

Abstract
Non-small cell lung cancers (NSCLC) are commonly treated with a platinum-based chemotherapy such as cisplatin (CDDP) in combination with ionizing radiation (IR). Although clinical trials have demonstrated that the combination of CDDP and IR appear to be synergistic in terms of therapeutic efficacy, the mechanism of synergism remains largely uncharacterized. We investigated the role of the DNA damage response (DDR) in CDDP radiosensitization using two NSCLC cell lines. Using clonogenic survival assays, we determined that the cooperative cytotoxicity of CDDP and IR treatment is sequence dependent, requiring administration of CDDP prior to IR (CDDP-IR). We identified and interrogated the unique time and agent-dependent activation of the DDR in NSCLC cells treated with cisplatin-IR combination therapy. Compared to treatment with CDDP or IR alone, CDDP-IR combination treatment led to persistence of γH2Ax foci, a marker of DNA double-strand breaks (DSB), for up to 24 hours after treatment. Interestingly, pharmacologic inhibition of DDR sensor kinases revealed the persistence of γ-H2Ax foci in CDDP-IR treated cells is independent of kinase activation. Taken together, our data suggest that delayed repair of DSBs in NSCLC cells treated with CDDP-IR contributes to CDDP radiosensitization and that alterations of the DDR pathways by inhibition of specific DDR kinases can augment CDDP-IR cytotoxicity by a complementary mechanism.
Keywords: lung cancer, cisplatin, radiation, damage response, ATR, ATM
1. Introduction
More than 200,000 people will be diagnosed with lung cancer in the United States this year, accounting for greater than 25% of all cancer deaths.1 Non-small cell lung carcinomas (NSCLC) are the most common lung cancers and are typically diagnosed at an advanced stage, having spread beyond the primary tumor site. Since at this stage curative surgical options are often limited,2 treatment of locally advanced disease typically includes administration of a platinum-containing drug, such as cisplatin cis-diamminedichloroplatinum II; CDDP) and ionizing radiation [IR].3,4 Treatment with a combination of both CDDP and IR improves survival over either treatment alone, with the greatest survival observed with concomitant rather than sequential treatment. 5–8 However, cancer model systems developed to investigate combination CDDP-IR treatment have yielded varying results, including reports of potential antagonistic interactions that are inconsistent with the clinical data.9,10 Therefore, a better understanding for the observed CDDP-IR clinical synergy is important.
Covalent binding of CDDP to DNA forms intra- and inter-strand DNA adducts which distort the double helical configuration. The DNA-CDDP intra-strand adducts are repaired by the nucleotide excision repair (NER) pathway while inter-strand adducts are repaired by the homologous recombination repair (HRR) pathway, and hypersensitivity to CDDP is often observed in cells deficient in either NER or HRR11–14. IR causes DNA nucleotide modifications, single and double strand DNA breaks (DSBs), both directly and indirectly via formation of oxygen free radicals. DSBs are particularly toxic to the cell, as a single DSB has been demonstrated to trigger cell death.15 IR-induced DSBs are repaired predominantly by the non-homologous end-joining (NHEJ) pathway, and NHEJ deficient cancer cells are hypersensitive to IR.16,17 DNA damage caused by both CDDP and IR activates DNA damage response (DDR) cascades which coordinate a complex interaction of downstream pathways to determine cell fate, including coordination of DNA repair, cell cycle arrest and apoptosis. The DDR is initiated at the site of DNA damage by the early (sensor) protein kinases: ataxia telangiectasia mutated (ATM), ATM and Rad3-related (ATR) and DNA-PKcs. While there is some overlap, ATM is primarily involved in the DDR to DSBs, such as those created by IR. DNA DSBs can be characterized by the detection of γ-H2Ax foci; downstream effectors of the DDR pathway which have been observed to correlate directly to the number of DSBs and persistence of which correlates with cellular survival.18–20 ATR is important in the DDR to single strand breaks, which are felt to develop on CDDP-damaged DNA through replication stress.21 Impaired function of ATM or DNA-PKcs leads to radiosensitization while inhibition of ATR has been shown to sensitize some cells to CDDP.12,22–27
The cooperative interaction of CDDP and IR is dependent on CDDP repair, as cells deficient in NER or HRR show increased radiosensitization to CDDP.9,17,28,29 The presence of a CDDP lesion on DNA inhibits NHEJ17,30,31 and we hypothesize that CDDP-IR synergy is determined by a CDDP lesion at close proximity to a DSB. However, despite the recognition of a likely role for DNA repair pathways in CDDP radiosensitization, little is known about the actual mechanism and role of the DDR in radiosensitization. This mechanism is of paramount importance, as drugs specifically targeting the DDR are currently under investigation in pre-clinical and early clinical trials. Here we investigate the impact of the DDR in CDDP-IR co-treatment in NSCLC. Our study supports a role for retained DSBs in CDDP radiosensitization and identifies a dissociation of DDR sensor kinase activation from sustained DSBs.
2. Materials and Methods
2.1. Materials
Compounds and reagents were purchased from Thermo-Fisher Scientific (Waltham, MA), unless otherwise stated.
2.2. Antibodies
Antibodies were obtained from the following commercial sources: anti-H2AxSer139 (Millipore; clone JBW301), anti-P53BP1Ser25 (Novus Biologicals; NB100-1803), anti-53BP1 (ThermoFisher Scientific; PA1-16565), anti-pChk1S345, anti-pChk2T68, anti-Chk1, anti-Chk2, anti-ATM (Cell Signaling Technology; 2348S, 2661S, 2G1D5, 1C12 and D2E2 respectively), anti-pATMS1981 (Rockland, NC9306342), anti-β-actin (Life Technologies, clone AC-15) and anti-vinculin (Abcam, clone ab18058).
2.3. Cell culture and treatment
Two NSCLC cell lines, A549 (CCL-185) and H460 (HTB-177), were obtained from the American Type Culture Collection and verified via STR testing (Manassas, VA). H460 and A549 cells were grown as previously described and incubated at 37°C in a humidified 5% CO2 atmosphere30. Cisplatin (Sigma) was added at the indicated concentrations to complete medium for 2 hours at 37°C. Following incubation with CDDP, cells were washed three times with PBS and replaced with fresh media lacking CDDP. Media was replaced with PBS prior to IR or mock IR treatments. For experiments using NU7441 (Tocris Bioscience), KU-55933 (Tocris Bioscience) or VE-821 (MedChem Express), cells were incubated with vehicle (DMSO) and/or the respective inhibitors at the concentrations listed (total of 0.1% DMSO) for 2 hours prior to treatment with IR. Incubation with the drug was continued until cell processing (0.5 or 24 hours) or for 24 hours after IR treatment (clonogenic survival assays).
IR treatments (Figures 1–5) were performed on ice using an HP Faxitron series X-ray generator (Faxitron Bioptics LLC). X-rays were filtered through a 0.5 mm aluminum filter at 160 kV resulting in a dose rate of 2.5 Gy/min. IR treatments (Figure 6) were performed in ice using a Precision X-RAD 320 X-ray generator (Precision X-Ray). X-rays were filtered using a 0.5mm aluminum filter at 160kV delivering 0.737 Gy/min. The devices undergo routine maintenance with dosimetry testing.
Figure 1. Differential Radiosensitization is Dependent on Time from Cisplatin Treatment.
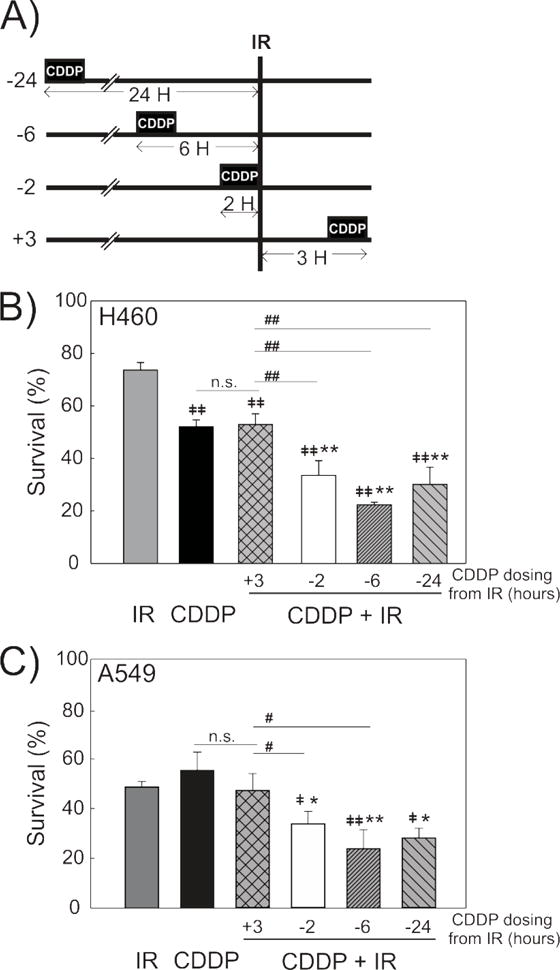
A schematic shows the treatments and time-course used to determine survival with CDDP-IR combination treatment (A). Clonogenic survival assays were used to determine cell survival in H460 (B) and A549 (C) NSCLC cells treated with CDDP (4 μM, 2 hours) at various time intervals from IR. Treatment with CDDP was 2, 6 or 24 hours before (−2, −6, −24) or 3 hours after (+3) IR treatment. N=3, mean +/− SD. *p<0.05, **p<0.001 compared to CDDP. ‡p<0.05, ‡‡p<0.001 compared to IR. #p<0.05, ##p<0.001 compared to +3
Figure 5. Effects of ATM inhibition on DNA damage response and γ-H2Ax foci formation.
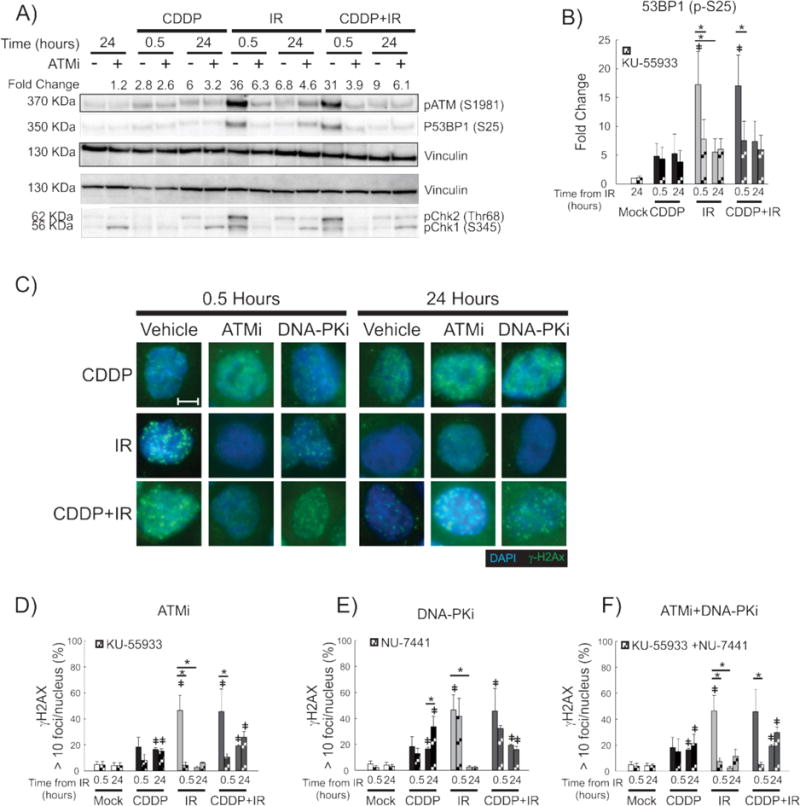
A representative immunoblot with vinculin used as a loading control (A) shows the impact of ATM inhibition following treatment with the ATM inhibitor, KU55933, on the DDR initiated by the treatments shown. Effect on the DDR is shown early (30 minutes) and late (24 hours) after IR/mock treatment with mean fold change in pATM expression shown above the immunoblot. Quantification of P53BP1 expression as a measure of treatment and time from IR is shown as fold-change over the untreated control (B). Representative fluorescence microscopy images of H460 cells are shown for the indicated treatments and time-points (C). Quantification of early and late γ-H2Ax-positive H460 cells treated with KU-55933 (ATMi; D), NU-7441 (DNA-PKi; E) and both inhibitors (F). 100× magnification. Size bar = 5 μm. *p<0.05, ‡ p<0.05 compared to untreated control.
Figure 6. Impact of ATR inhibition on DNA damage response, γ-H2Ax foci formation and cytotoxicity.
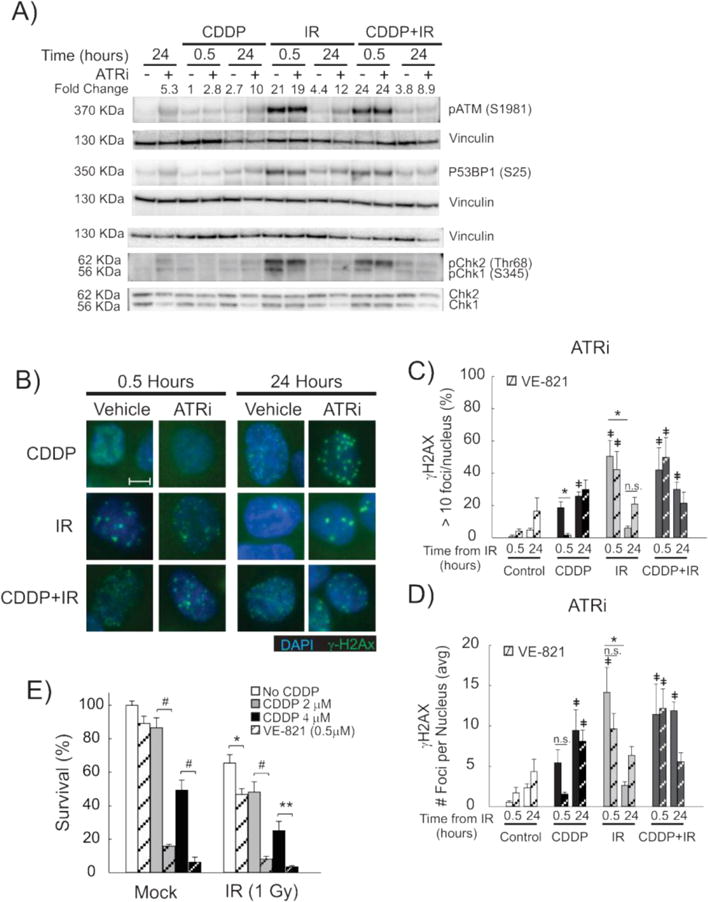
A representative immunoblot with vinculin used as a loading control (A) shows the impact of ATR inhibition (VE-821) on the early (0.5 hours) and late (24 hours) DDR to the indicated treatments in H460 cells. pATM fold change shown above the immunoblot (A). Representative fluorescence microscopy images of H460 cells are shown for the indicated treatments and time-points (B). 100× magnification, size bar = 5 μm. Quantification of γ-H2Ax positive H460 cells (C) and number of γ-H2Ax foci per nucleus (D) are shown for H460 cells treated with VE-821 (ATRi, striped bars) early (0.5 hours) and late (24 hours) after the indicated treatments. *p<0.05, ‡ p<0.05 compared to untreated control. Survival of H460 cells, as measured by clonogenic survival assay, are shown for the indicated combination treatment groups in the presence (hashed bars) or absence (solid bars) of the ATR inhibitor, VE-821 (E). Cell survival is shown in H460 cells treated with 4μM and 2 μM CDDP. Results are presented as average percent survival compared to control (vehicle) treatment +/− SEM. *p<0.05, **p<0.01, #p<0.001.
2.4. Cell viability analysis
Cell viability was assessed by a clonogenic survival assay as previously published.17,32 Briefly, cells were plated and treated as described. Following treatment, cells were removed from the plates with trypsin, suspended in complete media, and plated in triplicate. Plates were incubated at 37°C for 10–14 days, stained and fixed with a solution of 6% glutaraldehyde and 0.5% crystal violet in PBS, washed and colonies containing at least 50 cells were counted manually.
2.5. Immunofluorescence Microscopy
Immunofluorescence microscopy was performed as previously published33 with the following alterations. Cells were grown on chambered slides and mock-treated or treated with CDDP followed by either mock-IR or IR (1.5 Gy) treatment on ice. At the indicated times, cells were washed with PBS and fixed with 4% paraformaldehyde for 15 minutes. Permeabilizing solution (20 mM HEPES, 50 mM NaCl, 3 mM MgCl2, 300 mM sucrose and 0.5% Triton X-100) was added for 7 minutes followed by blocking with 5% goat serum (Jackson ImmunoResearch Laboratories). Slides were incubated with primary antibody to γ-H2Ax and 53BP1 overnight at 4°C. Bound antibody was detected using goat anti-mouse AlexaFluor 488 and anti-rabbit AlexaFluor594 (Life Technologies). Cell nuclei were counterstained using DAPI and the slides were mounted using ProlongGold anti-fade (Life Technologies). Fluorescence microscopy images were randomly obtained by identifying cell nuclei using DAPI filter. Images of foci were obtained under the FITC filter. Images were obtained and merged using a Nikon Eclipse 80i fluorescence microscope with camera using the 100× objective with oil immersion and NIS-Elements AR3.0 software (Nikon Instruments Inc.). Quantification of γ-H2Ax foci was performed using Image J software (NIH) and ITCN plugin (Image-based Tool for Counting Nuclei; Center for Bio-image Informatics, UC Santa Barbara).34 Briefly, each DAPI stained nucleus was defined as a region of interest (ROI). Uniform parameters defined the foci diameter, minimal distance and filter threshold for each merged FITC image and the number of foci per ROI was quantified using the ITCN plugin. Results are presented as percentage of cells with > 10 foci/nucleus or as the average number of foci per nucleus as indicated.
2.6. Flow cytometric analysis of γ-H2Ax
Flow cytometry was performed as previously described30 with the following alterations. Cells treated with CDDP, IR or both were collected by trypsinization at the indicated times, fixed with 4% paraformaldehyde and permeabilized with 70% ethanol. Cells were hydrated in PBS containing 1% BSA and 0.1% Triton X-100 and then incubated with antibody to γ-H2Ax overnight at 4°C,19 followed by detection by goat anti-mouse Alexa-Fluor 488. Cells were incubated in the dark with RNase A and PI stain for 30 minutes at room temperature followed by 1.5 hours at 4°C. Individual cellular absorbances were analyzed by ultraviolet and 488 nm lasers using a BD FACSCalibur flow cytometer. Data were analyzed using WinMDI software (Scripps Research Institute) for γ-H2Ax positivity. Processing, staining and analysis for cell cycle was performed as published.30
2.7. Protein Extraction and Western Blotting
Following treatment, cells were scraped and resuspended in RIPA buffer with protease and phosphatase inhibitors (Leupeptin 5μg/ml, Pepstatin A 1μL/ml, PMSF 1mM, EDTA 5mM, sodium orthovandate mM, sodium fluoride 10mM, β-glycerophosphate 10mM) on ice. Cells were lysed by passing 5 times through a 20 gauge syringe, then centrifuged and the supernatant recovered. Protein concentrations were determined using the Pierce BCA Protein Assay Kit (Life Technologies) according to the manufacturer instructions. Proteins were separated by electrophoresis on 4–12% Bis-Tris or 3–8% Tris-Acetate gels using a Criterion gel apparatus (BioRad Laboratories). Gels were blotted onto PVDF membranes using the Transblot Turbo Transfer system (BioRad) and blocked with 5% BSA in TBS-T. The membrane was incubated with primary antibodies, detected by peroxidase-coupled secondary antibodies (BioRad) and developed using enhanced chemiluminescence (BioRad). The chemiluminescence signals were quantified by densitometry (ImageJ) and normalized by housekeeping proteins (β-actin or vinculin), which generated arbitrary units.
2.8. Statistical Analysis
Data from at least 3 independent experiments are expressed as the means +/− SD or SEM. For statistical comparisons between two groups, student’s t-test was used, and between multiple groups, one- or two-way ANOVA was used. A p-value of < 0.05 was considered statistically significant.
3. Results
3.1. Cisplatin radiosensitization of NSCLC cells is sequence dependent
To identify NSCLC models that recapitulate clinical combination therapy, we identified the temporal and dose interactions between CDDP and IR in two well-characterized NSCLC cell lines of different histologic types, H460 and A549. Using clonogenic survival assays, we determined the toxicity of CDDP (LD50) to be similar between the two cell lines (4.5 μM, H460; 3.9 μM, A549, Supplemental Figure S1). Based on these results, H460 and A549 cells were treated with CDDP at 4μM for 2 hours at the indicated times, either preceding or following treatment with 1 Gy IR (Figure 1A). Treatment with CDDP prior to IR decreased cell survival, with the greatest cytotoxicity occurring when CDDP was administered 6 hours prior to IR in both H460 and A549 cells (Figure 1B and C). Importantly, treatment with CDDP following IR (3 hours later) resulted in no additional cytotoxicity compared to treatment with CDDP alone (Figure 1B and C). This indicates that CDDP–induced radiosensitization of H460 and A549 is sequence dependent, requiring treatment with CDDP prior to IR, effectively recapitulating the clinical observation in NSCLC.
3.2. Combination CDDP-IR treatment both induces and delays the resolution of γ-H2Ax foci
Having established a clinically relevant combination treatment model for NSCLC, we sought to understand the mechanism of CDDP-induced radiosensitization. We investigated how CDDP treatment impacts the DDR when given in combination with IR (CDDP-IR). We first ch aracterized the effect of CDDP-IR treatment on DNA damage by determination of H2Ax phosphorylation (γ-H2Ax) foci formation and morphology by immunofluorescence microscopy. Treatment of H460 cells with CDDP (4 μM), IR (1.5 Gy), or CDDP-IR each resulted in nuclear H2Ax phosphorylation at 30 minutes (Figure 2A). Consistent with previously published data, the morphologic appearance of the γ-H2Ax foci was dependent on the type of DNA damage induced by each treatment.35 IR treatment alone of H460 cells resulted in the development of large, distinct, punctate nuclear foci of γ-H2Ax staining. In contrast, CDDP treatment resulted in smaller, less distinct foci with more diffuse nuclear staining. Interestingly, treatment with CDDP-IR led to a nuclear γ-H2Ax pattern with characteristics of both CDDP and IR treatment when imaged 30 minutes post-IR. However, by 24 hours post-IR, treatment with CDDP-IR resulted in a persistence of foci which had a morphologic appearance of larger, more distinct foci, most similar to those formed early after IR treatment (Figure 2A). Similar morphologic changes were observed in A549 cells (Supplemental Figure S2).
Figure 2. Persistence of γ-H2Ax foci in Cisplatin-IR treated cells by Immunofluorescence.
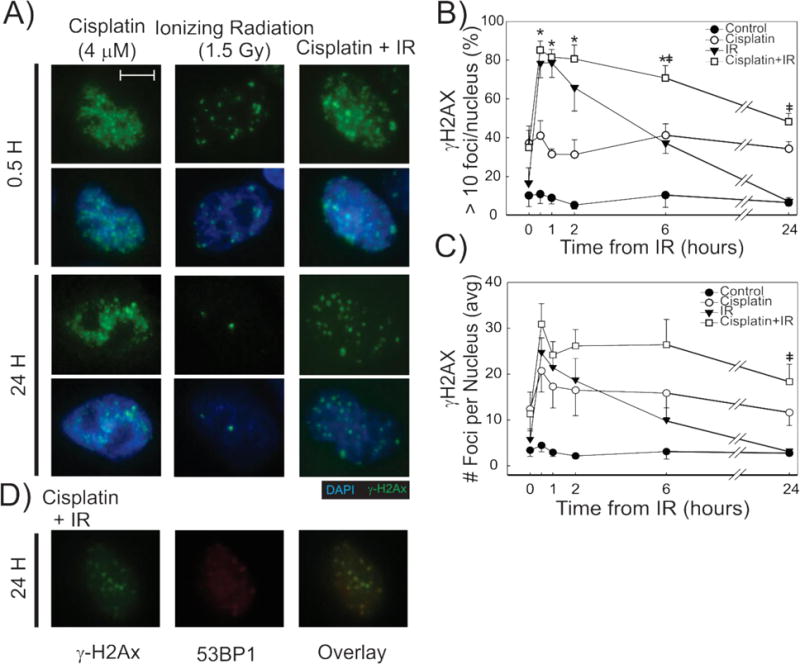
Representative fluorescence microscopy images for the indicated treatments and time-points (A), show differences in γH2Ax IF pattern. 100× magnification. Size bar = 5 μm. γ-H2Ax foci were quantified and the % cells with > 10 foci (B) and number of foci per nucleus (C) are plotted by time from IR for each of the treatments in H460 cells. N=3 biological replicates for each, +/− SEM. Differences are statistically significant in CDDP-IR treated cells when compared to CDDP (*) and IR (‡) using p<0.05. Representative fluorescence microscopy images show γ-H2Ax (green), 53BP1 (red) staining and an overlay of both in H460 cells 24-hours after treatment with combination CDDP-IR (D).
Quantification of our immunofluorescence results revealed an increase in γ-H2Ax positive H460 cells 30 minutes after treatment with IR alone and combination CDDP-IR (Figure 2B). Treatment with CDDP-IR did not result in an increase in γ-H2Ax positive cells or higher γ-H2Ax foci per cell when compared to those cells treated with IR alone when measured at 30 minutes (Figure 2B and C). However, cells treated with CDDP-IR displayed persistent γ-H2Ax foci at 24 hours when compared to those treated with IR alone, which by 24 hours displayed γ-H2Ax foci at background levels (Figure 2B). These results were mirrored in A549 cells, which also show ed similar numbers of γ-H2Ax positive cells early after treatment with IR and CDDP-IR and persistence of γ-H2Ax foci in only CDDP-IR treated cells (Supplemental Figure S2). Measurement of γ-H2Ax positive cells by flow cytometry confirmed 24 hour persistence of γ-H2Ax positive H460 cells in those treated with CDDP-IR, but not in those treated with IR alone (Supplemental Figure S3). We also show that CDDP-IR treatment causes persistent 53BP1 foci at 30 minutes (data not shown) and at 24 hours (Figure 2D). The 53bp1 foci co-localize with γ-H2Ax foci, further supporting the formation of persistent DSBs with combination cisplatin-IR treatment.
3.3. NSCLC cells treated with CDDP-IR are able to bypass CDDP-induced G2 arrest
DNA damage caused by both CDDP and IR are known to induce cell cycle checkpoint activation and cell cycle arrest in the G2 phase.30,36–38 Because cell cycle regulation is essential for determination of cell fate and is a measure of the DDR independent of γ-H2Ax, we determined the effect of treatment on our NSCLC model system. As expected, H460 cells treated with IR (1.5 Gy) alone showed a prominent G2 arrest at 6 hours and the majority of cells were able to progress through mitosis by 24 hours. Treatment with CDDP (4μM) alone caused a later G2/M arrest at 24 hours, as has been described.30,37,38 Surprisingly, though a G2 arrest was observed 6 hours following CDDP-IR treatment (consistent with that observed in cells treated with IR alone), most cells treated with CDDP-IR were able to complete mitosis and were in G1 phase at 24 hours, behaving in a similar fashion to those treated with IR alone (Figure 3A and B). These findings confirm that persistence of γ-H2Ax foci in CDDP-IR treated NSCLC cells is not due to increased DNA content associated with G2 phase arrest and that these cells treated with CDDP-IR can effectively bypass the CDDP-induced G2 arrest.
Figure 3. Cisplatin-IR treated H460 cells are able to bypass cisplatin-induced G2-arrest.
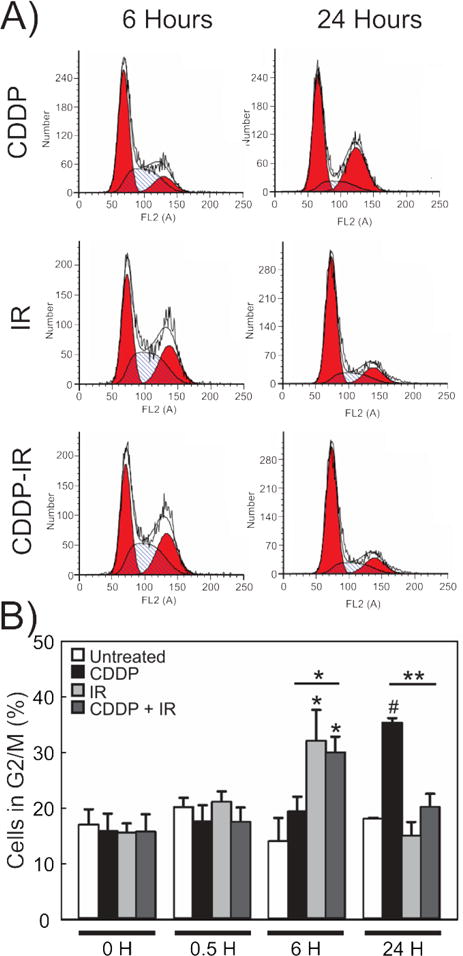
Representative histogram plots (A) and quantification of these results (B) in H460 cells treated with CDDP, IR or CDDP 2 hours prior to IR. N=3. Results are shown as percent cells in G2/M phase +/− SEM. *p<0.05, **p<0.01, #p<0.001.
3.4. Induction of early but not late γ-H2Ax foci is dependent on ATM activation in IR-treated H460 cells
To investigate the mechanism of the DDR pathway activation and its role in persistence of γ-H2Ax foci in CDDP-IR treated NSCLC cells, we assessed DDR kinase activation following treatment. B ecause of the important roles they play in initiating the DDR, phosphorylation of the early (sensor) kinase ATM (pATM) and activation by phosphorylation of the downstream effector proteins Chk2 (pChk2) and Chk1 (pChk1) were measured. We first selected a higher IR treatment dose (6 Gy), since pilot data in H460 cells showed that co-treatment with CDDP (4μM) and lower IR doses (1.5 Gy) caused only mild changes in measured DDR as measured by Western blotting for phosphorylated ATM and Chk2 in H460 cells (Supplemental Figure S4). As expected, treatment with IR and CDDP-IR caused an increase in the phosphorylation of ATM and Chk2. We also observed a time-dependent decrease in their phosphorylation (Figure 4A). Treatment with CDDP at this dose did not significantly alter ATM or Chk2 phosphorylation, but, as expected, did show a mild increase in Chk1 phosphorylation at 6 and 24 hours (Figure 4A). The analyses of combination CDDP-IR treatment at 6 and 24 hours post-treatment showed no significant differences in the magnitude, timing, or duration of ATM or Chk2 phosphorylation when compared to treatment with IR alone (Figure 4A). Gamma-H2Ax expression, as measured by Western blot densitometry, was increased in CDDP, IR and CDDP-IR treated cells when compared to untreated control cells (Figure 4A). There was a statistically significant increase in γ-H2Ax expression in IR and CDDP-IR treated cells 30 minutes after IR treatment, which was reduced by 6 hours (p<0.001, Figure 4B). Measurement of the kinetics of 53BP1 phosphorylation (a specific marker of DNA DSBs) was increased 30 minutes after both IR and CDDP-IR treatment and remained elevated at 6 hours only in CDDP-IR treated cells (Figure 4C), suggesting a possible persistence of DSBs in CDDP-IR treated cells but not in cells treated with IR alone.
Figure 4. DNA damage response to DNA damaging agents over time.
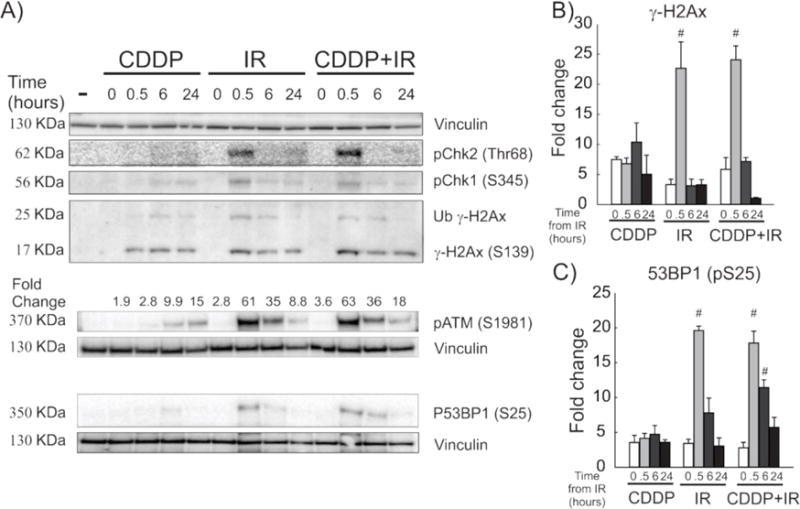
Representative immunoblots (A) show the DDR over time following treatment of H460 cells with CDDP, IR and CDDP-IR. Quantification of pATM (A), γ-H2Ax (B) and P53BP1 (C) expression by Western blot was controlled to vinculin expression and is shown as fold-change over the untreated control +/− SEM. #p<0.001 compared to Time 0 for each treatment type.
In order to further interrogate the role of the DDR in CDDP-IR toxicity, we manipulated the DDR pathways via pharmacologic inhibition. KU-55933 is a highly specific and potent ATP competitive inhibitor of ATM.25 Treatment of H460 cells with 10 μM KU-55933 was chosen for these assays, as this dosage has been previously shown to cause radiosensitization in H460 and A549 cells.10,39 As expected, treatment with KU-55933 resulted in inhibition of ATM phosphorylation as measured by Western blot analysis as well as decreased phosphorylation of the ATM-activated DDR protein, Chk2 (Figure 5A). Treatment with KU-55933 resulted in an altered early (30 minutes post-IR) DDR in cells treated with CDDP-IR and IR alone, but not in those treated with CDDP alone (Figure 5A). KU-55933 also decreased early IR and CDDP-IR induced pChk1, which is consistent with ATM-dependent activation of Chk1 early after IR treatment (Figure 5A). Treatment with KU-55933 also decreased 53BP1 phosphorylation early (30 minutes) following IR in cells treated with either CDDP-IR or with IR alone (Figure 5A and B). However, treatment with KU-55933 resulted in increased phosphorylation of Chk1 in all treated cells at 24 hours, which suggests late activation of the ATR pathway.
We wished to further investigate the role of ATM inhibition on the CDDP-IR induced DDR, ag ain evaluating γ-H2Ax foci development over time by immunofluorescence microscopy following treatment with CDDP, IR or CDDP-IR in the presence of KU-55933. Because of a redundant kinase role in IR-induced H2Ax phosphorylation,21 we also treated cells with the DNA-PK inhibitor, NU7441, at a dose previously shown to inhibit DSB repair.30,33 The ATM inhibitor, KU-55933, impaired γ-H2Ax foci formation in H460 cells early after IR treatment alone (Figure 5C and D, ATMi). ATM inhibition also resulted in fewer γ-H2Ax foci in CDDP-IR treated cells (Figure 5D), with the remaining nuclear γ-H2Ax expression more closely resembling cells treated with CDDP alone, illustrating smaller, less distinct foci (Figure 5C). Those treated with CDDP alone showed no alteration in γ-H2Ax appearance or in the number of γ-H2Ax positive cells when treated with KU-55933 (Figure 5C and D). These results confirm specificity of ATM to early γ-H2Ax foci development by IR-induced DSBs. When imaged at 24 hours, the ATM inhibitor, KU-55933, did not inhibit persistent γ-H2Ax foci in cells treated with CDDP and CDDP-IR (Figure 5C and D). Treatment with the DNA-PK inhibitor, NU7441, did not significantly alter the early or late γ-H2Ax response to treatment with CDDP, IR or CDDP-IR (Figure 5C and E). Inhibition of both ATM (KU-55933) and DNA-PK (NU7441) resulted in a statistically significant decrease in early but not late γ-H2Ax foci in IR and CDDP-IR treated H460 cells (Figure 5F, p<0.05). Similar results were obtained by quantifying average foci per nucleus (Supplemental Figure S5) and in A549 cells (not shown). Together, the data indicate that ATM is essential to the early DDR to CDDP-IR treatment, with induces H2Ax phosphorylation in response to DSBs. However, persistence of γ-H2Ax foci in CDDP-IR treated cells is not dependent on ATM or DNA-PK activation.
3.5. ATR sensitization to CDDP and combination CDDP-IR treatment is independent of early DDR
ATR has been shown to be particularly active in the DDR to CDDP treatment,21 and our Western blot data suggest a potential role for ATR in the late damage response, particularly in cells treated with an ATM inhibitor (Figure 5A). Therefore, we assessed the effect of ATR inhibition using the ATR inhibitor, VE-821. Pre-treatment with VE-821 two hours prior to IR and for 24 hours after IR treatment was performed at 10 μM, as has been recently studied and shown to modify the chemotherapeutic response to CDDP.40 Treatment with VE-821 at this dose was highly specific to inhibition of ATR, as the level of Chk1 phosphorylation, but not pATM or pChk2, was decreased early (30 minutes) after treatment with CDDP, IR and CDDP-IR (Figure 6A). Additionally, treatment with VE-821 did not alter 53BP1 phosphorylation in IR and CDDP-IR treated H460 cells, which demonstrates that ATR inhibition by VE-821 did not alter the early DDR to IR-induced DSBs (Figure 6A). A late increase in ATM and Chk2 phosphorylation was observed in all cells 24 hours after treatment with VE-821, along with a decrease in total Chk1, which suggests late activation of the ATM-dependent DDR pathways following ATR inhibition (Figure 6A).
In order to evaluate for alterations in response to VE-821 in H460 and A549 cells, we next investigated the effects of ATR inhibition of γ-H2Ax formation, as measured by immunofluorescence. Treatment with VE-821 did not inhibit γ-H2Ax formation in IR-treated cells, but decreased the number of nuclei positive for γ-H2Ax in CDDP treated cells at the early (30 minutes) time point (Figure 6B and C). However, early γ-H2Ax foci were still present with ATR inhibition in CDDP-IR treated cells, although the appearance was now punctate, with an appearance similar to cells treated with IR alone (Figure 6B). There was an increase in γ-H2Ax foci formation at 24 hours after treatment with CDDP and IR which did not reach statistical significance (Figures 6C and D); however, this effect was also observed in control cells treated with VE-821 at 24 hours, indicating that this effect is likely due to single-agent toxicity from VE-821. This suggests considerable cross-talk of DNA damage response pathways in H2Ax phosphorylation, as was also suggested by our Western blot analysis (Figure 6A). These results were confirmed in A549 NSCLC cells, with similar effects of ATR inhibition by VE-821 on γ-H2Ax nuclear staining observed in this NSCLC cell line (Supplemental Figure S6).
We next sought to determine the effect of VE-821 on NSCLC survival using clonogenic survival assays. Treatment of H460 cells with VE-821 resulted in dose-dependent single-agent toxicity (data not shown) and the LD10 dose (0.5 μM) was chosen for these assays. Treatment with VE-821 resulted in a dramatic sensitization of H460 cells, with 8 and 5.7 fold reduction in survival observed with VE-821 treatment over CDDP treatment alone (Figure 6E). Further cytotoxicity was observed in cells treated with CDDP-IR, with an 8.3 and 5.8 fold reduction in survival observed (Figure 6E). These results were independent of CDDP dose, as these effects were observed in H460 cells treated with 4 μM (LD50), 2μM (Figure 6E) and 0.5μM (data not shown) doses of CDDP as well. Evaluated independently, the decreased survival observed with the addition of VE-821 to IR treatment was not statistically significant, but combining the results of the six experiments resulted in a modest but statistically significant decrease in survival (p<0.05). However, in A549 cells, no increased cytotoxicity was observed with the addition of VE-821 across a number of different CDDP and VE-821 doses (data not shown). As both of these cells lines are p53 wild-type, and published data suggest that p53 mutational status can impact ATRi sensitization to chemotherapeutics,41,42 we assessed the effect of ATR inhibition on CDDP-IR sensitization in the p53 mutant NSCLC cell line, H1299. Interestingly, treatment of H1299 with VE-821 in combination with CDDP, IR and CDDP-IR gave results similar to those observed in A549 cells over a range of doses of VE-821 and CDDP (data note shown). Overall, these findings support that CDDP radiosensitization and the observed persistence of CDDP-IR induced DSBs is not directly dependent on ATR activation.
4. Discussion
Treatment of NSCLC with CDDP and IR remains a mainstay of therapy, but even with adherence to the best identified treatment regimens, survival remains low.3,4,43 Clarifying the cooperative interaction between CDDP and IR in NSCLC is essential to optimize current treatment regimens and to determine mechanisms which can be exploited to augment cancer toxicity. Our data support radiosensitization by CDDP in H460 and A549 NSCLC cells, which effectively recapitulates the clinical response to concomitant CDDP and IR treatment.5–8 The temporal nature of this interaction suggests that CDDP treatment primes the cell to IR-induced toxicity. Supporting our data are several studies which suggest a clear role for CDDP impairing DNA DSB repair. For instance, a deficiency in repair of CDDP lesions on DNA augments CDDP radiosensitization.17,28,29 Additionally, treatment of NHEJ-deficient cells with CDDP-IR show increased toxicity, but no longer show a synergistic interaction, highlighting the importance of NHEJ catalyzed DSB repair to the cooperative cytotoxicity of CDDP-IR co-treatment.17,28 We attribute this CDDP radiosensitization to an increase in CDDP residence time on the DNA such that upon treatment with IR there are increased opportunities for complex DNA damage. Additional support for CDDP inhibition of NHEJ-dependent DSB repair includes findings that CDDP lesions are able to inhibit translocation of Ku at DNA ends and are also able to inhibit DNA-PK activation.44,45 Furthermore, the presence of a CDDP lesion impairs NHEJ-dependent repair of adjacent DNA termini, suggesting CDDP inhibition of NHEJ-dependent repair of an adjacent IR-induced DSB as a potential mechanism for CDDP radiosensitization.17,30,31
Although typically a measure of DSBs, H2Ax phosphorylation has been described in response to other DNA damaging agents, including CDDP, and the differences in γ-H2Ax foci morphology we observed 30 minutes following CDDP and IR treatment are consistent with previously published work.19,35 Our study showed no difference at 30 minutes in the number of cells expressing γ-H2Ax foci or in the number of foci formed in NSCLC cells treated with IR alone compared to cells treated with CDDP-IR. This would seemingly contradict the findings of Sak et al, who described a decrease in γ-H2Ax foci formation in peripheral blood lymphocytes obtained from patients treated with CDDP and then exposed to IR in vitro.46 Our model uses NSCLC cells treated with clinically relevant doses of CDDP-IR. It is not surprising that the DDR induced by CDDP-IR differs between peripheral blood lymphocytes and clonally proliferating H460 and A549 NSCLC cells, which display the unchecked proliferation and cell cycle regulation characteristic of malignant cells. It is interesting that treatment of NSCLC cells with CDDP-IR in our treatment model caused abrogation of the CDDP-induced G2 arrest. While the mechanism behind is not fully defined, this is likely caused by the different kinetics of IR- and CDDP-induced DDR on cell cycle progression. As other agents that are able to abrogate chemotherapeutic-induced G2 arrest have demonstrated increased cytotoxicity, we suspect that this likely contributes to the increased cytotoxicity of CDDP-IR treatment.
The temporal difference in γ-H2Ax foci morphology observed in H460 and A549 at 30 minutes and 24 hours after CDDP-IR treatment is novel. Importantly, we are the first to describe persistence of γ-H2Ax foci only in those treated with CDDP-IR as compared to IR alone. This finding is critical, as persistence of γ-H2Ax positivity 24-hours following CDDP treatment correlates to reduced overall survival following CDDP and IR treatment.19,47,48 Measurement of γ-H2Ax via Western blot did not reveal a significant difference in intensity between cells treated with IR and CDDP-IR at 24 hours. This is consistent with publications showing that the relative intensity of γ-H2Ax as measured by Western blot does not correlate to the functional importance of γ-H2Ax when treated with DNA damaging agents.35 However, phosphorylation of 53BP1, a specific marker of DNA DSBs, was prolonged in CDDP-IR treated cells as compared to IR alone (Figure 4C), which would support a mechanism by which CDDP-IR treatment leads to persistence of DNA DSBs. Further supporting this mechanism is the finding that γ-H2Ax foci observed in CDDP-IR treated NSCLC cells at 24 hours displayed a punctate appearance, similar to those observed 30 minutes after IR treatment alone, and our observation that these persistent, punctate γ-H2Ax foci overlay with 53BP1 foci (Figure 2D). At least two possible mechanisms could explain this phenomenon. First, as the presence of a CDDP-DNA lesion in close proximity to a DSB impairs NHEJ-dependent DSB repair,30 it is possible that each of these γ-H2Ax foci represents an unresolved CDDP-DSB lesion. While the number of γ-H2Ax foci persisting at 24 hours is likely more than could be explained by the random distribution of complex CDDP-DSB lesions, the presence of a CDDP lesion could differentially influence the position of DSB distribution so that complex CDDP-DSB lesions are more frequent than would be expected by random distribution, leading to persistent γ-H2Ax foci at the site of unrepaired DSBs. This is supported by studies suggesting CDDP causes differential distribution of DSBs following treatment with IR and bleomycin49,50 and an increased number of IR-induced DSBs51, although this remains controversial.52 Additionally, H460 cells treated with CDDP-IR were able to progress through the G2/M checkpoint prior to the G2 arrest induced by CDDP treatment alone (Figure 3). It is known that daughter cells inherit residual γ-H2Ax foci,48 but this alone does not explain γ-H2Ax persistence following CDDP-IR, as treatment with IR caused similar progression through the cell cycle as treatment with CDDP-IR, but with resolution of γ-H2Ax foci. Progression through the cell cycle without repair of the CDDP-DNA lesions could result in the development of complex CDDP-DSBs at the site of stalled replication forks or non-DSB cluster lesions (comprised of two or more DNA SSBs on opposing DNA strands) in close proximity to CDDP lesions, as has recently been suggested by Sahbani et al.52 These findings further support a potential mechanism by which cooperative cytotoxicity observed in CDDP-IR treated NSCLC cells is caused by persistence of DNA DSBs.
Our data show that persistence of γ-H2Ax foci in CDDP-IR treated NSCLC cells occurs independently of persistent or prolonged activation of the sensory and effector DDR kinases, including pATM, pChk1 and pChk2. However, activation of the sensor DDR kinases, ATM and ATR, is important in the early γ-H2Ax response to CDDP, IR and CDDP-IR treatments. As expected, inhibition of ATM (KU-55933) impairs IR-induced γ-H2Ax foci development and inhibition of ATR (VE-821) specifically impairs CDDP-induced γ-H2Ax staining. Here we show that both ATM and ATR inhibitors cause distinct alterations in γ-H2Ax nuclear staining patterns in CDDP-IR treated cells by inhibition of the early IR- or CDDP-specific staining patterns respectively. However, both KU-55933 and VE-821 failed to inhibit γ-H2Ax foci persistence in CDDP-IR treated cells, again highlighting the importance of CDDP to IR sensitization and supporting cross-activation of DDR pathways.21 Synergy between VE-821 and CDDP has been previously reported23,24 and treatment with the ATR inhibitor VE-821 at a dose resulting in low single-agent toxicity resulted in selective synergy in H460 cells treated with CDDP and CDDP-IR with only a moderate decrease in survival in those treated with IR alone, consistent with an ATR-dependent DDR by CDDP.21 However, treatment of A549 and H1299 NSCLC cells with VE-821 did not result in synergy with CDDP or CDDP-IR. Variable responses of cancer cells to VE-821 have been described, and, at least in part, ascribed to differences in ATM-p53 activation.41,42 However, our model showed a lack of sensitization in a p53 wild-type (A549) and mutant (H1299) cell lines. Many additional factors have been shown to impact VE-821 chemo- and radiosensitization, including duration of VE-821 dosing, with longer treatment associated with decreased survival,53 and other measures of cytotoxicity such as MTT, have been used to support these findings. Our findings are consistent with the large variability in chemosensitization observed with ATR inhibitors, which are likely due to a combination of differences in cancer cell characteristics, treatment conditions and outcome measures in vitro. However, persistence of γ-H2Ax foci in CDDP-IR treated cells is independent of sensitization by ATR inhibition, which further supports a mechanism by which CDDP radiosensitization is due to impaired DSB repair and independent of the impact of DDR activation by CDDP-IR treatment.
5. Conclusion
In conclusion, our data indicate that the cooperative cytotoxicity observed with CDDP and IR treatment is sequence dependent, requiring treatment with CDDP prior to IR. Combination treatment with CDDP followed by IR leads to persistence of γ-H2Ax foci at 24 hours, which persist even with inhibitors of the DDR sensor kinases, ATM, ATR and DNA-PK. This supports a mechanism by which CDDP-IR cytotoxicity is due to persistence of unrepaired DSBs. The further characterization of CDDP radiosensitization and the role of DDR sensor kinase inhibitors in NSCLC CDDP-IR cytotoxicity are important and timely. Selection of ATM and ATR inhibitors based on the specific type of DNA damaging treatment and identified genetic make-up of the tumor may lead to optimization of tumor-specific cytotoxicity, particularly as ATR inhibitors are entering early clinical trials (Clinical Trials.gov: NCT02157792 and NCT02223923). Our results signal the need for further evaluation of VE-821 and similar drugs to identify specific tumor characteristics which would predict response to these therapies. Furthermore, it supports a potential role of small molecular inhibitors of proteins involved in NER and HRR, the primary pathways involved in repair of CDDP lesions on DNA, in potentiation of CDDP-IR cytotoxicity.
Supplementary Material
Highlights.
Combination cisplatin–radiation causes sequence dependent cytotoxicity in non-small cell lung cancer
Treatment with combination cisplatin-ionizing radiation results in persistent DNA double strand breaks
Cells treated with ionizing radiation following cisplatin abrogate cisplatin mediated G2 arrest
Persistence of DNA double strand breaks caused by cisplatin-radiation therapy is independent of early DNA damage response kinases
Acknowledgments
We thank Dr. Irina Petrache for the use of her fluorescence microscope. We appreciate the helpful discussions and comments from members of the Turchi, Petrache and Jalal laboratories regarding this manuscript. This work was supported by the Indiana University-Health Values Grant (VHF-370-SEARS), the Ralph and Grace M. Showalter Research Fund and funding from the American Cancer Society to the Indiana University Simon Cancer Center (IRG-84-002-28) to C.R. Sears and by the National Institutes of Health R01 CA180710 to J.J. Turchi. The funding sources listed had no involvement in study design, data collection, analysis, or interpretation or in the writing of this report.
Abbreviations
- NSCLC
non-small cell lung cancer
- CDDP
cisplatin
- IR
ionizing radiation
- CDDP-IR
combination treatment with cisplatin followed by ionizing radiation
- DDR
DNA damage response
- DSB
double strand break
- NER
nucleotide excision repair
- HRR
homologous recombination repair
- NHEJ
non-homologous end-joining
- Gy
Gray
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure of Potential Conflicts of Interest: All authors report no conflict of interest and do not have any financial relationships with commercial entities that have an interest in the subject of this manuscript.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 2.Wakelee HA, Bernardo P, Johnson DH, Schiller JH. Changes in the natural history of nonsmall cell lung cancer (NSCLC)—Comparison of outcomes and characteristics in patients with advanced NSCLC entered in Eastern Cooperative Oncology Group trials before and after 1990. Cancer. 2006;106:2208–2217. doi: 10.1002/cncr.21869. [DOI] [PubMed] [Google Scholar]
- 3.Jett JR, Schild SE, Keith RL, Kesler KA. Treatment of Non-small Cell Lung Cancer, Stage IIIB: ACCP Evidence-Based Clinical Practice Guidelines (2nd Edition) Chest. 2007;132:266S–276S. doi: 10.1378/chest.07-1380. [DOI] [PubMed] [Google Scholar]
- 4.Stinchcombe TE, Socinski MA. Current Treatments for Advanced Stage Non-Small Cell Lung Cancer. Proc Am Thorac Soc. 2009;6:233–241. doi: 10.1513/pats.200809-110LC. [DOI] [PubMed] [Google Scholar]
- 5.O’Rourke N, Roqué i Figuls M, Farré Bernadó N, Macbeth F. Concurrent chemoradiotherapy in non-small cell lung cancer. Cochrane Database of Systemic Reviews. 2010 doi: 10.1002/14651858.CD002140.pub3. Art. No.: CD002140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Auperin A, et al. Meta-Analysis of Concomitant Versus Sequential Radiochemotherapy in Locally Advanced Non-Small-Cell Lung Cancer. J Clin Oncol. 2010;28:2181–2190. doi: 10.1200/jco.2009.26.2543. [DOI] [PubMed] [Google Scholar]
- 7.Groen H, et al. Carboplatin- and cisplatin-induced potentiation of moderate-dose radiation cytotoxicity in human lung cancer cell lines. Br J Cancer. 1995;1995:1406–1411. doi: 10.1038/bjc.1995.522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Curran WJ, et al. Sequential vs Concurrent Chemoradiation for Stage III Non–Small Cell Lung Cancer: Randomized Phase III Trial RTOG 9410. J Natl Cancer Inst. 2011;103:1452–1460. doi: 10.1093/jnci/djr325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dolling J-A, Boreham DR, Brown DL, Raaphorst GP, Mitchel REJ. Cisplatin-modifications of DNA repair and ionizing radiation lethality in yeast, Saccharomyces cerevisiae. Mutat Res. 1999;433:127–136. doi: 10.1016/s0921-8777(98)00069-x. [DOI] [PubMed] [Google Scholar]
- 10.Toulany M, Mihatsch J, Holler M, Chaachouay H, Rodemann HP. Cisplatin-mediated radiosensitization of non-small cell lung cancer cells is stimulated by ATM inhibition. Radiotherapy and oncology: journal of the European Society for Therapeutic Radiology and Oncology. 2014;111:228–236. doi: 10.1016/j.radonc.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 11.Sakai W, et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. 2008;451:1116–1120. doi: 10.1038/nature06633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kartalou M, Essigmann JM. Mechanisms of resistance to cisplatin. Mutat Res. 2001;478:23–43. doi: 10.1016/s0027-5107(01)00141-5. [DOI] [PubMed] [Google Scholar]
- 13.McKay BC, Becerril C, Ljungman M. P53 plays a protective role against UV- and cisplatin-induced apoptosis in transcription-coupled repair proficient fibroblasts. Oncogene. 2001;20:6805–6808. doi: 10.1038/sj.onc.1204901. [DOI] [PubMed] [Google Scholar]
- 14.Koberle B, Masters JRW, Hartley JA, Wood RD. Defective repair of cisplatin-induced DNA damage caused by reduced XPA protein in testicular germ cell tumors. Curr Biol. 1999;9:273–276. doi: 10.1016/s0960-9822(99)80118-3. [DOI] [PubMed] [Google Scholar]
- 15.Ho KSY. Induction of DNA Double-Strand Breaks by X-Rays in a Radiosensitive Strain of the Yeast Saccharomyces Cerevisiae. Mutat Res. 1975;30:327–334. [PubMed] [Google Scholar]
- 16.Jeggo P, Lavin MF. Cellular radiosensitivity: How much better do we understand it? Int J Radiat Biol. 2009;85:1061–1081. doi: 10.3109/09553000903261263. [DOI] [PubMed] [Google Scholar]
- 17.Boeckman HJ, Trego KS, Turchi JJ. Cisplatin Sensitizes Cancer Cells to Ionizing Radiation via Inhibition of Nonhomologous End Joining. Mol Cancer Res. 2005;3:277–285. doi: 10.1158/1541-7786.mcr-04-0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rogakou EP, Pilch AH, Ivanova VS, Bonner WM. DNA Double-stranded Breaks Induce Histone H2AX Phosphorylation on Serine 139. J Biol Chem. 1998;273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 19.Olive PL, Banath JP. Kinetics of H2AX phosphorylation after exposure to cisplatin. Cytometry B Clin Cytom. 2009;76:79–90. doi: 10.1002/cyto.b.20450. [DOI] [PubMed] [Google Scholar]
- 20.Sedelnikova OA, Rogakou EP, Panyutin IG, Bonner WM. Quantitative Detection of 125IdU-Induced DNA Double-Strand Breaks with γ-H2AX Antibody. Radiat Res. 2002;158:486–492. doi: 10.1667/0033-7587(2002)158[0486:qdoiid]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 21.Woods D, Turchi JJ. Chemotherapy induced DNA damage response: convergence of drugs and pathways. Cancer Biol Ther. 2013;14:379–389. doi: 10.4161/cbt.23761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ahmad S. Platinum-DNA Interactions and Subsequent Cellular Processes Controlling Sensitivity to Anticancer Platinum Complexes. Chem Biodivers. 2010;7:543–565. doi: 10.1002/cbdv.200800340. [DOI] [PubMed] [Google Scholar]
- 23.Huntoon CJ, et al. ATR inhibition broadly sensitizes ovarian cancer cells to chemotherapy independent of BRCA status. Cancer Res. 2013;73:3683–3691. doi: 10.1158/0008-5472.CAN-13-0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hall AB, et al. Potentiation of tumor responses to DNA damaging therapy by the selective ATR inhibitor VX-970. Oncotarget. 2014;5:5674–5685. doi: 10.18632/oncotarget.2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hickson I, et al. Identification and Characterization of a Novel and Specific Inhibitor of the Ataxia-Telangiectasia Mutated Kinase ATM. Cancer Res. 2004;64:9152–9159. doi: 10.1158/0008-5472.CAN-04-2727. [DOI] [PubMed] [Google Scholar]
- 26.Taccioli GE, et al. Targeted Disruption of the Catalytic Subunit of the DNA-PK Gene in Mice Confers Severe Combined Immunodeficiency and Radiosensitivity. Immunity. 1998;9:355–366. doi: 10.1016/s1074-7613(00)80618-4. [DOI] [PubMed] [Google Scholar]
- 27.Rosenzweig KE, Youmell MB, Palayoor ST, Price BD. Radiosensitization of Human Tumor Cells by the Phosphatidylinositol 3-Kinase Inhibitors Wortmannin and LY294002 Correlates with Inhibition of DNA-dependent Protein Kinase and Prolongued G2-M Delay. Clin Cancer Res. 1997;3:1149–1156. [PubMed] [Google Scholar]
- 28.Myint WK, Cheng NG, Raaphorst GP. Examining the non-homologous repair process following cisplatin and radiation treatments. Int J Radiat Biol. 2002;78:417–424. doi: 10.1080/0955300011011304. [DOI] [PubMed] [Google Scholar]
- 29.Raaphorst G, LeBlanc J-M, Li LF. A Comparison of Response to Cisplatin, Radiation and Combined Treatment for Cells Deficient in Recombination Repair Pathways. Anticancer Res. 2005;25:53–58. [PubMed] [Google Scholar]
- 30.Sears CR, Turchi JJ. Complex cisplatin-double strand break (DSB) lesions directly impair cellular non-homologous end-joining (NHEJ) independent of downstream damage response (DDR) pathways. The Journal of biological chemistry. 2012;287:24263–24272. doi: 10.1074/jbc.M112.344911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Diggle CP. Inhibition of double-strand break non-homologous end-joining by cisplatin adducts in human cell extracts. Nucleic Acids Res. 2005;33:2531–2539. doi: 10.1093/nar/gki528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mendonca MS, et al. Differential mechanisms of x-ray-induced cell death in human endothelial progenitor cells isolated from cord blood and adults. Radiat Res. 2011;176:208–216. doi: 10.1667/rr2427.1. [DOI] [PubMed] [Google Scholar]
- 33.Stiff T, et al. ATM and DNA-PK Function Redundantly to Phosphorylate H2AX after Exposure to Ionizing Radiation. Cancer Res. 2004;64:2390–2396. doi: 10.1158/0008-5472.can-03-3207. [DOI] [PubMed] [Google Scholar]
- 34.Byun J, et al. Automated tool for the detection of cell nuclei in digital microscopic images: Application to retinal images. Mol Vis. 2006;2006 [PubMed] [Google Scholar]
- 35.Revet I, et al. Functional relevance of the histone gammaH2Ax in the response to DNA damaging agents. Proc Natl Acad Sci U S A. 2011;108:8663–8667. doi: 10.1073/pnas.1105866108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maity A, McKenna WG, Muschel RJ. The molecular basis for cell cycle delays following ionizing radiation: a review. Radiother Oncol. 1994;31:1–13. doi: 10.1016/0167-8140(94)90408-1. [DOI] [PubMed] [Google Scholar]
- 37.Almeida GM, Duarte TL, Farmer PB, Steward WP, Jones GDD. Multiple end-point analysis reveals cisplatin damage tolerance to be a chemoresistance mechanism in a NSCLC model: Implications for predictive testing. Int J Cancer. 2008;122:1810–1819. doi: 10.1002/ijc.23188. [DOI] [PubMed] [Google Scholar]
- 38.Fujikane T, et al. Flow Cytometric Analysis of the Kinetic Effects of Cisplatin on Lung Cancer Cells. Cytometry. 1989;10:788–795. doi: 10.1002/cyto.990100617. [DOI] [PubMed] [Google Scholar]
- 39.Shaheen FS, et al. Targeting the DNA Double Strand Break Repair Machinery in Prostate Cancer. PLoS ONE. 2011;6:e20311. doi: 10.1371/journal.pone.0020311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ashley AK, et al. DNA-PK phosphorylation of RPA32 Ser4/Ser8 regulates replication stress checkpoint activation, fork restart, homologous recombination and mitotic catastrophe. DNA Repair. 2014;21:131–139. doi: 10.1016/j.dnarep.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fokas E, et al. Targeting ATR in vivo using the novel inhibitor VE-822 results in selective sensitization of pancreatic tumors to radiation. Cell Death Dis. 2012;3:e441. doi: 10.1038/cddis.2012.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reaper PM, et al. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat Chem Biol. 2011;7:428–430. doi: 10.1038/nchembio.573. 10.1038/NCHEMBIO.573. [DOI] [PubMed] [Google Scholar]
- 43.Detterbeck FC, Boffa DJ, Tanoue LT. The New Lung Cancer Staging System. Chest. 2009;136:260–271. doi: 10.1378/chest.08-0978. [DOI] [PubMed] [Google Scholar]
- 44.Turchi JJ, Henkels KM. Human Ku Autoantigen Binds Cisplatin-damaged DNA but Fails to Stimulate Human DNA-activated Protein Kinase. J Biol Chem. 1996;271:13861–13867. doi: 10.1074/jbc.271.23.13861. [DOI] [PubMed] [Google Scholar]
- 45.Turchi JJ, Henkels KM, Zhou Y. Cisplatin-DNA adducts inhibit translocation subunits of DNA-PK. Nucleic Acids Res. 2000;28:4634–4641. doi: 10.1093/nar/28.23.4634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sak A, et al. Long-Term In vivo Effects of Cisplatin on γ-H2AX Foci Signaling in Peripheral Lymphocytes of Tumor Patients After Irradiation. Clin Cancer Res. 2009;15:2927–2934. doi: 10.1158/1078-0432.ccr-08-0650. [DOI] [PubMed] [Google Scholar]
- 47.Banuelos CA, Banath JP, Kim JY, Aquino-Parsons C, Olive PL. gammaH2AX expression in tumors exposed to cisplatin and fractionated irradiation. Clinical cancer research: an official journal of the American Association for Cancer Research. 2009;15:3344–3353. doi: 10.1158/1078-0432.CCR-08-3114. [DOI] [PubMed] [Google Scholar]
- 48.Klokov D, MacPhail SM, Banath JP, Byrne JP, Olive PL. Phosphorylated histone H2AX in relation to cell survival in tumor cells and xenografts exposed to single and fractionated doses of X-rays. Radiotherapy and oncology: journal of the European Society for Therapeutic Radiology and Oncology. 2006;80:223–229. doi: 10.1016/j.radonc.2006.07.026. [DOI] [PubMed] [Google Scholar]
- 49.Mascharak PK, Sugiura Y, Kuwahara J, Suzuki T, Lippard SJ. Alteration and activation of sequence-specific cleavage of DNA by bleomycin in the presence of the antitumor drug cis-diamminedichloroplatinum(II) Proc Natl Acad Sci U S A. 1983;80:6795–6798. doi: 10.1073/pnas.80.22.6795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vràna O, Brabec V. The effect of combined treatment with platinum complexes and ionizing radiation on DNA in vitro. Int J Radiat Biol Relat Stud Phys Chem Med. 1986;50:995–1007. doi: 10.1080/09553008614551411. [DOI] [PubMed] [Google Scholar]
- 51.Rezaee M, Sanche L, Hunting DJ. Cisplatin enhances the formation of DNA single- and double-strand breaks by hydrated electrons and hydroxyl radicals. Radiat Res. 2013;179:323–331. doi: 10.1667/RR3185.1. [DOI] [PubMed] [Google Scholar]
- 52.Kouass Sahbani S, Rezaee M, Cloutier P, Sanche L, Hunting DJ. Non-DSB clustered DNA lesions induced by ionizing radiation are largely responsible for the loss of plasmid DNA functionality in the presence of cisplatin. Chem Biol Interact. 2014;217:9–18. doi: 10.1016/j.cbi.2014.04.004. [DOI] [PubMed] [Google Scholar]
- 53.Prevo R, et al. The novel ATR inhibitor VE-821 increases sensitivity of pancreatic cancer cells to radiation and chemotherapy. Cancer Biol Ther. 2012;13:1072–1081. doi: 10.4161/cbt.21093. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.