

Abstract
A recent single cell mRNA sequencing study by Dueck et al. compares neuronal transcriptomes to the transcriptomes of adipocytes and cardiomyocytes. Single cell ‘omic approaches such as those used by the authors are at the leading edge of molecular and biophysical measurement. Many groups are currently employing single cell sequencing approaches to understand cellular heterogeneity in cancer and during normal development. These single cell approaches also are beginning to address long‐standing questions regarding nervous system diversity. Beyond an innate interest in cataloging cell type diversity in the brain, single cell neuronal diversity has important implications for neurotypic neural circuit function and for neurological disease. Herein, we review the authors’ methods and findings, which most notably include evidence of unique expression profiles in some single neurons.
Keywords: brain somatic mosaicism, neuronal diversity, single cell genomics
Abbreviation
- TIVA
transcriptome in vivo analysis
Introduction
Neuronal diversity has been evident since the first drawings by Cajal in the late 1800s 1, 2. These traces revealed neuronal morphologies that are reminiscent of snowflakes: every neuron looks different. Electrophysiological recordings of single neurons beginning in the early 1900s, and the subsequent development of the patch‐clamp technique in the 1970s 3, furthered our appreciation of neuronal diversity. Many neurons are excitatory, others are inhibitory, and still others modulate another neuron's response to excitation and inhibition. Molecular biology and transgenic animal technologies have driven molecular characterization of neuronal diversity even further 4, 5. Neuroscientists now have molecular markers for many subtypes of excitatory, inhibitory, and modulatory neurons; moreover, two decades of work have delineated transcription factor codes that drive neuronal diversity in different developmental lineages, including the spinal cord 6 and cerebral cortex 7, 8. Single neuronal transcriptomes clearly identify expected neuronal cell types, and extend our appreciation of neuronal diversity by identifying additional neuronal subtypes 9, 10, 11.
Current working knowledge of neural circuit function is framed by the observations that some neurons (or small ensembles of neurons) encode memories of time and space 12, 13, while other neurons seem to hold our knowledge of media celebrities 14. Indeed, Tonegawa and colleagues have recently used optogenetic activation to demonstrate the existence of memory engrams in defined neural circuits 15, 16, 17. Collectively, these findings motivate a deeper question for single cell transcriptome analysis: is the engram represented in single cell transcriptomes? Given the vast diversity of expected engrams in a brain, a first prediction of this hypothesis may be that neuronal transcriptomes are more variable than the transcriptomes of other cell types.
A recent paper by Kim, Eberwine, and colleagues explores the question of neuronal transcriptome variability by comparing almost one hundred single cell transcriptomes from various developmental lineages in mice 18. As expected, the transcriptomes clearly distinguish single neurons from single cardiomyocytes and from single adipocytes; likewise, single cardiomyocytes and single adipocytes also have distinct transcriptomes. This study supports a central expectation derived from bulk tissue analysis, that single neurons express a larger number of genes than the other single cells examined. Further, the authors identify a core subset of 404 universally expressed genes that are detected in all 91 cells. As might be expected, these genes are annotated for housekeeping functions such as translation and metabolism, and mutations in these genes are highly associated with human disease. Although not profound findings on their own, these measurements demonstrate the veracity of the authors' single cell approach.
Dueck et al. then investigate cell type‐specific functions within single cell transcriptomes by selecting the 400 most highly expressed genes in each cell type (excluding the universally expressed genes). Enrichment analysis of these genes agrees with the expectation that the cell's phenotype can be inferred from its transcriptome. They find genes annotated for synaptic transmission to be enriched in neurons, genes annotated for cardiac muscle development to be enriched in embryonic cardiomyocytes, and genes annotated for lipid metabolism to be enriched in adipocytes. Moreover, when the same approach is applied to rat neurons, the collection of neuron‐specific, highly expressed genes is largely conserved between species. Intriguingly, the authors note that the majority of commonly expressed genes, both within and across cell types, do not have published phenotypic associations, and, thus, this single cell study has identified new candidate genes for subsequent disease‐association studies.
Neuronal transcriptomes vary from non‐neuronal transcriptomes not just in size, but also in character. Dueck et al. detect significantly more non‐exonic sequence in cortical and hippocampal pyramidal neurons than in the other cell types studied. There is evidence that introns retained in expressed mRNA have important roles in trafficking mRNA to dendrites 19, 20, but other functional roles for this additional expression are largely unexplored. Nevertheless, there remains an open question regarding whether the neuronal transcriptome complexity revealed by single cell studies can reveal even more neuronal diversity than we already appreciate.
Single cell measurement
Confidence in single cell measurements is currently limited by a dilemma analogous to Schrödinger's cat: how can you know the state of a single cell before measuring it? It is very challenging to measure the state of a single cell because single cells have picograms of DNA and RNA, while next generation sequencing typically requires hundreds of nanograms of input material to construct a sequencing library. Thus, amplification of nucleic acids is a necessary prerequisite that leaves one with few means to know what the transcriptome was before it was amplified (see 21, 22 for technical details of single cell transcriptome amplification). This technical concern has been allayed in part by detecting known cell type‐specific genes. Nonetheless, as pointed out by Dueck et al., single cell measurements of various cell types are further confounded by the distinct phenotypes of different cell populations. Cell type‐specific differences in lipid content, cell size, and cell cycle state (e.g. G0 or proliferating) may affect amplification approaches and must all be carefully considered as caveats to any conclusions drawn from single cell data.
Yet, to their credit, Dueck et al. approach single cell heterogeneity head on. They set a baseline for technical noise by analyzing bulk mRNA samples from millions of single cells diluted to single cell levels (10, 50, and 100 pg of material) and performing twelve replicate mRNA amplifications on these samples. From this data, they estimate technical variation in single cell mRNA amplification for each transcript and, in turn, begin to separate technical variation from biological variation in expression levels by calculating F‐statistics for a subset of genes. The F‐statistic is calculated for each gene from the variance observed in a given gene's single cell expression level relative to the variation observed at that expression level among dilution replicates.
Neuronal transcriptome variability
Surprisingly, the Dueck et al. study reports that serotonergic neurons have the highest mean F statistic of all cell types examined, while cortical and hippocampal pyramidal neurons have lower F‐statistics, and thus less cell–cell transcriptome variability, than the other cell types examined. Notably, the nature of the serotonergic neuron transcriptomes is unique among the cell types examined in that they were isolated from a freshly prepared adult brain slice. All other cell types were examined after having been dissociated from embryonic tissue and cultured in vitro prior to analysis. Thus, the developmental maturity of each cell type is another caveat that requires careful consideration. An alternative hypothesis, however, is that neuronal transcriptome complexity is a reflection of the diverse excitatory, inhibitory, and modulatory inputs that each neuron is receiving and interpreting in vivo.
Transcriptomes are dynamic; neuronal activity leads to gene expression changes that can occur on very rapid time scales. Diverse connectivity and activity patterns for any single neuron lead to the expectation of neuron–neuron transcriptional differences. It follows that a more direct comparison of in vivo and in vitro neurons of the same type might have very different transcriptome complexity due to ongoing activity‐dependent mechanisms. Another single cell study from Eberwine, Kim and colleagues reports just this experiment using transcriptome in vivo analysis (TIVA).
The TIVA single cell approach uses photoactivatable, cell‐permeant oligo‐dT probes to capture a single cell's transcriptome in its native environment 23. Using TIVA, marked differences were detected between the transcriptome of single 1‐week‐old hippocampal neurons in acute slices relative to single perinatally isolated hippocampal neurons after one week in culture. Specifically, the TIVA approach identified 645 bi‐modally expressed genes (i.e. genes that were either on or off in single cells) in vivo but only 27 in vitro. The majority of these also displayed sparse expression in CA1 in the Allen Brain Atlas. One explanation of these data is that some CA1 neurons are in the “on” state for bimodal genes, while others are in the “off” state. Of course, from these single time point measurements, it is not known whether single neurons maintain a single state or whether these neurons are oscillating between states. Furthermore, it is not clear whether bimodal expression patterns mark distinct subtypes of CA1 neurons, or whether neurons in the “on” state convey distinct information from neurons in the “off” state.
The Dueck et al. study also reveals a number of neurons with unique transcriptomes; they report private genes that were expressed in only one of the 91 single cells analyzed. Remarkably, 334 of 371 private genes were expressed only in neurons, although this may be partially accounted for by the higher number of genes found in neurons compared to other cell types. Of these private genes, 50 were olfactory receptor transcripts. This is surprising for two reasons: first, these receptors are not known to be expressed in cortical or hippocampal neurons, and, second, stochastic regulation is central to restricting the expression of these large gene families so that only one allele is expressed in single olfactory receptor neuron 24. Here, of 19 single cortical and 18 single hippocampal neurons, five of each type were found to privately express olfactory receptor genes; eight of these ten neurons were found to co‐express more than one olfactory receptor gene, and five expressed four or five different olfactory receptor genes (Dueck and Kim, personal communication). Private genes were not detected in serotonergic neurons, nor have they been reported for other single cell analysis of in vivo isolated cortical or hippocampal neurons. Yet, it is important to note that few in vitro cultured cardiomyocytes and adipocytes express private genes, so this intriguing finding may be as likely to reflect something specific to neuronal genome regulation and structure as much as it reflects something specific to neuronal phenotype.
Nervous system diversity may extend to the level where every neuronal transcriptome is unique. Indeed, the hypothesis that neuronal diversity can enhance information coding and neural synchronization has already been put forward based on computational models 25, 26. However, interpretation of single neuron transcriptional profiles is further confounded by promiscuous transcription without protein detection; for example, un‐rearranged T‐cell receptor transcripts 27. Conversely, Marder and colleagues have shown that similar circuit behavior can arise from widely variable transcript levels in single neurons in the same circuit 28. But two major questions arise from this knowledge. Would neural circuit performance be either improved or impaired if neuronal transcriptomes were identical? And do neurodevelopmental programs exist that specifically bring about transcriptional diversity? What is clear from the Dueck et al. study; however, is that we would not be able to ask these questions without single cell analysis.
Dynamic neuronal genomes and epigenomes
Transcriptomic comparisons among single neurons are implicitly based on the assumption that all neurons have the same genome and that neuronal epigenomes are stable (Fig. 1, current model). It is now clear that many neurons have different genomes and that neuronal epigenomes are dynamic.
Figure 1.
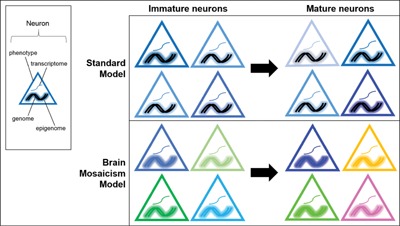
Single cell transcriptomes reflect many aspects of cell state. Cell intrinsic and extrinsic developmental cues lead to some level of neuronal diversity in immature neurons. Activity‐dependent plasticity then further diversifies individual neurons as the brain matures. Standard models of neurodevelopment are based on the assumption that neuronal genomes are static. We propose a brain mosaicism model where neuronal genomes change during development and then further through life, leading to greater levels of neuron–neuron diversity. In tandem with epigenomic modifications and transcriptional regulation, this diversity could perhaps approach a level where every neuron is unique. Schematic of various aspects of cell state: the transcriptome is represented as a single curved line, the genome is represented a double helix, the epigenome is represented as a glow around the genome, and the neuron's phenotype is represented by the border color of each triangle.
Neurotypic neuronal genomes vary from one another due to whole and subchromosomal duplications and deletions (Fig. 1) 29, 30, 31, 32, 33, 34, 35. Endogenous mobile elements also “jump” and mutate new loci 36, 37, 38, 39, 40, although estimates of the prevalence of de novo insertions varies widely 38, 39. Very recent studies report single nucleotide variants (SNVs) and small insertion/deletion polymorphisms that mark neuronal lineages 41. Some neurons with variant genomes have been shown to express distinct transcriptomes 36, 42 in addition neuronal genomes may accumulate additional variants over their lifetime 43, 44. Accumulating evidence has demonstrated that one functional consequence of brain somatic mosaicism can be neurological disease 45, 46. For example, somatic mutations in the mTOR signaling pathway lead to overgrowth of one hemisphere in hemimegalencephaly 47, 48. Likewise, schizophrenia has been linked with additional chromosome one mosaicism in cortical neurons 49 and, separately, elevated mobile element activity in hippocampal neurons 50. Mosaic amplification of the amyloid precursor protein (APP) locus 51 and other somatic mutations 52 have also been reported in cortical neurons from Alzheimer's disease patients. Taken together, multiple lines of evidence make it clear that brain mosaicism exists in neurotypic human brains.
Epigenomic patterns of DNA methylation were also generally thought to be very stable among cells of the same type; the epigenome is programmed during development leading to increasingly restricted cell fate potential. However, this concept has also been overturned in neurons 53, 54. DNA methylation patterns are re‐written by TET‐mediated conversion of 5‐methycytosine to 5‐hydroxymethyl cytosine 55, 56. Neurons have roughly fivefold higher levels of 5‐hydroxymethylcytosine than other cell types examined 57, as well as frequent and widespread de novo demethylation and methylation 58 and, perhaps, neuron–neuron epigenomic differences.
Nevertheless, epigenomic modifications can only be performed on the genome that is present in any single neuron and every neuron may have a unique genome. Thus, not only is a single cell transcriptome a snapshot of ongoing neuronal activity, it is also likely to be a reflection of the neuron's genome and epigenome at that point in time (Fig. 1, brain mosaicism model).
We propose that meaningful experiments to test the importance of neuronal transcriptional diversity for neural circuit form and function need to account for neuronal genomic and epigenomic diversity. Technological advances that allow one to measure multiple ‘omes from the same single cell are on the rapidly advancing horizon 59, 60. Farther into the future, we should be able to obtain time‐series measurements from the same single cell (http://commonfund.nih.gov/singlecell/challenge). With measurement of these parameters in hundreds of neuronal phenotypes, we will know if correlative evidence supports a broad role for genetic and epigenetic changes in neuron‐to‐neuron transcriptional diversity. Moreover, manipulation of genetic and epigenetic parameters should elucidate the cell biological mechanisms that control transcriptional diversity in neurons. The key experiments will combine single cell ‘omic measurements and novel computational approaches 22, 61 with electrophysiological recordings and behavioral analysis. With these tools at hand, it will be possible to test the hypothesis that minimizing transcriptional diversity will not change the mean performance of a population of circuits or individuals; rather it may diminish the variance in the population 62.
Conclusion
Our understanding of the brain has advanced in fits and starts alongside technological innovations that have enabled increasingly discrete measurement of neuron identity and function. During the past 10 years microscopy has advanced so that we can now image single neurons with resolution that is beyond the diffraction limit of light 63, 64. During the past five years, advances in next generation sequencing and single cell amplification have brought single cell genome, epigenome, and transcriptome measurements to begin to address fundamental questions in neuroscience. We don't yet know if “Jennifer Aniston” neurons have a distinct transcriptome from “Bill Clinton” neurons. Moreover, we don't know if every person's “Jennifer Aniston” neuron has the same transcriptome. What we do know is that we do not know, yet rapidly advancing single cell approaches like those developed by Dueck et al. and many others seem poised to begin to answer these questions.
Note added in proof
Since the acceptance of this manuscript, additional relevant manuscripts have been published. First, in a recent review Dueck et al. discuss with great insight how single cell transcriptome variability may be advantageous for higher‐level function of multi‐cellular systems, such as neural circuits 65. And second, recent single cell studies from the Buck and Xie laboratories show that immature olfactory sensory neurons can express multiple olfactory receptor genes 66, 67. Thus, the observation by Dueck et al. that multiple olfactory receptor genes are among the unique private genes detected in some cultured neurons may indicate some level of reversion to an immature or de‐differentiated state in these cells.
The authors have declared no conflict of interest.
Acknowledgments
We thank two anonymous reviewers for constructive comments on an initial version of the manuscript. MJM thanks Fred H. Gage for many insightful conversations about neuronal diversity at the single cell level. This work was supported by T32 GM008328‐24 to LJH, T32 GM008136‐30 to WDC, and U01 MH106882 to MJM.
References
- 1. Cajal SRy. 1890. Textura de las circunvoluticiones cerebrales de los mamiferos inferiores. Nota preventiva. Gaceta Medica Catalana 22–31. [Google Scholar]
- 2. Cajal SRy. 1891. Sur la structure de l'ecorce cerebale de quelques mammiferes. La Cellule 125–76. [Google Scholar]
- 3. Neher E, Sakmann B. 1976. Single‐channel currents recorded from membrane of denervated frog muscle fibres. Nature 260: 799–802. [DOI] [PubMed] [Google Scholar]
- 4. Masland RH. 2001. Neuronal diversity in the retina. Curr Opin Neurobiol 11: 431–6. [DOI] [PubMed] [Google Scholar]
- 5. Sanes JR, Masland RH. 2015. The types of retinal ganglion cells: current status and implications for neuronal classification. Annu Rev Neurosci 38: 221–46. [DOI] [PubMed] [Google Scholar]
- 6. Alaynick WA, Jessell TM, Pfaff SL. 2011. SnapShot: spinal cord development. Cell 146: 178–e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Molyneaux BJ, Arlotta P, Menezes JR, Macklis JD. 2007. Neuronal subtype specification in the cerebral cortex. Nat Rev Neurosci 8: 427–37. [DOI] [PubMed] [Google Scholar]
- 8. Sugino K, Hempel CM, Miller MN, Hattox AM, et al. 2006. Molecular taxonomy of major neuronal classes in the adult mouse forebrain. Nat Neurosci 9: 99–107. [DOI] [PubMed] [Google Scholar]
- 9. Zeisel A, Munoz‐Manchado AB, Codeluppi S, Lonnerberg P, et al. 2015. Brain structure. Cell types in the mouse cortex and hippocampus revealed by single‐cell RNA‐seq. Science 347: 1138–42. [DOI] [PubMed] [Google Scholar]
- 10. Macosko EZ, Basu A, Satija R, Nemesh J, et al. 2015. Highly parallel genome‐wide expression profiling of individual cells using nanoliter droplets. Cell 161: 1202–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pollen AA, Nowakowski TJ, Shuga J, Wang X, et al. 2014. Low‐coverage single‐cell mRNA sequencing reveals cellular heterogeneity and activated signaling pathways in developing cerebral cortex. Nat Biotechnol 32: 1053–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Aimone JB, Wiles J, Gage FH. 2006. Potential role for adult neurogenesis in the encoding of time in new memories. Nat Neurosci 9: 723–7. [DOI] [PubMed] [Google Scholar]
- 13. Moser EI, Kropff E, Moser MB. 2008. Place cells, grid cells, and the brain's spatial representation system. Annu Rev Neurosci 31: 69–89. [DOI] [PubMed] [Google Scholar]
- 14. Quiroga RQ, Reddy L, Kreiman G, Koch C, et al. 2005. Invariant visual representation by single neurons in the human brain. Nature 435: 1102–7. [DOI] [PubMed] [Google Scholar]
- 15. Redondo RL, Kim J, Arons AL, Ramirez S, et al. 2014. Bidirectional switch of the valence associated with a hippocampal contextual memory engram. Nature 513: 426–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ramirez S, Liu X, Lin PA, Suh J, et al. 2013. Creating a false memory in the hippocampus. Science 341: 387–91. [DOI] [PubMed] [Google Scholar]
- 17. Liu X, Ramirez S, Pang PT, Puryear CB, et al. 2012. Optogenetic stimulation of a hippocampal engram activates fear memory recall. Nature 484: 381–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dueck H, Khaladkar M, Kim TK, Spaethling JM, et al. 2015. Deep sequencing reveals cell‐type‐specific patterns of single‐cell transcriptome variation. Genome Biol 16: 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Buckley PT, Lee MT, Sul JY, Miyashiro KY, et al. 2011. Cytoplasmic intron sequence‐retaining transcripts can be dendritically targeted via ID element retrotransposons. Neuron 69: 877–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Khaladkar M, Buckley PT, Lee MT, Francis C, et al. 2013. Subcellular RNA sequencing reveals broad presence of cytoplasmic intron‐sequence retaining transcripts in mouse and rat neurons. PLoS ONE 8: e76194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Macaulay IC, Voet T. 2014. Single cell genomics: advances and future perspectives. PLoS Genet 10: e1004126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Grun D, van Oudenaarden A. 2015. Design and analysis of single‐cell sequencing experiments. Cell 163: 799–810. [DOI] [PubMed] [Google Scholar]
- 23. Lovatt D, Ruble BK, Lee J, Dueck H, et al. 2014. Transcriptome in vivo analysis (TIVA) of spatially defined single cells in live tissue. Nat Methods 11: 190–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Magklara A, Lomvardas S. 2013. Stochastic gene expression in mammals: lessons from olfaction. Trends Cell Biol 23: 449–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Padmanabhan K, Urban NN. 2010. Intrinsic biophysical diversity decorrelates neuronal firing while increasing information content. Nat Neurosci 13: 1276–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mejias JF, Longtin A. 2012. Optimal heterogeneity for coding in spiking neural networks. Phys Rev Lett 108: 228102. [DOI] [PubMed] [Google Scholar]
- 27. Syken J, Shatz CJ. 2003. Expression of T cell receptor beta locus in central nervous system neurons. Proc Natl Acad Sci USA 100: 13048–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Marder E, Goeritz ML, Otopalik AG. 2015. Robust circuit rhythms in small circuits arise from variable circuit components and mechanisms. Curr Opin Neurobiol 31: 156–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rehen SK, McConnell MJ, Kaushal D, Kingsbury MA, et al. 2001. Chromosomal variation in neurons of the developing and adult mammalian nervous system. Proc Natl Acad Sci USA 98: 13361–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rehen SK, Yung YC, McCreight MP, Kaushal D, et al. 2005. Constitutional aneuploidy in the normal human brain. J Neurosci 25: 2176–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. McConnell MJ, Lindberg MR, Brennand KJ, Piper JC, et al. 2013. Mosaic copy number variation in human neurons. Science 342: 632–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cai X, Evrony GD, Lehmann HS, Elhosary PC, et al. 2014. Single‐cell, genome‐wide sequencing identifies clonal somatic copy‐number variation in the human brain. Cell Rep 8: 1280–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yurov YB, Iourov IY, Vorsanova SG, Liehr T, et al. 2007. Aneuploidy and confined chromosomal mosaicism in the developing human brain. PLoS ONE 2: e558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Westra JW, Rivera RR, Bushman DM, Yung YC, et al. 2010. Neuronal DNA content variation (DCV) with regional and individual differences in the human brain. J Comp Neurol 518: 3981–4000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mosch B, Morawski M, Mittag A, Lenz D, et al. 2007. Aneuploidy and DNA replication in the normal human brain and Alzheimer's disease. J Neurosci 27: 6859–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Muotri AR, Chu VT, Marchetto MC, Deng W, et al. 2005. Somatic mosaicism in neuronal precursor cells mediated by L1 retrotransposition. Nature 435: 903–10. [DOI] [PubMed] [Google Scholar]
- 37. Coufal NG, Garcia‐Perez JL, Peng GE, Yeo GW, et al. 2009. L1 retrotransposition in human neural progenitor cells. Nature 460: 1127–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Upton KR, Gerhardt DJ, Jesuadian JS, Richardson SR, et al. 2015. Ubiquitous L1 mosaicism in hippocampal neurons. Cell 161: 228–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Evrony GD, Cai X, Lee E, Hills LB, et al. 2012. Single‐neuron sequencing analysis of L1 retrotransposition and somatic mutation in the human brain. Cell 151: 483–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Baillie JK, Barnett MW, Upton KR, Gerhardt DJ, et al. 2011. Somatic retrotransposition alters the genetic landscape of the human brain. Nature 479: 534–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lodato MA, Woodworth MB, Lee S, Evrony GD, et al. 2015. Somatic mutation in single human neurons tracks developmental and transcriptional history. Science 350: 94–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kaushal D, Contos JJ, Treuner K, Yang AH, et al. 2003. Alteration of gene expression by chromosome loss in the postnatal mouse brain. J Neurosci 23: 5599–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Suberbielle E, Sanchez PE, Kravitz AV, Wang X, et al. 2013. Physiologic brain activity causes DNA double‐strand breaks in neurons, with exacerbation by amyloid‐beta. Nat Neurosci 16: 613–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Madabhushi R, Gao F, Pfenning AR, Pan L, et al. 2015. Activity‐induced DNA breaks govern the expression of neuronal early‐response genes. Cell 161: 1592–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Insel TR. 2014. Brain somatic mutations: the dark matter of psychiatric genetics? Mol Psychiatry 19: 156–8. [DOI] [PubMed] [Google Scholar]
- 46. Poduri A, Evrony GD, Cai X, Walsh CA. 2013. Somatic mutation, genomic variation, and neurological disease. Science 341: 1237758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lee JH, Huynh M, Silhavy JL, Kim S, et al. 2012. De novo somatic mutations in components of the PI3K‐AKT3‐mTOR pathway cause hemimegalencephaly. Nat Genet 44: 941–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Poduri A, Evrony GD, Cai X, Elhosary PC, et al. 2012. Somatic activation of AKT3 causes hemispheric developmental brain malformations. Neuron 74: 41–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yurov YB, Iourov IY, Vorsanova SG, Demidova IA, et al. 2008. The schizophrenia brain exhibits low‐level aneuploidy involving chromosome 1. Schizophr Res 98: 139–47. [DOI] [PubMed] [Google Scholar]
- 50. Bundo M, Toyoshima M, Okada Y, Akamatsu W, et al. 2014. Increased l1 retrotransposition in the neuronal genome in schizophrenia. Neuron 81: 306–13. [DOI] [PubMed] [Google Scholar]
- 51. Bushman DM, Kaeser GE, Siddoway B, Westra JW, et al. 2015. Genomic mosaicism with increased amyloid precursor protein (APP) gene copy number in single neurons from sporadic Alzheimer's disease brains. eLife 4: e05116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sala Frigerio C, Lau P, Troakes C, Deramecourt V, et al. 2015. On the identification of low allele frequency mosaic mutations in the brains of Alzheimer's disease patients. Alzheimers Dement 11: 1265–76. [DOI] [PubMed] [Google Scholar]
- 53. Lister R, Mukamel EA, Nery JR, Urich M, et al. 2013. Global epigenomic reconfiguration during mammalian brain development. Science 341: 1237905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mo A, Mukamel EA, Davis FP, Luo C, et al. 2015. Epigenomic signatures of neuronal diversity in the mammalian brain. Neuron 86: 1369–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tahiliani M, Koh KP, Shen Y, Pastor WA, et al. 2009. Conversion of 5‐methylcytosine to 5‐hydroxymethylcytosine in mammalian DNA by MLL partner T ET1. Science 324: 930–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kriaucionis S, Heintz N. 2009. The nuclear DNA base 5‐hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 324: 929–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Globisch D, Munzel M, Muller M, Michalakis S, et al. 2010. Tissue distribution of 5‐hydroxymethylcytosine and search for active demethylation intermediates. PLoS ONE 5: e15367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Guo JU, Su Y, Shin JH, Shin J, et al. 2014. Distribution, recognition and regulation of non‐CpG methylation in the adult mammalian brain. Nat Neurosci 17: 215–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Dey SS, Kester L, Spanjaard B, Bienko M, et al. 2015. Integrated genome and transcriptome sequencing of the same cell. Nat Biotechnol 33: 285–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Macaulay IC, Haerty W, Kumar P, Li YI, et al. 2015. G&T‐seq: parallel sequencing of single‐cell genomes and transcriptomes. Nat Methods 12: 519–22. [DOI] [PubMed] [Google Scholar]
- 61. Stegle O, Teichmann SA, Marioni JC. 2015. Computational and analytical challenges in single‐cell transcriptomics. Nat Rev Genet 16: 133–45. [DOI] [PubMed] [Google Scholar]
- 62. Singer T, McConnell MJ, Marchetto MC, Coufal NG, et al. 2010. LINE‐1 retrotransposons: mediators of somatic variation in neuronal genomes? Trends Neurosci 33: 345–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hell SW. 2009. Microscopy and its focal switch. Nat Methods 6: 24–32. [DOI] [PubMed] [Google Scholar]
- 64. Ji N, Shroff H, Zhong H, Betzig E. 2008. Advances in the speed and resolution of light microscopy. Curr Opin Neurobiol 18: 605–16. [DOI] [PubMed] [Google Scholar]
- 65. Dueck H, Eberwine J, Kim J. 2015. Variation is function: are single cell differences functionally important? BioEssays in press DOI: 10.1002/bies.201500124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hanchate NK, Kondoh K, Lu Z, Kuang D, et al. 2015. Single‐cell transcriptomics reveals receptor transformations during olfactory neurogenesis. Science 350: 1251–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tan L, Li Q, Xie XS. 2015. Olfactory sensory neurons transiently express multiple olfactory receptors during development. Mol Syst Biol 11: 844. [DOI] [PMC free article] [PubMed] [Google Scholar]