 2011) and B. dolosa (Lieberman et al. 2011) isolates from different CF patients. Warren et al. (2011) found mutations in different individual isolates that decrease the invasion ability of P. aeruginosa in the host and enhance biofilm formation and the occurrence of multidrug efflux pumps. Additionally, both the loss of ability to catabolize 4-hydroxyphenylacetic acid and the increased resistance to ciprofloxacin evolved in individual isolates (Yang et al. 2011). Furthermore, Smith et al. (2006) and Cramer et al. (2011) both found mutations in genes encoding for multidrug efflux pumps, chemotaxis, motility, virulence or QS in different isolates. Finally, transcriptome analysis indicated that change in gene expression between in vivo P. aeruginosa isolates also displayed parallelism. Examples include downregulation of pili synthesis and upregulation of biofilm formation (Huse et al. 2010) and upregulation of proteins involved in microaerobic growth (Hoboth et al. 2009).
Consequences of diversification
The insurance hypothesis
The self-generated diversity during biofilm growth might offer protection against changing and adverse environmental conditions. Populations composed of diverse subpopulations are in general expected to perform better because of the likelihood that some subpopulations will thrive as prevailing conditions change. Monospecies forests are for example more susceptible to environmental perturbations than mixed woodlands (McCann 2000). This principle is known as the ‘insurance hypothesis’ (Yachi and Loreau 1999; McCann 2000; Boles, Thoendel and Singh 2004). In contrast to what the term suggests, the insurance hypothesis does not imply that diversity evolves ‘because’ it helps the population to survive; increased survival is rather a lucky consequence of diversity. Boles et al. (2004) found evidence for this process in P. aeruginosa biofilms that underwent diversification during growth in different types of biofilm setups. Two main morphotypes (mini and wrinkly) with specialized biofilm phenotypes i.e. accelerated detachment (mini) and hyperbiofilm formation (wrinkly) evolved in these setups through a RecA-dependent mechanism (see above). Although not evolved in the presence of antimicrobials, biofilms inoculated with the wrinkly morphotype alone showed an increased resistance to hydrogen peroxide, hypochlorite and tobramycin. Wild-type biofilms that produced variant subpopulations showed increased resistance to hydrogen peroxide as compared to biofilms formed by the recA mutant that did not generate variants. Similarly, Tyerman et al. (2013) observed a fast emergence of heritable variation for broad-spectrum antibiotic resistance in E. coli biofilms, which were evolved in the absence of antibiotics. Other examples consistent with an insurance effect are Allegrucci and Sauer (2007) and Ryder, Chopra and O'Neill (2012) who found much higher survival of cells isolated from biofilms than planktonic cultures of S. pneumoniae and Staphylococcus respectively after plating them on antibiotic agar. The occurrence of trade-offs was not directly tested in these study and therefore the term ‘insurance effect’ should be used with caution, as argued by Cooper, Beaumont and Rainey (2005).
Insight into a mechanism by which biofilm diversification might provide an insurance effect was obtained by Koch et al. (2014). When grown in colony biofilms, methicillin-resistant S. aureus isolates were found to diversify into two new strains. The first strain competed with the parent strain via secretion of a toxic bacteriocin, but the parent strain counter-adapted to this challenge by generating a second new strain which is resistant to the bacteriocin. This same resistance mechanism also provides cross-protection against vancomycin, the prefered antibiotic to treat MRSA. Although these biofilms have not been evolved in the presence of vancomycin, this coevolutionary arms race results in the emergence of a vancomycin resistant variant, which provides an insurance for survival when treatment starts. Importantly, this strain diversification was also shown to occur in vivo and both coevolved phenotypes resemble strains commonly found in clinic, emphasizing the relevance of this mechanism (Koch et al. 2014).
In sum, evidence suggests that biofilms can serve as hotbeds of diversity that promote adaptation to harsh conditions. Such adaptation could be particularly important in chronic infections inside the host during which bacteria need to withstand severe and fluctuating conditions to persist.
Altered biofilm productivity
Another important consequence of diversification is that it can affect total biofilm productivity. Character displacement between coevolving genotypes can optimize the use of available resources and as such increase total productivity (Hector et al. 1999; Tilman et al. 2001; Bell et al. 2005). For example, in the B. cenocepacia bead transfer model, increased biofilm productivity was observed over time. As described above, the observed process of ecological succession through niche construction and character displacement coincided with a reduced competition between the evolving variants, the emergence of positive interactions and enhanced community productivity (Poltak and Cooper 2011; Ellis et al. 2015). Another example was provided by Brockhurst et al. (2006). As described above, in the P. fluorescens radiation in static microcosms a WS WS morphotype evolved, which forms a biofilm mat at the air–liquid interface. Brockhurst et al. (2006) studied further diversification within this biofilm mat and showed that character displacement between coevolving WS variants takes place within the mat in order to reduce resource competition. As expected, the extent of character displacement between pairs of coevolved WS (measured by the strength of negative frequency-dependent selection) was then found to be positively correlated with the biofilm productivity.
Ecological competition between strains then can cause character displacement, which as a by-product can promote total community productivity. However, this is not guaranteed as character displacement may also reduce productivity of a strain. Moreover, the evolution of interference competition and antibiotic warfare (as exemplified in the previous paragraph (Koch et al. 2014)) is likely common and will tend to decrease productivity.
Trade-offs between biofilm and free-living state
Another potential consequence of adaptive diversification in biofilms is that evolved cells are no longer adapted to non-biofilm conditions. In a P. aeruginosa biofilm evolution experiment, for example, culture-impaired variants arose (Penterman et al. 2014). These variants were found to have an altered outer-membrane lipopolysaccharide structure (lack of B-band LPS) compared to the wild type, due to nonsynonymous mutations in wbpJ, which increased their fitness within the biofilm but sensitized them to killing by a self-produced antimicrobial outside the biofilm. Proposed explanations of why the mutant has an advantage in biofilm environment were that energy might be saved by not producing B-band EPS or that this leads to an advantage by enhancing the cell's ability to aggregate or adhere. Similar trade-off mechanisms might operate in natural biofilms since it has been shown that in CF infections P. aeruginosa evolves mutations inactivating B-band LPS biosynthesis at high frequencies.
These findings suggest that the transition between biofilm growth and free-living state can be costly for bacteria and also raise the possibility that biofilms in natural settings produce large culture-resistant subpopulations that thrive in situ but fail to be detected by culture-based sampling (‘viable-but non-culturable’ phenotype). Another example includes the competition-colonization trade-offs between EPS producing and non-producing strains in a V. cholerae biofilm. Producers have an advantage in the biofilm because they can move to the top of the biofilm and suffocate their neighboring cells. However, EPS-producers have a decreased dispersion capacity compared to EPS deficient strains (Nadell and Bassler 2011). Local competition is thus dominated by EPS producers, while non-producers have an advantage regarding dispersion capacity. This enhanced ability of non-producers to disperse might explain why certain bacterial species, among which V. cholerae, use QS to terminate polymer secretion at high cell density (Nadell et al. 2008).
EVOLUTION OF COOPERATIVE TRAITS IN BIOFILMS
A key outcome of evolution in biofilms that we have not yet discussed is the emergence of cooperative traits. Cooperative traits are phenotypes that increase the fitness of another cell and have at least in part evolved because of this effect (West et al. 2006; Mitri and Foster 2013). Simple cooperative traits, such as extracellular enzymes, iron-scavenging siderophores and extracellular polymers, are widespread in the microbial world and in biofilms. Niche specialization is often mediated by the development of traits that are cooperative in nature and as such cooperation plays an important role in adaptive diversification. For example, cellulose overproduction by the WS emerged in the P. fluorescens radiation is a cooperative trait, which allows them to form a biofilm mat and obtain—as a group-superior access to oxygen (Rainey and Rainey 2003).
Explaining the evolution of cooperative traits can be challenging because they are prone to exploitation by ‘cheaters’, rapidly growing cell-lines that lack the cooperative trait but benefit from the cooperation of others. There is evidence that cheating occurs within biofilms (Boyle et al. 2013). Indeed, as already touched upon above, cellulose production by the WS in the P. fluorescens biofilm mat is vulnerable to SM cheaters, which do not produce cellulose but still reap the advantage of residing in the mat. The SM cheaters however disrupt the structural integrity of the mat, which causes it to sink earlier (Rainey and Rainey 2003). Moreover, Popat et al. (2012) reported that QS) cheating can occur in P. aeruginosa biofilm populations owing to exploitation of QS-regulated public goods. Similarly, Savage, Chopra and O'Neill (2013) reported the emergence of a white variant during S. aureus biofilm growth, which due to activation of the agr QS system shows an overproduction of a-haemolysin and other extracellular compounds and has an advantage in the early stages of biofilm formation. The presence of a large subpopulation of cells in the biofilm exhibiting enhanced activity of the agr system again allowed the emergence of a cheating large pale variant, which is deficient in the QS system and exploits the exoproducts of QS proficient cells.
Social evolution theory, which deals with understanding the evolutionary trajectories of cooperative traits, predicts that, for a given cost-to-benefit ratio of cooperation, the benefits of a cooperative behavior must be sufficiently directed to other cooperating individuals for cooperation to remain evolutionarily stable against exploitation (Hamilton 1964; West et al. 2006; Mitri and Foster 2013). That is, cooperation is favored when interaction preferentially occurs between cells that carry the same genotype at the locus driving a social trait, such as clonemates (the genotypic view of microbial interactions; Mitri and Foster 2013).
Mechanisms that stabilize within genotype cooperation in biofilms
Evolution experiments and competition experiments in biofilms have provided insights in how biofilm growth and structure can reduce the extent of interaction between cooperative and non-cooperative cells and as such promote cooperation (Nadell, Xavier and Foster 2009). Indeed, the ecological diversification we focused on in the last section can help to stabilize cooperation. Diverse groups may be less susceptible to invasion by cheaters, both because it can allow a cooperator to escape the niche of cheat and because a diverse population leaves fewer resources unexploited (Brockhurst et al. 2006). Consistent with this Brockhurst et al. (2006) found that diverse populations of WS were more resistant to cheating strains than monocultures of WS strains. However, character displacement is just one of several processes in biofilms that can promote cooperation within a genotype. Here, we give an overview of additional processes (Fig. 3). The first four processes act by spatially segregating cooperators and non-cooperator, while the latter two reduce the distance over which the benefit of cooperation acts.
Figure 3.
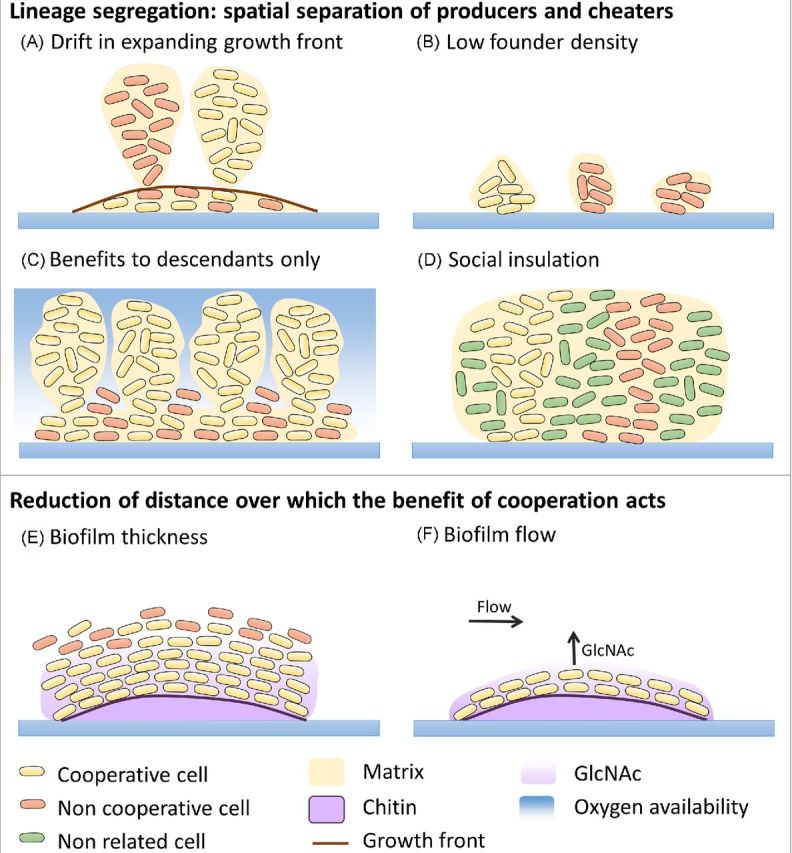
Overview of mechanisms that stabilize cooperation in biofilms. There are two general types of mechanisms: the spatial separation of producers and cheaters (A–D) and reduction of distance over which the benefit of cooperation acts (E and F). (A) ‘Drift in expanding growth front’: random drift in the thin active growth layers causes lineage segregation (Nadell, Foster and Xavier 2010). (B) ‘Low founder density’: at low founder cell density, the cells are initially more separated from each other at the surface, allowing more cell divisions before the growing cell clusters get into contact and as such increasing lineage segregation (van Gestel et al. 2014). (C) ‘Benefits to descendants only’: polymer production causes the descendants of the producers to be pushed up to areas with an increased oxygen availability, while non-producers are being suffocated (Nadell and Bassler 2011). (D) ‘Social insulation’: at high-nutrient conditions, the addition of another species causes the producers and non-producers to be separated from each other (Mitri, Xavier and Foster 2011). (E) ‘Biofilm thickness’: producers of chitinase (yellow), an enzyme to degrade chitin (blue) to usable GlcNAc, are located at places with a high biofilm density. Due to the thick biofilm, diffusion is low and non-producers (red) are not able take advantage of the GlcNAc as it will be depleted by neighboring producers cells before it can reach the non-producers (Drescher et al. 2014). (F) ‘Biofilm flow’: under static conditions, the GlcNAc produced from chitin (blue) by chitinase producers (yellow) can also be used by non-producers (red). However, under flow conditions, the GlcNAc will be transported away from the biofilm surface, causing only neighboring producers cells to benefit from the GlcNAc (Drescher et al. 2014).
Drift in expanding growth fronts favors lineage segregation and cooperation
Biofilm cells often proliferate into larger cell groups. As explained above, experiments with bacterial colonies have indicated that expanding cell groups can segregate into sectors, each dominated by a single genetic lineage (Golding, Cohen and Ben-Jacob 1999; Hallatschek et al. 2007). Random genetic drift at the expanding frontiers (i.e. the thin active layer of growing cells at the outside of the colony) is thought to be at the basis of this sectoring (Klopfstein, Currat and Excoffier 2006; Hallatschek et al. 2007; Hallatschek and Nelson 2008). By using an agent-based biofilm model that employs mechanistic descriptions of solute diffusion and cell growth, Nadell, Foster and Xavier (2010) showed that the extent of lineage segregation within biofilms is inversely related to the depth of the active layer of growing cells. The active layer depth itself increases with nutrient levels, substrate diffusivity and slower cell growth rate (Mitri, Clarke and Foster 2015). Thick active layers promote lineage mixing, while decreasing active layer depth generates increasingly strong lineage segregation, ultimately leading to the formation of towers consisting of single genotypes.
In order to explore a potential connection between lineage segregation and the evolution of social phenotypes, a cooperative phenotype was incorporated in the model in the form of a diffusible extracellular enzyme that is costly for the cooperative individuals to produce, but benefits all cells in the local area. Competition between a cooperating cell line and an exploitative line was then studied for different active layer depths and thus different levels of segregation. When active layers are thick, leading to well-mixed cell lineages, the enzyme is homogenously distributed through cell groups and the non-cooperative cell line is therefore able to exploit the cooperative line (Nadell, Foster and Xavier 2010). This result is consistent with observations of exploitation in liquid cultures (West et al. 2006). With decreasing active layer depth, the cooperative cells and exploitative cells no longer remain well mixed, which results in an increasingly asymmetric distribution of the benefits of the cooperative secreted enzyme to cooperative cells. This allows cooperative cells to outcompete exploitative cells. In summary, this model thus suggests that clusters of genetically related cells can emerge quite easily in spatially constrained cell groups allowing stable persistence of cooperative phenotypes. In vitro evidence was provided by Van Dyken et al. (2013), who found that expanding colonies of fluorescently labeled Saccharomyces cerevisiae cells on agar show genetic demixing of cooperators and cheaters, followed by increase in cooperator frequency as cooperator sectors overtake neighboring defector sectors.
Low founder cell density promotes lineage segregation and cooperation
Simulations with an agent-based model revealed that founder cell density also clearly affects spatial segregation within biofilms (van Gestel et al. 2014). The degree of spatial segregation is inversely related to the density of cells at the onset of biofilm formation. At low founder cell density, the cells are initially more separated from each other at the surface, allowing more cell divisions before the growing cell clusters get into contact and as such increasing spatial structure. EPS can be considered as a public good as they are shared, provide an advantage to the surrounding cells and are costly to produce. Consistent with the effect of spatial segregation described above, the model predicts that at high founder cell density cooperation by sharing the public good EPS is unstable, while at lower founder cell density EPS producers outcompete non-producers. Similar results were obtained in vitro when B. subtilis biofilms were inoculated with different inoculum sizes: the lower the inoculum the more spatial segregation and the less exploitation of EPS production.
EPS secretion pushes cells up and directs benefit to descendants
An additional mechanism resulting in spatial segregation of cooperators and non-cooperators was described by Xavier and Foster (2007), who applied an agent-based model to study the outcome of evolutionary competitions between strains that differ in their level of EPS production. This study indicated that polymer production is altruistic for cells lying above a focal cell, since it pushes descendants up and out into better oxygen conditions. At the same time, it provides a strong advantage at the scale of the cell lineages by suffocating neighboring non-producers. The upward expansion of the biofilm provides thus a mechanism by which the advantages of polymer production are specifically directed toward the descendants, as such stabilizing cooperation through EPS. In addition, EPS-producing cells better adhere to each other, resulting in strong mother–daughter interactions that may also contribute to stabilization of cooperation (Schluter et al. 2015). An interesting side note is that the adhesion properties of EPS can provide additional evolutionary advantages such as resistance to shear stress and an active displacement of non-producing cells near the growth surface. This has been predicted by agent-based models and validated in in vitro V. cholera biofilms (Nadell and Bassler 2011; Schluter et al. 2015).
Social insulation in multispecies biofilms promotes within species cooperation
Metagenomic sequencing has made us aware of the vast level of species diversity associated with biofilms. Mitri, Xavier and Foster (2011) used an agent-based model to study the role of species diversity within biofilm communities on the evolution of social phenotypes, more particularly growth-promoting secretions. At high nutrient concentrations, the additional species were found to act as social insulators that keep non-secretor genotypes away from secretor genotypes and as such promote cooperation within the focal species. Social insulation thus provides a final mechanism causing spatial segregation. Under conditions of strong nutrient competition, however, the addition of new species to a focal species was found to inhibit cooperation within the focal species by eradicating secreting genotypes before they could become established. The potential for ecological competition to preferentially harm cooperators was also shown in an in vitro study in which adding S. aureus promoted P. aeruginosa siderophore non-producers over producers (Harrison et al. 2008).
Thick biofilms limit diffusion and confine public goods to cooperators
Spatial segregation is not sufficient to stabilize cooperation in all cases. Indeed, Drescher et al. (2014) studied factors that could stabilize the costly production of chitinases by V. cholera. These are extracellular enzymes that degrade the solid polymer chitin into soluble nutrient-rich GlcNAc oligomers. It was found that biofilm growth of V. cholerae on chitin flakes in non-mixed liquid could not completely prevent outcompetition of the wild type by a chitinase non-producer, even not when extending the distance between producers and non-producers by lowering the inoculation size. Strong diffusion effects that homogenize the GlcNAc concentrations and make GlcNAc equally available to non-producers were shown to be at the basis. However, attention was drawn to the spontaneous emergence of matrix hyperproducers that occasionally occurred during these experiments. Matrix hyperproducers were then shown to produce very thick biofilms, in which the public good dilemma was completely solved. In these thick biofilms, cells are multilayered and densely packed, which strongly reduces the diffusion of GLcNAc oligomers because of fast uptake by neighboring cells. As such, by limiting diffusion, production of thick biofilms provides a means to reduce the distance over which the benefit of cooperation acts and to preferentially direct it to cooperators.
Fluid flow removes public goods, as such denying access to cheaters
Drescher et al. (2014) described an additional mechanism that reduces the distance over which the benefit of cooperation acts. Natural biofilms are often subjected to liquid flow above their surface. It was found that even low flow rates could stabilize chitinase production in V. cholera biofilms. Flow facilitates transport of GlcNAc away from the surface of the biofilm, which reduces the concentration available to all cells. This is selectively disadvantageous to chitinase non-producers, because these cells do not benefit from chitinases digesting chitin in their immediate neighborhood. As such, similar to thick biofilms, flow reduces the distance over which the benefit provided by cooperators acts.
Cooperation among species and strains
Whereas the stabilizing mechanisms explained above focus on cooperation between cells of the same genotype, far less effort has been invested in the study of cooperation between genotypes. The genotypic view of social interactions states that cooperation within genotypes should be common, whereas the conditions for cooperation between genotypes are much more restricted making competition between genotypes the rule (Mitri and Foster 2013). The reason that cooperation within a single genotype should be common is that genetically identical bacterial cells have the same evolutionary interest (Hamilton 1964; West et al. 2006; Mitri and Foster 2013). However, different genotypes have their own evolutionary interests. For example, a focal species is only expected to increase the fitness of another species (i.e. evolve cooperative traits) when the return benefits outweigh any costs of ecological competition between the genotypes. Moreover, relatedness within the focal species needs to be high for these return benefits to fall back only on cooperating cells, and not on cheating strains, which would destabilize cooperation.
As described above the emergence of spatiogenetic structure in biofilms is expected to promote cooperation within genotypes (Nadell, Foster and Xavier 2010; Van Dyken et al. 2013; van Gestel et al. 2014). However, evolutionary models suggest that the effect of spatiogenetic structure on cooperation between genotypes is more complex (Mitri, Xavier and Foster 2011; Oliveira, Niehus and Foster 2014). Indeed, it proves difficult to find conditions that simultaneously favor both within- and among-genotype cooperation. At low nutrient concentrations, spatial segregation prevents the genotypes from interacting with each other. At high nutrient concentrations, genotypic mixing allows the genotypes to interact with each other. However, under the same conditions cheaters also outcompete cooperators. Consistently with the genotypic view, relatively few examples of cooperation between genotypes have been described so far literature. Hansen et al. (2007), for example, described the evolution of an exploitative—not cooperative—interaction during biofilm formation, between an Acinetobacter strain and a P. putida strain that feeds on the benzoate produced by the former. For a more detailed discussion, we refer to Mitri and Foster (2013).
HOW RELEVANT IS LABORATORY BIOFILM EVOLUTION TO NATURAL CONDITIONS?
An important question is whether observations made for evolution in biofilms in laboratory settings are relevant for evolution in natural biofilms. It can be expected that part of the fast adaptation observed in laboratory evolution experiments is a consequence of growing the bacteria in a novel environment that they have not encountered before. This was illustrated by McElroy et al. (2014) who sequenced the metagenome of populations of two P. aeruginosa strains (the lab strain PAO1 and the clinical isolate 18A) at two time points during short-term biofilm evolution. Here, it appeared that the better preadaptation of PAO1 to the laboratory environment resulted in a lower number of variants observed when PAO1 was evolved in a biofilm as compared to 18A. It should be noted however that PAO1 only went through half of the number of generations (∼5.3 generations in total) compared to 18A (∼10.3 generations), which could also be a reason for its lower number of variants. Despite these observations, at least two lines of evidence indicate that evolution in laboratory biofilms is not solely adaptation to the laboratory environment and is relevant for natural biofilms. First, many evolution experiments show a much higher level of diversification when the bacteria are grown in biofilms as compared to free-living state, under very similar laboratory conditions. Diversity is quickly lost when the biofilm communities are subjected to planktonic growth (Rainey and Travisano 1998; Boles, Thoendel and Singh 2004; Koh et al. 2007; Allegrucci and Sauer 2008; Poltak and Cooper 2011; Savage, Chopra and O'Neill 2013). This indicates that large part of the diversification is a specific consequence of growth under biofilm conditions rather than adaptation to the laboratory environment. Second, the exceptional parallelism found between laboratory-derived variants and in vivo isolates indicates that some of the same forces that drive biofilm adaptation in vitro also contribute to adaptation during chronic infections (Kirisits et al. 2005; Smith et al. 2006; Cramer et al. 2011; Lieberman et al. 2011; Traverse et al. 2012; Savage, Chopra and O'Neill 2013; Penterman et al. 2014).
CONCLUSION
Despite the predominance of biofilm growth in nature, only a relatively limited number of evolution experiments have been performed with biofilm populations. Some of these studies employed simple models to identify general principles. For example, a static test tube, called ‘microcosm’, in which P. fluorescens is able to diversify and form a biofilm-like mat, has been used in a large number of studies to obtain broad insight into many aspects of the evolutionary process of adaptive radiation (Rainey and Travisano 1998). On the other hand, more complex in vitro experiments and even in vivo experiments, in which isolates from the same patients were analyzed over time, were performed to gain better understanding of the course of chronic infections and persistent contaminations per se, and to aid the design of improved therapeutics and disinfectants.
The majority of biofilm evolution studies have focused on the fast emergence of morphotypic, phenotypic and genotypic variation within biofilms. Several evolutionary and ecological processes have been proposed to underlie this diversification, although the relative role of each of them remains to be determined and is likely strongly dependent on the nature of the specific biofilm under study. First, the environmental heterogeneity within biofilms provides ecological opportunity, which can result from one strain modifying the environment in a manner that allows another to thrive (Poltak and Cooper 2011). Diversifying selection, generated by resource competition, favors the emergence of diverse ecological niche specialists. Additionally, biofilm structure can subdivide the population into a number of more or less independently evolving subpopulations, a phenomenon called population fragmentation, which may promote the fixation of diverse mutations of smaller effect through an increased influence of drift and a lower access to rare beneficial mutations of large effect (Habets et al. 2006; Hallatschek et al. 2007). Similarly, the presence of gradients may provide stepping stones for diverse mutations of smaller effect. Another source of diversification in biofilms comes from the increased fixation time associated with clonal interference, which itself is a consequence of the slow wave-like spread of beneficial mutations through space in structured populations (Martens and Hallatschek 2011). Finally, biofilms experience an increased level of HGT and an increased mutation rate through (oxygen) stress induced mutagenesis and the occurrence of hypermutator phenotypes. These provide the genetic variation for drift and diversifying selection to act upon.
A growing body of knowledge is being generated on the genetic mechanisms underlying diversification within biofilms. The evolution of a stable, diverse community requires the occurrence of fitness trade-offs between niches, so that no single genotype is able to outcompete the others in all niches. Analysis of variants of the P. fluorescens radiation in static microcosms indicated antagonistic pleiotropy, i.e. alleles beneficial in one environment being deleterious in others, as an important molecular mechanism behind this fitness trade-offs across niches (MacLean, Bell and Rainey 2004). Intriguingly, Traverse et al. (2012) found, through metagenome sequencing of their diversifying B. cenocepacia biofilm population, that competition between genotypes cannot only act within niches, but that recurrent invasion of niches by novel genotypes originated in other niches is possible as well.
A striking amount of parallelism has been observed in biofilms at the morphotypic, phenotypic and genotypic level, both between replicate lineages within the same evolution experiment and across different evolution experiments. Moreover, the exceptional parallelism found between laboratory-derived variants and in vivo isolates indicates that some of the same forces that drive biofilm adaptation also contribute to adaptation during chronic infections (Traverse et al. 2012; Lind, Farr and Rainey 2015).
Biofilm diversification has important consequences for bacterial survival and productivity and its study is therefore of key importance to design improved antimicrobial strategies and diagnostic techniques. First, the self-generated diversity during biofilm growth might offer protection against changing and adverse environmental conditions, a principle known as the ‘insurance hypothesis’ (Boles, Thoendel and Singh 2004). Most worrisome, it has been shown that biofilm diversification can lead to increased antibiotic resistance, even when strains evolve in the absence of antibiotics (Koch et al. 2014). Additionally, diversification can increase biofilm productivity because of a more efficient use of all available resources (Ellis et al. 2015). Finally, some of the evolved variants can experience stark trade-offs with opposing effects on fitness inside and outside biofilms (Penterman et al. 2014). This might result in biofilm subpopulations which are non-culturable under standard laboratory conditions, and as such impede diagnostics.
Diversification in biofilms can also stabilize cooperative traits, not in the least because diverse groups are likely less susceptible to invasion by non-cooperative cheaters (Brockhurst et al. 2006). More generally, there appear to be many routes to the evolution of cooperation within biofilms, particularly between cells of the same genotype that have the same evolutionary interests (Hamilton 1964; West et al. 2006; Mitri and Foster 2013). Evolution experiments have provided insights in how biofilm growth and structure can reduce the extent of interaction between cooperative and non-cooperative cells and as such promote cooperation (Nadell, Xavier and Foster 2009). These strategies either act by spatially segregating cooperators and non-cooperator, or by reducing the distance over which the benefit of cooperation acts. First, random genetic drift at the active layer of growing cells can favor segregation between cooperating and non-cooperating lineages, as such reducing the interaction and stabilizing the cooperation between both (Nadell, Foster and Xavier 2010; Van Dyken et al. 2013). Similar lineage segregation can be caused by inoculating strains at low density (van Gestel et al. 2014). Indeed, at low founder cell density, the cells are initially more separated from each other at the surface, allowing more cell divisions before the growing cell clusters get into contact.
The evolution of EPS production can push each other up into better oxygen conditions. The upward expansion of the biofilm provides a mechanism by which EPS producers are segregated from non-producers and the advantages of EPS production are specifically directed toward the descendants (Xavier and Foster 2007). Moreover, EPS producing cells adhere better to each other, resulting in strong mother–daughter interactions that may also contribute to stabilization of cooperation (Nadell and Bassler 2011; Schluter et al. 2015). Social insulation by additional genotypes in multispecies biofilms provides a final mechanism causing spatial segregation to keep non-secretor genotypes away from secretor genotypes and as such promote cooperation within the focal species (Mitri, Xavier and Foster 2011). Another strategy to stabilize cooperation is the production of thick biofilms, since this can limit the diffusion of shared extracellular products and as such confine these public goods to the cooperators (Drescher et al. 2014). Similarly, fluid flow above the biofilm can direct public goods to cooperators by rapidly removing the public goods, as such denying access to cheaters (Drescher et al. 2014). The genotypic view of social interactions predicts that cooperation between genotypes is much less common because the focal genotype is expected to increase the fitness of another genotype (i.e. evolve cooperative traits) only when the return benefits outweigh any costs of ecological competition between the genotypes (Mitri and Foster 2013).
Whereas most of the biofilm evolution studies up till now focused on the analysis of characteristics and interactions of a few isolated variants at the endpoint of the experiment, the advent of whole-genome and whole-population sequencing techniques (Barrick and Lenski 2013), facilitated by improving haplotype reconstruction protocols (Pulido-Tamayo et al. 2015), will undoubtedly lead to an explosion of information on many aspects of evolutionary dynamics in biofilms in the coming years. Moreover, the continually improving techniques to precisely isolate and sequence single cells and to study gene expression at the single cell level in situ will make it possible to accurately correlate specific genotypes with specific biofilm niches. For a detailed overview of upcoming techniques to study genomes and gene expression at the single cell level we refer to our recent review on this topic (Roberfroid, Vanderleyden and Steenackers 2016).
Whereas most research on biofilm evolution until now has been centered around mechanisms of diversification and cooperation, our analysis of the literature reveals that at least two research topics deserve increasing attention in the future. A first topic is development of resistance against antimicrobials in biofilms. Indeed, it can be envisioned that several of the ecological and evolutionary processes behind diversification can also promote a faster resistance development against antimicrobials, especially the higher mutation rate and HGT within biofilms and the stepping stones provided by gradients of antimicrobials. Moreover, biofilm growth might promote the evolution of cooperative resistance mechanisms, such as extracellular enzymes that degrade antibiotics, which are not stable in planktonic cultures. Second, the vast majority of studies focused on diversification or cooperation within monospecies biofilms, whereas evolution within more realistic multispecies biofilms is a major area that remains to be explored.
Supplementary Material
Acknowledgments
We would like to thank Sarah Hammarlund and three anonymous reviewers for their comments on the manuscript. We acknowledge Stijn Robijns for providing the micrograph in the graphical abstract.
FUNDING
This work was supported by the KU Leuven Research Fund (IDO/11/008), by the Institute for the Promotion of Innovation through Science and Technology in Flanders under grant IWT-SBO 120050 (NEMOA), by FWO-Vlaanderen (W0.020.11N) and by and the European Commission's Seventh Framework Programme (FP7/2007(2013) under the grant agreement COATIM (Project no. 278425). HS is a post-doctoral researcher of the FWO-Vlaanderen. IP is a research assistant of the IWT-Vlaanderen. KRF is supported by European Research Council Grant 242670.
Conflict of interest. None declared.
REFERENCES
- Ackermann M, Schauerte A, Stearns SC, et al. Experimental evolution of aging in a bacterium. BMC Evol Biol. 2007;7:126. doi: 10.1186/1471-2148-7-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akers KS, Cardile AP, Wenke JC, et al. Biofilm formation by clinical isolates and its relevance to clinical infections. Adv Exp Med Biol. 2015;830:1–28. doi: 10.1007/978-3-319-11038-7_1. [DOI] [PubMed] [Google Scholar]
- Allegrucci M, Sauer K. Characterization of colony morphology variants isolated from Streptococcus pneumoniae biofilms. J Bacteriol. 2007;189:2030–8. doi: 10.1128/JB.01369-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allegrucci M, Sauer K. Formation of Streptococcus pneumoniae non-phase-variable colony variants is due to increased mutation frequency present under biofilm growth conditions. J Bacteriol. 2008;190:6330–9. doi: 10.1128/JB.00707-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson DI. The biological cost of mutational antibiotic resistance: any practical conclusions? Curr Opin Microbiol 20069461–5. [DOI] [PubMed] [Google Scholar]
- Antonova ES, Hammer BK. Quorum-sensing autoinducer molecules produced by members of a multispecies biofilm promote horizontal gene transfer to Vibrio cholerae. FEMS Microbiol Lett. 2011;322:68–76. doi: 10.1111/j.1574-6968.2011.02328.x. [DOI] [PubMed] [Google Scholar]
- Bantinaki E, Kassen R, Knight CG, et al. Adaptive divergence in experimental populations of Pseudomonas fluorescens. III. Mutational origins of wrinkly spreader diversity. Genetics. 2007;176:441–53. doi: 10.1534/genetics.106.069906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baquero F, Coque TM. Widening the spaces of selection: evolution along sublethal antimicrobial gradients. mBio. 2014;5:e02270. doi: 10.1128/mBio.02270-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baquero F, Negri MC, Morosini MI, et al. Antibiotic-selective environments. Clin Infect Dis. 1998;27(Suppl 1):S5–11. doi: 10.1086/514916. [DOI] [PubMed] [Google Scholar]
- Barrett RD, MacLean RC, Bell G. Experimental evolution of Pseudomonas fluorescens in simple and complex environments. Am Nat. 2005;166:470–80. doi: 10.1086/444440. [DOI] [PubMed] [Google Scholar]
- Barrick JE, Lenski RE. Genome dynamics during experimental evolution. Nat Rev Genet. 2013;14:827–39. doi: 10.1038/nrg3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behe MJ. Experimental evolution, loss-of-function mutations, and ‘the first rule of adaptive evolution’. Q Rev Biol. 2010;85:419–45. doi: 10.1086/656902. [DOI] [PubMed] [Google Scholar]
- Bell T, Newman JA, Silverman BW, et al. The contribution of species richness and composition to bacterial services. Nature. 2005;436:1157–60. doi: 10.1038/nature03891. [DOI] [PubMed] [Google Scholar]
- Bjarnsholt T, Alhede M, Eickhardt-Sorensen SR, et al. The in vivo biofilm. Trends Microbiol. 2013;21:466–74. doi: 10.1016/j.tim.2013.06.002. [DOI] [PubMed] [Google Scholar]
- Bjedov I, Tenaillon O, Souza V, et al. Stress-induced mutagenesis in bacteria. Science. 2003;300:1404–9. doi: 10.1126/science.1082240. [DOI] [PubMed] [Google Scholar]
- Boles BR, Singh PK. Endogenous oxidative stress produces diversity and adaptability in biofilm communities. P Natl Acad Sci USA. 2008;105:12503–8. doi: 10.1073/pnas.0801499105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boles BR, Thoendel M, Singh PK. Self-generated diversity produces insurance effects in biofilm communities. P Natl Acad Sci USA. 2004;101:16630–5. doi: 10.1073/pnas.0407460101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle KE, Heilmann S, van Ditmarsch D, et al. Exploiting social evolution in biofilms. Curr Opin Microbiol. 2013;16:207–12. doi: 10.1016/j.mib.2013.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockhurst MA, Colegrave N, Hodgson DJ, et al. Niche occupation limits adaptive radiation in experimental microcosms. PLoS One. 2007;2:1–4. doi: 10.1371/journal.pone.0000193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockhurst MA, Colegrave N, Rozen DE. Next-generation sequencing as a tool to study microbial evolution. Mol Ecol. 2011;20:972–80. doi: 10.1111/j.1365-294X.2010.04835.x. [DOI] [PubMed] [Google Scholar]
- Brockhurst MA, Hochberg ME, Bell T, et al. Character displacement promotes cooperation in bacterial biofilms. Curr Biol. 2006;16:2030–4. doi: 10.1016/j.cub.2006.08.068. [DOI] [PubMed] [Google Scholar]
- Buckling A, Kassen R, Bell G, et al. Disturbance and diversity in experimental microcosms. Nature. 2000;408:961–4. doi: 10.1038/35050080. [DOI] [PubMed] [Google Scholar]
- Burmolle M, Ren D, Bjarnsholt T, et al. Interactions in multispecies biofilms: do they actually matter? Trends Microbiol. 2014;22:84–91. doi: 10.1016/j.tim.2013.12.004. [DOI] [PubMed] [Google Scholar]
- Chase JM, Leibold MA. Ecological Niches: Linking Classical and Contemporary Approaches. Chicago: Univ Chicago Press; 2003. [Google Scholar]
- Ciofu O, Tolker-Nielsen T, Jensen PO, et al. Antimicrobial resistance, respiratory tract infections and role of biofilms in lung infections in cystic fibrosis patients. Adv Drug Deliv Rev. 2015;85:7–23. doi: 10.1016/j.addr.2014.11.017. [DOI] [PubMed] [Google Scholar]
- Coenye T, Nelis HJ. In vitro and in vivo model systems to study microbial biofilm formation. J Microbiol Methods. 2010;83:89–105. doi: 10.1016/j.mimet.2010.08.018. [DOI] [PubMed] [Google Scholar]
- Conibear TCR, Collins SL, Webb JS. Role of mutation in Pseudomonas aeruginosa biofilm development. PLoS One. 2009;4:e6289. doi: 10.1371/journal.pone.0006289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper TF, Beaumont HJE, Rainey PB. Biofilm diversity as a test of the insurance hypothesis. Microbiology. 2005;151:2815–6. doi: 10.1099/mic.0.28026-0. [DOI] [PubMed] [Google Scholar]
- Cooper TF, Lenski RE. Experimental evolution with E. coli in diverse resource environments. I. Fluctuating environments promote divergence of replicate populations. BMC Evol Biol. 2010;10:11. doi: 10.1186/1471-2148-10-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper VS, Staples RK, Traverse CC, et al. Parallel evolution of small colony variants in Burkholderia cenocepacia biofilms. Genomics. 2014;104:447–52. doi: 10.1016/j.ygeno.2014.09.007. [DOI] [PubMed] [Google Scholar]
- Costerton JW, Lewandowski Z, Caldwell DE, et al. Microbial biofilms. Annu Rev Microbiol. 1995;49:711–45. doi: 10.1146/annurev.mi.49.100195.003431. [DOI] [PubMed] [Google Scholar]
- Cramer N, Klockgether J, Wrasman K, et al. Microevolution of the major common Pseudomonas aeruginosa clones C and PA14 in cystic fibrosis lungs. Environ Microbiol. 2011;13:1690–704. doi: 10.1111/j.1462-2920.2011.02483.x. [DOI] [PubMed] [Google Scholar]
- Davies D. Understanding biofilm resistance to antibacterial agents. Nat Rev Drug Discov. 2003;2:114–22. doi: 10.1038/nrd1008. [DOI] [PubMed] [Google Scholar]
- Davis MR, Muszynski A, Lollett IV, et al. Identification of the mutation responsible for the temperature-sensitive lipopolysaccharide O-antigen defect in the Pseudomonas aeruginosa cystic fibrosis isolate 2192. J Bacteriol. 2013;195:1504–14. doi: 10.1128/JB.01999-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day RL, Laland KN, Odling-Smee FJ. Rethinking adaptation: the niche-construction perspective. Perspect Biol Med. 2003;46:80–95. doi: 10.1353/pbm.2003.0003. [DOI] [PubMed] [Google Scholar]
- Day T, Young KA. Competitive and facilitative evolutionary diversification. Bioscience. 2004;54:101–9. [Google Scholar]
- Denamur E, Matic I. Evolution of mutation rates in bacteria. Mol Microbiol. 2006;60:820–7. doi: 10.1111/j.1365-2958.2006.05150.x. [DOI] [PubMed] [Google Scholar]
- Dettman JR, Rodrigue N, Melnyk AH, et al. Evolutionary insight from whole-genome sequencing of experimentally evolved microbes. Mol Ecol. 2012;21:2058–77. doi: 10.1111/j.1365-294X.2012.05484.x. [DOI] [PubMed] [Google Scholar]
- Deziel E, Comeau Y, Villemur R. Initiation of biofilm formation by Pseudomonas aeruginosa 57RP correlates with emergence of hyperpiliated and highly adherent phenotypic variants deficient in swimming, swarming, and twitching motilities. J Bacteriol. 2001;183:1195–204. doi: 10.1128/JB.183.4.1195-1204.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake JW, Charlesworth B, Charlesworth D, et al. Rates of Spontaneous Mutation. Genetics. 1998;146:1667–86. doi: 10.1093/genetics/148.4.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drescher K, Nadell CD, Stone HA, et al. Solutions to the public goods dilemma in bacterial biofilms. Curr Biol. 2014;24:50–5. doi: 10.1016/j.cub.2013.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driffield K, Miller K, Bostock JM, et al. Increased mutability of Pseudomonas aeruginosa in biofilms. J Antimicrob Chemoth. 2008;61:1053–6. doi: 10.1093/jac/dkn044. [DOI] [PubMed] [Google Scholar]
- Dunham MJ, Badrane H, Ferea T, et al. Characteristic genome rearrangements in experimental evolution of Saccharomyces cerevisiae. P Natl Acad Sci USA. 2002;99:16144–9. doi: 10.1073/pnas.242624799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elena SF, Lenski RE. Evolution experiments with microorganisms: the dynamics and genetic bases of adaptation. Nat Rev Genet. 2003;4:457–69. doi: 10.1038/nrg1088. [DOI] [PubMed] [Google Scholar]
- Ellis CN, Traverse CC, Mayo-Smith L, et al. Character displacement and the evolution of niche complementarity in a model biofilm community. Evolution. 2015;69:283–93. doi: 10.1111/evo.12581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson GC, Bertels F, Rainey PB. Adaptive divergence in experimental populations of Pseudomonas fluorescens. V. insight into the niche specialist fuzzy spreader compels revision of the model Pseudomonas radiation. Genetics. 2013;195:1319–35. doi: 10.1534/genetics.113.154948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkesson A, Jelsbak L, Yang L, et al. Adaptation of Pseudomonas aeruginosa to the cystic fibrosis airway: an evolutionary perspective. Nat Rev Microbiol. 2012;10:841–51. doi: 10.1038/nrmicro2907. [DOI] [PubMed] [Google Scholar]
- Fukami T, Beaumont HJ, Zhang XX, et al. Immigration history controls diversification in experimental adaptive radiation. Nature. 2007;446:436–9. doi: 10.1038/nature05629. [DOI] [PubMed] [Google Scholar]
- Galhardo RS, Hastings PJ, Rosenberg SM. Mutation as a stress response and the regulation of evolvability. Crit Rev Biochem Mol. 2007;42:399–435. doi: 10.1080/10409230701648502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein AC, Lo DS, Otto SP. Parallel genetic changes and nonparallel gene-environment interactions characterize the evolution of drug resistance in yeast. Genetics. 2012;192:241–52. doi: 10.1534/genetics.112.142620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golding I, Cohen I, Ben-Jacob E. Studies of sector formation in expanding bacterial colonies. EPL-Europhys Lett. 1999;48:587–93. [Google Scholar]
- Goller CC, Romeo T. Environmental influences on biofilm development. Curr Top Microbiol. 2008;322:37–66. doi: 10.1007/978-3-540-75418-3_3. [DOI] [PubMed] [Google Scholar]
- Gomez P, Buckling A. Real-time microbial adaptive diversification in soil. Ecol Lett. 2013;16:650–5. doi: 10.1111/ele.12093. [DOI] [PubMed] [Google Scholar]
- Goymer P, Kahn SG, Malone JG, et al. Adaptive divergence in experimental populations of Pseudomonas fluorescens. II. Role of the GGDEF regulator WspR in evolution and development of the wrinkly spreader phenotype. Genetics. 2006;173:515–26. doi: 10.1534/genetics.106.055863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012;481:306–13. doi: 10.1038/nature10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guggenheim B, Giertsen E, Schupbach P, et al. Validation of an in vitro biofilm model of supragingival plaque. J Dent Res. 2001;80:363–70. doi: 10.1177/00220345010800011201. [DOI] [PubMed] [Google Scholar]
- Habets MG, Rozen DE, Hoekstra RF, et al. The effect of population structure on the adaptive radiation of microbial populations evolving in spatially structured environments. Ecol Lett. 2006;9:1041–8. doi: 10.1111/j.1461-0248.2006.00955.x. [DOI] [PubMed] [Google Scholar]
- Hall AR, Miller AD, Leggett HC, et al. Diversity-disturbance relationships: frequency and intensity interact. Biol Lett. 2012;8:768–71. doi: 10.1098/rsbl.2012.0282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallatschek O, Hersen P, Ramanathan S, et al. Genetic drift at expanding frontiers promotes gene segregation. P Natl Acad Sci USA. 2007;104:19926–30. doi: 10.1073/pnas.0710150104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallatschek O, Nelson DR. Gene surfing in expanding populations. Theor Popul Biol. 2008;73:158–70. doi: 10.1016/j.tpb.2007.08.008. [DOI] [PubMed] [Google Scholar]
- Hall-Stoodley L, Costerton JW, Stoodley P. Bacterial biofilms: from the natural environment to infectious diseases. Nat Rev Microbiol. 2004;2:95–108. doi: 10.1038/nrmicro821. [DOI] [PubMed] [Google Scholar]
- Hall-Stoodley L, Stoodley P. Evolving concepts in biofilm infections. Cell Microbiol. 2009;11:1034–43. doi: 10.1111/j.1462-5822.2009.01323.x. [DOI] [PubMed] [Google Scholar]
- Hamilton WD. The genetical evolution of social behaviour. I. J Theor Biol. 1964;7:1–16. doi: 10.1016/0022-5193(64)90038-4. [DOI] [PubMed] [Google Scholar]
- Hansen SK, Rainey PB, Haagensen JA, et al. Evolution of species interactions in a biofilm community. Nature. 2007;445:533–6. doi: 10.1038/nature05514. [DOI] [PubMed] [Google Scholar]
- Hardin G. The competitive exclusion principle. Science. 1960;131:1292–7. doi: 10.1126/science.131.3409.1292. [DOI] [PubMed] [Google Scholar]
- Harrison F, Paul J, Massey RC, et al. Interspecific competition and siderophore-mediated cooperation in Pseudomonas aeruginosa. Isme J. 2008;2:49–55. doi: 10.1038/ismej.2007.96. [DOI] [PubMed] [Google Scholar]
- Haussler S, Lehmann C, Breselge C, et al. Fatal outcome of lung transplantation in cystic fibrosis patients due to small-colony variants of the Burkholderia cepacia complex. Eur J Clin Microbiol. 2003;22:249–53. doi: 10.1007/s10096-003-0901-y. [DOI] [PubMed] [Google Scholar]
- Hector A, Schmid B, Beierkuhnlein C, et al. Plant diversity and productivity experiments in european grasslands. Science. 1999;286:1123–7. doi: 10.1126/science.286.5442.1123. [DOI] [PubMed] [Google Scholar]
- Hegreness M, Shoresh N, Hartl D, et al. An equivalence principle for the incorporation of favorable mutations in asexual populations. Science. 2006;311:1615–7. doi: 10.1126/science.1122469. [DOI] [PubMed] [Google Scholar]
- Hengge R. Principles of c-di-GMP signalling in bacteria. Nat Rev Microbiol. 2009;7:263–73. doi: 10.1038/nrmicro2109. [DOI] [PubMed] [Google Scholar]
- Hermsen R, Deris JB, Hwa T. On the rapidity of antibiotic resistance evolution facilitated by a concentration gradient. P Natl Acad Sci USA. 2012;109:10775–80. doi: 10.1073/pnas.1117716109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herron MD, Doebeli M. Parallel evolutionary dynamics of adaptive diversification in Escherichia coli. PLoS Biol. 2013;11:e1001490. doi: 10.1371/journal.pbio.1001490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobley L, Harkins C, MacPhee CE, et al. Giving structure to the biofilm matrix: an overview of individual strategies and emerging common themes. FEMS Microbiol Rev. 2015;39:649–69. doi: 10.1093/femsre/fuv015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoboth C, Hoffmann R, Eichner A, et al. Dynamics of adaptive microevolution of hypermutable Pseudomonas aeruginosa during chronic pulmonary infection in patients with cystic fibrosis. J Infect Dis. 2009;200:118–30. doi: 10.1086/599360. [DOI] [PubMed] [Google Scholar]
- Hogardt M, Heesemann J. Adaptation of Pseudomonas aeruginosa during persistence in the cystic fibrosis lung. Int J Med Microbiol. 2010;300:557–62. doi: 10.1016/j.ijmm.2010.08.008. [DOI] [PubMed] [Google Scholar]
- Huse HK, Kwon T, Zlosnik JE, et al. Parallel evolution in Pseudomonas aeruginosa over 39 000 generations in vivo. mBio. 2010;1:e00199–10. doi: 10.1128/mBio.00199-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson-Kanth A, Tegmark-Wisell K, Arvidson S, et al. Natural human isolates of Staphylococcus aureus selected for high production of proteases and alpha-hemolysin are sigmaB deficient. Int J Med Microbiol. 2006;296:229–36. doi: 10.1016/j.ijmm.2006.01.067. [DOI] [PubMed] [Google Scholar]
- Kassen R. The evolution of specialists, generalists and the maintenance of diversity. J Evol Biol. 2002;15:173–90. [Google Scholar]
- Kassen R. Toward a general theory of adaptive radiation: insights from microbial experimental evolution. Ann NY Acad Sci. 2009;1168:3–22. doi: 10.1111/j.1749-6632.2009.04574.x. [DOI] [PubMed] [Google Scholar]
- Kassen R. Experimental Evolution and the Nature of Biodiversity. Greenwood Village, CO: Roberts and Company, Publishers; 2014. [Google Scholar]
- Kassen R, Llewellyn M, Rainey PB. Ecological constraints on diversification in a model adaptive radiation. Nature. 2004;431:984–8. doi: 10.1038/nature02923. [DOI] [PubMed] [Google Scholar]
- Kassen R, Rainey PB. The ecology and genetics of microbial diversity. Annu Rev Microbiol. 2004;58:207–31. doi: 10.1146/annurev.micro.58.030603.123654. [DOI] [PubMed] [Google Scholar]
- Kawecki TJ, Lenski RE, Ebert D, et al. Experimental evolution. Trends Ecol Evol. 2012;27:547–60. doi: 10.1016/j.tree.2012.06.001. [DOI] [PubMed] [Google Scholar]
- Kim W, Racimo F, Schluter J, et al. Importance of positioning for microbial evolution. P Natl Acad Sci USA. 2014;111:E1639–47. doi: 10.1073/pnas.1323632111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirisits MJ, Prost L, Starkey M, et al. Characterization of colony morphology variants isolated from Pseudomonas aeruginosa biofilms. Appl Environ Microb. 2005;71:4809–21. doi: 10.1128/AEM.71.8.4809-4821.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klopfstein S, Currat M, Excoffier L. The fate of mutations surfing on the wave of a range expansion. Mol Biol Evol. 2006;23:482–90. doi: 10.1093/molbev/msj057. [DOI] [PubMed] [Google Scholar]
- Knight CG, Zitzmann N, Prabhakar S, et al. Unraveling adaptive evolution: how a single point mutation affects the protein coregulation network. Nat Genet. 2006;38:1015–22. doi: 10.1038/ng1867. [DOI] [PubMed] [Google Scholar]
- Koch G, Yepes A, Forstner KU, et al. Evolution of resistance to a last-resort antibiotic in Staphylococcus aureus via bacterial competition. Cell. 2014;158:1060–71. doi: 10.1016/j.cell.2014.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh KS, Lam KW, Alhede M, et al. Phenotypic diversification and adaptation of Serratia marcescens MG1 biofilm-derived morphotypes. J Bacteriol. 2007;189:119–30. doi: 10.1128/JB.00930-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler T, Buckling A, van Delden C. Cooperation and virulence of clinical Pseudomonas aeruginosa populations. P Natl Acad Sci USA. 2009;106:6339–44. doi: 10.1073/pnas.0811741106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korona R, Nakatsu CH, Forney LJ, et al. Evidence for multiple adaptive peaks from populations of bacteria evolving in a structured habitat. P Natl Acad Sci USA. 1994;91:9037–41. doi: 10.1073/pnas.91.19.9037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koza A, Moshynets O, Otten W, et al. Environmental modification and niche construction: developing O2 gradients drive the evolution of the Wrinkly Spreader. Isme J. 2011;5:665–73. doi: 10.1038/ismej.2010.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraigsley AM, Finkel SE. Adaptive evolution in single species bacterial biofilms. FEMS Microbiol Lett. 2009;293:135–40. doi: 10.1111/j.1574-6968.2009.01526.x. [DOI] [PubMed] [Google Scholar]
- Kreft JU, Picioreanu C, Wimpenny JW, et al. Individual-based modelling of biofilms. Microbiology. 2001;147:2897–912. doi: 10.1099/00221287-147-11-2897. [DOI] [PubMed] [Google Scholar]
- Lang GI, Rice DP, Hickman MJ, et al. Pervasive genetic hitchhiking and clonal interference in forty evolving yeast populations. Nature. 2013;500:571–4. doi: 10.1038/nature12344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenski RE, Rose MR, Simpson SC, et al. Long-term experimental evolution in Escherichia coli. 1. Adaptation and divergence during 2000 generations. Am Nat. 1991;138:1315–41. [Google Scholar]
- Lieberman TD, Michel JB, Aingaran M, et al. Parallel bacterial evolution within multiple patients identifies candidate pathogenicity genes. Nat Genet. 2011;43:1275–80. doi: 10.1038/ng.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lind PA, Andersson DI. Whole-genome mutational biases in bacteria. PNatl Acad Sci USA. 2008;105:17878–83. doi: 10.1073/pnas.0804445105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lind PA, Farr AD, Rainey PB. Experimental evolution reveals hidden diversity in evolutionary pathways. eLife. 2015;4:e07074. doi: 10.7554/eLife.07074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipsitch M. The rise and fall of antimicrobial resistance. Trends Microbiol. 2001;9:438–44. doi: 10.1016/s0966-842x(01)02130-8. [DOI] [PubMed] [Google Scholar]
- Loewe L, Textor V, Scherer S. High deleterious genomic mutation rate in stationary phase of Escherichia coli. Science. 2003;302:1558–60. doi: 10.1126/science.1087911. [DOI] [PubMed] [Google Scholar]
- Lujan AM, Macia MD, Yang L, et al. Evolution and adaptation in Pseudomonas aeruginosa biofilms driven by mismatch repair system-deficient mutators. PLoS One. 2011;6:e27842. doi: 10.1371/journal.pone.0027842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macarthur R, Levins R. Competition, habitat selection, and character displacement in a patchy environment. P Natl Acad Sci USA. 1964;51:1207–10. doi: 10.1073/pnas.51.6.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBain AJ. Chapter 4 In Vitro biofilm models. Adv Appl Microbiol. 2009;69:99–132. doi: 10.1016/S0065-2164(09)69004-3. [DOI] [PubMed] [Google Scholar]
- McCann KS. The diversity-stability debate. Nature. 2000;405:228–33. doi: 10.1038/35012234. [DOI] [PubMed] [Google Scholar]
- McDonald MJ, Gehrig SM, Meintjes PL, et al. Adaptive divergence in experimental populations of Pseudomonas fluorescens. IV. Genetic constraints guide evolutionary trajectories in a parallel adaptive radiation. Genetics. 2009;183:1041–53. doi: 10.1534/genetics.109.107110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElroy KE, Hui JG, Woo JK, et al. Strain-specific parallel evolution drives short-term diversification during Pseudomonas aeruginosa biofilm formation. P Natl Acad Sci USA. 2014;111:E1419–27. doi: 10.1073/pnas.1314340111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean RC, Bell G, Rainey PB. The evolution of a pleiotropic fitness tradeoff in Pseudomonas fluorescens. P Natl Acad Sci USA. 2004;101:8072–7. doi: 10.1073/pnas.0307195101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddamsetti R, Lenski RE, Barrick JE. Adaptation, clonal interference, and frequency dependent interactions in a long term evolution experiment with Escherichia coli. Genetics. 2015;200:619–31. doi: 10.1534/genetics.115.176677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen JS, Burmolle M, Hansen LH, et al. The interconnection between biofilm formation and horizontal gene transfer. FEMS Immunol Med Mic. 2012;65:183–95. doi: 10.1111/j.1574-695X.2012.00960.x. [DOI] [PubMed] [Google Scholar]
- Martens EA, Hallatschek O. Interfering waves of adaptation promote spatial mixing. Genetics. 2011;189:1045–60. doi: 10.1534/genetics.111.130112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens EA, Kostadinov R, Maley CC, et al. Spatial structure increases the waiting time for cancer. New J Phys. 2011;13:115014. doi: 10.1088/1367-2630/13/11/115014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez JL, Baquero F. Mutation frequencies and antibiotic resistance. Antimicrob Agents Ch. 2000;44:1771–7. doi: 10.1128/aac.44.7.1771-1777.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mena A, Smith EE, Burns JL, et al. Genetic adaptation of Pseudomonas aeruginosa to the airways of cystic fibrosis patients is catalyzed by hypermutation. J Bacteriol. 2008;190:7910–7. doi: 10.1128/JB.01147-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzgar D, Wills C. Evidence for the adaptive evolution of mutation rates. Cell. 2000;101:581–4. doi: 10.1016/s0092-8674(00)80869-7. [DOI] [PubMed] [Google Scholar]
- Meyer JR, Dobias DT, Weitz JS, et al. Repeatability and contingency in the evolution of a key innovation in phage lambda. Science. 2012;335:428–32. doi: 10.1126/science.1214449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer JR, Schoustra SE, Lachapelle J, et al. Overshooting dynamics in a model adaptive radiation. P Roy Soc B-Biol Sci. 2011;278:392–8. doi: 10.1098/rspb.2010.0640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miralles R, Gerrish PJ, Moya A, et al. Clonal interference and the evolution of RNA viruses. Science. 1999;285:1745–7. doi: 10.1126/science.285.5434.1745. [DOI] [PubMed] [Google Scholar]
- Mitri S, Clarke E, Foster KR. Resource limitation drives spatial organization in microbial groups. ISME J. 2015:1–12. doi: 10.1038/ismej.2015.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitri S, Foster KR. The genotypic view of social interactions in microbial communities. Annu Rev Genet. 2013;47:247–73. doi: 10.1146/annurev-genet-111212-133307. [DOI] [PubMed] [Google Scholar]
- Mitri S, Xavier JB, Foster KR. Social evolution in multispecies biofilms. P Natl Acad Sci USA. 2011;108(Suppl 2):10839–46. doi: 10.1073/pnas.1100292108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadell CD, Bassler BL. A fitness trade-off between local competition and dispersal in Vibrio cholerae biofilms. P Natl Acad Sci USA. 2011;108:14181–5. doi: 10.1073/pnas.1111147108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadell CD, Foster KR, Xavier JB. Emergence of spatial structure in cell groups and the evolution of cooperation. PLoS Comput Biol. 2010;6:e1000716. doi: 10.1371/journal.pcbi.1000716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadell CD, Xavier JB, Foster KR. The sociobiology of biofilms. FEMS Microbiol Rev. 2009;33:206–24. doi: 10.1111/j.1574-6976.2008.00150.x. [DOI] [PubMed] [Google Scholar]
- Nadell CD, Xavier JB, Levin SA, et al. The evolution of quorum sensing in bacterial biofilms. PLoS Biol. 2008;6:e14. doi: 10.1371/journal.pbio.0060014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahum JR, Godfrey-Smith P, Harding BN, et al. A tortoise–hare pattern seen in adapting structured and unstructured populations suggests a rugged fitness landscape in bacteria. P Natl Acad Sci USA. 2015;112:201410631. doi: 10.1073/pnas.1410631112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson AI, Koskiniemi S, Eriksson S, et al. Bacterial genome size reduction by experimental evolution. P Natl Acad Sci USA. 2005;102:12112–6. doi: 10.1073/pnas.0503654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odum EP. Ecology. 2nd edn. New York: Holt, Rinehart, and Winston; 1975. [Google Scholar]
- Oliveira NM, Niehus R, Foster KR. Evolutionary limits to cooperation in microbial communities. P Natl Acad Sci USA. 2014;111:17941–6. doi: 10.1073/pnas.1412673111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Rourke D, FitzGerald CE, Traverse CC, et al. There and back again: consequences of biofilm specialization under selection for dispersal. Front Genet. 2015;6:18. doi: 10.3389/fgene.2015.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SC, Krug J. Clonal interference in large populations. P Natl Acad Sci USA. 2007;104:18135–40. doi: 10.1073/pnas.0705778104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penterman J, Nguyen D, Anderson E, et al. Rapid evolution of culture-impaired bacteria during adaptation to biofilm growth. Cell Rep. 2014;6:293–300. doi: 10.1016/j.celrep.2013.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perfeito L, Pereira MI, Campos PR, et al. The effect of spatial structure on adaptation in Escherichia coli. Biol Lett. 2008;4:57–59. doi: 10.1098/rsbl.2007.0481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perron GG, Gonzalez A, Buckling A. Source-sink dynamics shape the evolution of antibiotic resistance and its pleiotropic fitness cost. P Roy Soc B-Biol Sci. 2007;274:2351–6. doi: 10.1098/rspb.2007.0640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picioreanu C, Kreft JU, Van Loosdrecht MC. Particle-based multidimensional multispecies biofilm model. Appl Environ Microb. 2004;70:3024–40. doi: 10.1128/AEM.70.5.3024-3040.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poltak SR, Cooper VS. Ecological succession in long-term experimentally evolved biofilms produces synergistic communities. ISME J. 2011;5:369–78. doi: 10.1038/ismej.2010.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponciano JM, La HJ, Joyce P, et al. Evolution of diversity in spatially structured Escherichia coli populations. Appl Environ Microb. 2009;75:6047–54. doi: 10.1128/AEM.00063-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popat R, Crusz SA, Messina M, et al. Quorum-sensing and cheating in bacterial biofilms. P Roy Soc B-Biol Sci. 2012;279:4765–71. doi: 10.1098/rspb.2012.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulido-Tamayo S, Sanchez-Rodriguez A, Swings T, et al. Frequency-based haplotype reconstruction from deep sequencing data of bacterial populations. Nucleic Acids Res. 2015;43:e105. doi: 10.1093/nar/gkv478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainey PB, Rainey K. Evolution of cooperation and conflict in experimental bacterial populations. Nature. 2003;425:69–72. doi: 10.1038/nature01906. [DOI] [PubMed] [Google Scholar]
- Rainey PB, Travisano M. Adaptive radiation in a heterogeneous environment. Nature. 1998;31:69–72. doi: 10.1038/27900. [DOI] [PubMed] [Google Scholar]
- Rani SA, Pitts B, Stewart PS. Rapid diffusion of fluorescent tracers into Staphylococcus epidermidis biofilms visualized by time lapse microscopy. Antimicrob Agents Ch. 2005;49:728–32. doi: 10.1128/AAC.49.2.728-732.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberfroid S, Vanderleyden J, Steenackers HP. Gene expression variability in clonal populations: causes and consequences. Crit Rev Microbiol. 2016;5:1–16. doi: 10.3109/1040841X.2015.1122571. [DOI] [PubMed] [Google Scholar]
- Romling U, Galperin MY, Gomelsky M. Cyclic di-GMP: the first 25 years of a universal bacterial second messenger. Microbiol Mol Biol Rev. 2013;77:1–52. doi: 10.1128/MMBR.00043-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg SM. Evolving responsively adaptive mutation. Nat Rev Genet. 2001;2:504–15. doi: 10.1038/35080556. [DOI] [PubMed] [Google Scholar]
- Rosenberg SM, Queitsch C. Combating evolution to fight disease. Science. 2014;343:1088–9. doi: 10.1126/science.1247472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozen DE, de Visser JA, Gerrish PJ. Fitness effects of fixed beneficial mutations in microbial populations. Curr Biol. 2002;12:1040–5. doi: 10.1016/s0960-9822(02)00896-5. [DOI] [PubMed] [Google Scholar]
- Rozen DE, Habets MGJL, Handel A, et al. Heterogeneous adaptive trajectories of small populations on complex fitness landscapes. PLoS One. 2008;3:e1715. doi: 10.1371/journal.pone.0001715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumbaugh KP, Carty NL. In vivo models of biofilm infection. In: Bjarnsholt T, editor. Biofilm Infections. New York: Springer-Verlag; 2011. pp. 267–29. [Google Scholar]
- Ryder VJ, Chopra I, O'Neill AJ. Increased mutability of Staphylococci in biofilms as a consequence of oxidative stress. PLoS One. 2012;7:e47695. doi: 10.1371/journal.pone.0047695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saint-Ruf C, Garfa-Traore M, Collin V, et al. Massive diversification in aging colonies of Escherichia coli. J Bacteriol. 2014;196:3059–73. doi: 10.1128/JB.01421-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage VJ, Chopra I, O'Neill AJ. Population diversification in Staphylococcus aureus biofilms may promote dissemination and persistence. PLoS One. 2013;8:e62513. doi: 10.1371/journal.pone.0062513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schluter D. The Ecology of Adaptive Radiation. New York: Oxford University Press; 2000. [Google Scholar]
- Schluter J, Nadell CD, Bassler BL, et al. Adhesion as a weapon in microbial competition. ISME J. 2015;9:139–49. doi: 10.1038/ismej.2014.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson G. The Major Features of Evolution. New York: Columbia University Press; 1953. [Google Scholar]
- Smith EE, Buckley DG, Wu Z, et al. Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. P Natl Acad Sci USA. 2006;103:8487–92. doi: 10.1073/pnas.0602138103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sniegowski PD, Gerrish PJ. Beneficial mutations and the dynamics of adaptation in asexual populations. Philos T Roy Soc B. 2010;365:1255–63. doi: 10.1098/rstb.2009.0290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer P, Greenman J, McKenzie C, et al. In vitro biofilm model for studying tongue flora and malodour. J Appl Microbiol. 2007;103:985–92. doi: 10.1111/j.1365-2672.2007.03344.x. [DOI] [PubMed] [Google Scholar]
- Spiers AJ. A mechanistic explanation linking adaptive mutation, niche change, and fitness advantage for the wrinkly spreader. Int J Evol Biology. 2014;2014:675432. doi: 10.1155/2014/675432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiers AJ, Bohannon J, Gehrig SM, et al. Biofilm formation at the air–liquid interface by the Pseudomonas fluorescens SBW25 wrinkly spreader requires an acetylated form of cellulose. Mol Microbiol. 2003;50:15–27. doi: 10.1046/j.1365-2958.2003.03670.x. [DOI] [PubMed] [Google Scholar]
- Spiers AJ, Kahn SG, Bohannon J, et al. Adaptive divergence in experimental populations of Pseudomonas fluorescens. I. Genetic and phenotypic bases of wrinkly spreader fitness. Genetics. 2002;161:33–46. doi: 10.1093/genetics/161.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starkey M, Hickman JH, Ma L, et al. Pseudomonas aeruginosa rugose small-colony variants have adaptations that likely promote persistence in the cystic fibrosis lung. J Bacteriol. 2009;191:3492–503. doi: 10.1128/JB.00119-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steenackers H, Hermans K, Vanderleyden J, et al. Salmonella biofilms: an overview on occurrence, structure, regulation and eradication. Food Res Int. 2012;45:502–31. [Google Scholar]
- Stern DL. The genetic causes of convergent evolution. Nat Rev Genet. 2013;14:751–64. doi: 10.1038/nrg3483. [DOI] [PubMed] [Google Scholar]
- Stewart PS, Franklin MJ. Physiological heterogeneity in biofilms. Nat Rev Microbiol. 2008;6:199–210. doi: 10.1038/nrmicro1838. [DOI] [PubMed] [Google Scholar]
- Taddei F, Matic I, Radman M. cAMP-dependent SOS induction and mutagenesis in resting bacterial populations. P Natl Acad Sci USA. 1995;92:11736–40. doi: 10.1073/pnas.92.25.11736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilman D, Reich PB, Knops J, et al. Diversity and productivity in a long-term grassland experiment. Science. 2001;294:843–5. doi: 10.1126/science.1060391. [DOI] [PubMed] [Google Scholar]
- Traber KE, Lee E, Benson S, et al. agr function in clinical Staphylococcus aureus isolates. Microbiology. 2008;154:2265–74. doi: 10.1099/mic.0.2007/011874-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traverse CC, Mayo-Smith LM, Poltak SR, et al. Tangled bank of experimentally evolved Burkholderia biofilms reflects selection during chronic infections. P Natl Acad Sci USA. 2012;110:E250–9. doi: 10.1073/pnas.1207025110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travis E, Travis J. Mutators in space: the dynamics of high-mutability clones in a two-patch model. Genetics. 2004;167:513–22. doi: 10.1534/genetics.167.1.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyerman JG, Ponciano JM, Joyce P, et al. The evolution of antibiotic susceptibility and resistance during the formation of Escherichia coli biofilms in the absence of antibiotics. BMC Evol Biol. 2013;13:22. doi: 10.1186/1471-2148-13-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udall YC, Deeni Y, Hapca SM, et al. The evolution of biofilm-forming Wrinkly Spreaders in static microcosms and drip-fed columns selects for subtle differences in wrinkleality and fitness. FEMS Microbiol Ecol. 2015;91 doi: 10.1093/femsec/fiv057. [DOI] [PubMed] [Google Scholar]
- Van Dyken JD, Muller MJ, Mack KM, et al. Spatial population expansion promotes the evolution of cooperation in an experimental Prisoner's Dilemma. Curr Biol. 2013;23:919–23. doi: 10.1016/j.cub.2013.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gestel J, Weissing FJ, Kuipers OP, et al. Density of founder cells affects spatial pattern formation and cooperation in Bacillus subtilis biofilms. ISME J. 2014;8:2069–79. doi: 10.1038/ismej.2014.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waite RD, Struthers JK, Dowson CG. Spontaneous sequence duplication within an open reading frame of the pneumococcal type 3 capsule locus causes high-frequency phase variation. Mol Microbiol. 2001;42:1223–32. doi: 10.1046/j.1365-2958.2001.02674.x. [DOI] [PubMed] [Google Scholar]
- Warren AE, Boulianne-Larsen CM, Chandler CB, et al. Genotypic and phenotypic variation in Pseudomonas aeruginosa reveals signatures of secondary infection and mutator activity in certain cystic fibrosis patients with chronic lung infections. Infect Immun. 2011;79:4802–18. doi: 10.1128/IAI.05282-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber SD, Ludwig W, Schleifer KH, et al. Microbial composition and structure of aerobic granular sewage biofilms. Appl Environ Microb. 2007;73:6233–40. doi: 10.1128/AEM.01002-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West SA, Griffin AS, Gardner A, et al. Social evolution theory for microorganisms. Nat Rev Microbiol. 2006;4:597–607. doi: 10.1038/nrmicro1461. [DOI] [PubMed] [Google Scholar]
- Wong A, Rodrigue N, Kassen R. Genomics of adaptation during experimental evolution of the opportunistic pathogen Pseudomonas aeruginosa. PLos Genet. 2012;8:e1002928. doi: 10.1371/journal.pgen.1002928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrande M, Roth JR, Hughes D. Accumulation of mutants in “aging” bacterial colonies is due to growth under selection, not stress-induced mutagenesis. P Natl Acad Sci USA. 2008;105:11863–8. doi: 10.1073/pnas.0804739105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright S. Evolution in Mendelian populations. Genetics. 1931;16:97–159. doi: 10.1093/genetics/16.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xavier JB, Foster KR. Cooperation and conflict in microbial biofilms. P Natl Acad Sci USA. 2007;104:876–81. doi: 10.1073/pnas.0607651104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xavier JB, Martinez-Garcia E, Foster KR. Social evolution of spatial patterns in bacterial biofilms: when conflict drives disorder. Am Nat. 2009;174:1–12. doi: 10.1086/599297. [DOI] [PubMed] [Google Scholar]
- Xavier JB, Picioreanu C, van Loosdrecht MC. A framework for multidimensional modelling of activity and structure of multispecies biofilms. Environ Microbiol. 2005;7:1085–103. doi: 10.1111/j.1462-2920.2005.00787.x. [DOI] [PubMed] [Google Scholar]
- Yachi S, Loreau M. Biodiversity and ecosystem productivity in a fluctuating environment: the insurance hypothesis. P Natl Acad Sci USA. 1999;96:1463–8. doi: 10.1073/pnas.96.4.1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Jelsbak L, Marvig RL, et al. Evolutionary dynamics of bacteria in a human host environment. P Natl Acad Sci USA. 2011;108:7481–6. doi: 10.1073/pnas.1018249108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarwood JM, Paquette KM, Tikh IB, et al. Generation of virulence factor variants in Staphylococcus aureus biofilms. J Bacteriol. 2007;189:7961–7. doi: 10.1128/JB.00789-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoder JB, Clancey E, Des Roches S, et al. Ecological opportunity and the origin of adaptive radiations. J Evol Biol. 2010;23:1581–96. doi: 10.1111/j.1420-9101.2010.02029.x. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Lambert G, Liao D, et al. Acceleration of emergence of bacterial antibiotic resistance in connected microenvironments. Science. 2011;333:1764–7. doi: 10.1126/science.1208747. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.