

Abstract
Background
Drugs that inhibit cyclooxygenase-2 (COX-2) while sparing cyclooxygenase-1 (COX-1) represent a new attractive therapeutic development and offer new perspective for further use of COX-2 inhibitors. Intention of this work is to develop safer, selective COX-2 inhibitors that do not produce harmful effects.
Results
A series of 55 tyrosine derivatives were designed for evaluation as selective COX-2 inhibitors and investigated by in silico for their anti-inflammatory activities using C-Docker. The results of docking study showed that 35 molecules were found to selectively inhibit the enzyme COX-2. These molecules formed stable π hydrophobic and additional van der Waals interactions in the active site side pocket of COX-2. The molecules selected from docking studies were examined through ADMET descriptors and Osiris property explorer to find its safety profile as well. The tyrosine derivatives containing toxic fragments were eliminated.
Conclusion
The results conclude that out of 55, 19 molecules possessed best binding energy (< −3.333 kcal/mol) and these molecules had more selective and safer COX-2 inhibitor profile compared to the standard celecoxib.
Graphical abstract.
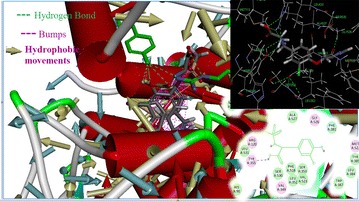
3-D structural interactions of COX-2 inhibiting tyrosine derivatives.
Keywords: Anti-inflammatory, Tyrosine derivatives, Docking, ADMET descriptors, Osiris
Background
Cyclooxygenase-1 (COX-1) and Cyclooxygenase-2 (COX-2) are two discrete isoforms of cyclooxygenase enzyme. These enzymes play a catalytic role in transfiguration of arachidonic acid to prostaglandins in the cyclic pathway of arachidonic acid [1, 2]. Prostaglandins (PGs) are involved in various pathophysiological conditions such as inflammation, carcinogenesis, cardiovascular activity etc. Generally, COX-2 is not detectable in most normal tissues, but it is induced by pro-inflammatory cytokines, growth factors and carcinogens. This fact indicates the role of COX-2 in inflammation [3]. Rheumatoid synovium expression of COX-2 is up regulated in inflammatory tissues resulting in the production of prostaglandin precursors which ultimately gets converted into PGs [4].
Some of the coxib derivatives, Rofecoxib, Celecoxib, Etoricoxib and Valdecoxib are selective COX-2 inhibitors that act by blocking COX-2 enzyme responsible for inflammation and pain [5]. Most of these coxib derivatives have been voluntarily withdrawn from the worldwide market due to safety concerns of an increased risk of cardiovascular events in patients. Due to greater therapeutic effect, Celecoxib is remaining in the market, even though it have a risk of serious and potentially fatal adverse cardiovascular thrombotic events, myocardial infarction and stroke [6].
Importantly, design of agents with higher anti-inflammatory potential and less side effects is one of the most challenging areas in the inflammation. On review of literature, researchers have proved anti-inflammatory effects for dibromotyrosine derivatives [7]. In this concern, we searched for tyrosine scaffold from the natural sources since the biologically active natural compounds are composed of very complex structures. This complexity makes the compounds extremely novel. The marine sponges such as Psammaplysilla purpurea and Ianthella basta are known to produce biogenetically related bromotyrosine derived secondary metabolites [8, 9]. These observations prompted us to design and develop analogue(s) of bromotyrosine derivatives which specifically inhibits COX-2 with improved biological activity. As part of this drug development, an effort has been made to develop higher-quality drug candidates through computational techniques.
Methods
Ligand preparation
A library of novel 55 tyrosine molecules were designed based on the SAR studies of known anti-inflammatory drugs. These molecules were generated with tyrosine as a basic skeleton. The 15 (R1) and 16 (R2) position of aromatic ring hydrogen was substituted with different electronegative groups such us, –I, –Br, –Cl and –NO2. Further, one hydrogen atom of –NH2 group in 14 (R3) position was replaced by –SO2CH3 group. The eighth position (R4) of phenolic –OH group hydrogen was replaced by diverse heterocyclic fragments (Fig. 1). The structures of these molecules were drawn in Hyperchem molecular modeling and visualization tool (version 7.5) and the energies were minimized using ADS. The minimized ligands and proteins were saved in structure data (.sd) and.pdb format (Fig. 2) respectively for further studies.
Fig. 1.
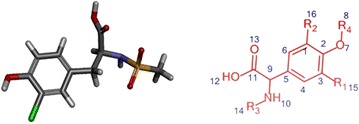
3D and 2D structure of energy minimized tyrosine derivatives
Fig. 2.
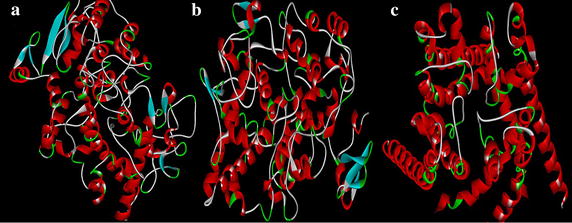
Minimized secondary structure of a COX-2 (3NT1) b COX-1 (3KK6) c hERG protein (homology model)
Docking study
The docking study was performed using Accelyrs Discovery Studio client version 2.5 software (Accelyrs Inc., http://www.accelrys.com). The X-ray crystallographic structure of COX-2 (PDB ID 3NT1) protein bound with naproxen was acquired from the protein data bank (PDB) at a resolution of 1.73 Å (Table 1). The active site was defined with a 8.500 (Å) radius around the bound inhibitor which covered all the active site amino acids of the COX-2 protein. A grid-based molecular docking method, C-DOCKER algorithm was used to dock the small molecules into the protein active site. The designed structures were submitted to CHARMm (Chemistry at HARvard Macromolecular Mechanics) force field for structure refinement. All water molecules, bound inhibitor and other hetero atoms were removed from the macromolecule and polar hydrogen atoms were added. The designed structures were also verified for its valency, missing hydrogen and any structural disorders like connectivity and bond orders. Energy minimization was carried out for all compounds using CHARMm force field to make stable conformation of protein with an energy gradient of 0.01 kcal/mol/A°. A final minimization of the ligand in the rigid receptor using non-softened potential was performed. For each final pose, the CHARMm energy (interaction energy plus ligand strain) and the interaction energy alone were calculated. The poses were sorted by CHARMm energy and the top scoring (most negative, thus favorable to binding) poses. The energy minimized individual proteins and the designed structures along with the binding site sphere radius (Table 2; Fig. 3) and the X, Y and Z coordinates (Table 3) were submitted to the C-Docker job parameter. The docked conformation which had the lowest C-Docker energy was selected to analyze the mode of binding pattern. The C-Docker energy score, hydrogen bond and VDW interactions were visualized in C-Docker report and used for further analysis.
Table 1.
Protein resolution and its stable conformational energy
| PDB ID | Description | Resolution (Å) | Initial potential energy (kcal/mol) |
Final potential energy (kcal/mol) |
|---|---|---|---|---|
| 3NT1 | High resolution structure of naproxen:COX-2 complex | 1.73 | −492,721 | −500,025 |
| 3KK6 | Crystal structure of COX-1 in complex with celecoxib | 2.75 | 248,964,312.95 | −34,200.97 |
| HMa | hERG IFD S terfenadine model 1 | – | −15,609 | −21,445.6 |
a Homology modeling
Table 2.
Binding sphere radius and X, Y and Z coordinate values of defined protein binding site
| Protein PDB ID | Binding sphere radius (Å) | Coordinates (Å) | ||
|---|---|---|---|---|
| X | Y | Z | ||
| 3NT1 | 8.50067 | −40.406 | −51.829 | −22.502 |
| 3KK6 | 6.98804 | −32.413 | −51.829 | −5.617 |
| hERG_IFD_S−terfenadine_model_1 | 7.41161 | 189.526 | −0.442 | 40.737 |
Fig. 3.
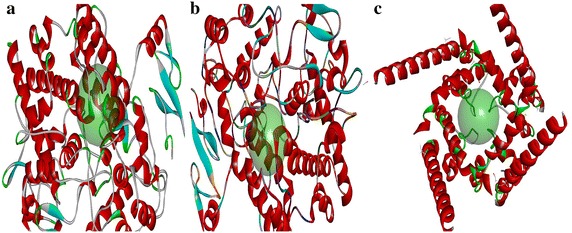
Binding site representation of proteins a COX-2 b COX-1 c hERG_IFD_S- terfenadine_model_1
Table 3.
C-Docker docking protocol parameters
| Parameters | Inputs |
|---|---|
| Input receptor | ../Input/3NT1.dsv |
| Input ligands | C:\Users\g\Desktop\all 55 new.sd |
| Input site sphere | −40.4058, −51.8288, −22.5019, 8.50067 |
| Top hits | 1 |
| Random conformations | 10 |
| Random conformations dynamics steps | 1000 |
| Random conformations dynamics target temperature | 1000 |
| Include electrostatic interactions | True |
| Orientations to refine | 10 |
| Maximum bad orientations | 800 |
| Orientation VDW energy threshold | 300 |
| Simulated annealing | True |
| Heating steps | 2000 |
| Heating target temperature | 700 |
| Cooling steps | 5000 |
| Cooling target temperature | 300 |
| Force field | CHARMm |
| Use full potential | TRUE |
| Grid extension | 8 |
| Ligand partial charge method | CHARMm |
| Random number seed | 314,159 |
| Final minimization | Full potential |
| Random dynamics time step | 0.002 |
The potential fatal adverse effects viz ulcerogenecity and cardiotoxicity were determined by C-Docker using the crystal structures of COX-1 in complex with celecoxib (3KK6:2.75 Å) and hERG_IFD_S-terfenadine_model_1 [Homology model (HM)] (Table 1) which were chosen from the PDB and Schrodinger website respectively. The binding sites of the COX-1 (3KK6) and hERG proteins were defined with the radii of 6.988 and 7.411 Ǻ respectively. The novelty of the final hits was confirmed using SciFinder [10] and PubChem [11] structure search tools.
Docking protocol validation
The validation of the docking protocol is essential to analyse the prediction ability of the proposed method [12]. In this study, validation is performed by two methods to verify whether our docking protocol is able to discriminate selective and non-selective COX-2 inhibitors. To start with, four native co-crystallised ligands of selective and non-selective COX-2 inhibitors were identified and kept as reference template. The structures of these ligands were drawn separately and its energies were minimized. RMSD values were calculated and analysed by redocking the energy minimised ligand on reference template by molecular overlay technique in ADS. In the second method, the structures of various selective and non-selective inhibitors were drawn and the potential energies of the molecules were minimized with the help of conjugated gradient algorithm. Further, these molecules were docked with the COX-2 (3NT1) protein to calculate the binding energies. The experimental IC50 activity values of these molecules were compared with its corresponding predicted C-Docker energy values and the point plot is graphed to identify the correlation between the IC50 and C-Docker energy.
Toxicity study
ADMET descriptors
Most of the failure of drug candidates during clinical trials is due to its poor pharmacokinetic and toxicity properties [13]. Hence, prediction of ADMET properties prior to expensive experimental procedures is considered to be essential for the selection of successful candidates. In this work, in silico ADMET studies were done using ADMET descriptors algorithm of ADS. This protocol uses the six pharmacokinetic parameters like Human Intestinal Absorption (HIA), Blood–Brain–Barrier (BBB) penetration, aqueous solubility, hepatotoxicity levels, cytochrome P450 2D6 inhibition and Plasma Protein Binding (PPB) to quantitatively predict the molecular properties of selected 35 ligands.
Osiris property explorer
Toxicity risks (mutagenicity, tumorigenicity, skin irritation, reproduction) and physicochemical properties (drug likeness and drug score) of the selected 35 tyrosine derivatives were calculated using OSIRIS Property Explorer (free web-based program). The drug likeness (d) was calculated with the following equation by summing up the scores of molecular fragments (Vi) and n indicates the number of molecular fragments [14].
| 1 |
The fragment list was created by shredding 3300 traded drug as well as 1500 commercially available chemicals.
The drug score (ds) combines drug-likeness, cLogP, logS, molecular weight and toxicity risks in one handy value that may be used to judge the compound’s overall potential to be qualified as a drug. This value was calculated by multiplying the contributions of individual properties with Eq. (1) [15].
| 2 |
ds is the drug score. si are the contributions calculated directly from of cLogP, logS, molecular weight and drug-likeness ti is the contribution taken from the four toxicity risk types via the Eq. (2) which describes a spline curve.
Results and discussion
Docking
The results of C-Docker protocol run were analysed. These results have provided essential information relating to the orientation of the tyrosine derivatives in the active site of proteins (3NT1, 3KK6, hERG).
Molecular docking
In this study, 35 drug-like hit compounds were selected from the designed 55 tyrosine derivatives based on their better binding affinity (–C-Docker energy) compared to the standard celecoxib (Table 4). The active site was defined based on the bound inhibitor, naproxen, in a crystal structure of COX-2 (PDB code 3NT1). The important criteria considered in the selection of best hit compounds was binding modes, molecular interactions with the active site components and fitness scores. Evaluation of the interaction pattern of tyrosine derivatives makes clear that the molecule 8 (Fig. 4) have six folds higher affinity (−78.7003) in the COX-2 active site compared to standard celecoxib (17.3339). This interaction affinity is due to the 24th oxygen atom of the carboxylic group in tyrosine moiety has formed two site point interactions with the binding site residue of Arg120 and Tyr355 residue. The 25th oxygen atom of the molecule produced one ligand point interaction with Arg120 residue which allows major interaction impact of the tyrosine derivatives on catalytic domain of COX-2 protein. Besides, aromatic ring of the tyrosine skeleton make π-cationic interaction with Arg120. This created a stable conformation of the molecule 8 in the hydrophobic binding site of the COX-2 protein. This long hydrophobic channel creates cyclooxygenase active site that inhibits the inflammation via non-steroidal anti-inflammatory drugs. This active site lengthen from the membrane binding domain to the region where the catalyzed chemical reaction takes place [16, 17]. In addition, R1 and R2 bromine substitution had generated VDW interaction with Val523 and Phe518 that permitted the molecule 8 to access an additional side pocket which is a pre-requisite for COX-2 drug selectivity. This structural modification may be attributed to the interchange of valine at position of 523 in COX-2 for a relatively bulky isoleucine residue in COX-1 [5]. The substitution of 1, 3-thiazole ring at –OH (R4) position of tyrosine induced the VDW and electrostatic interactions with the active site amino acids. It created conducive chemical environment in the COX-2 binding site. Substitution of electronegative sulfonyl group at R3 position enhanced the binding potential of the molecule by interacting with Ser353 (Figs. 5, 6). It is confirmed from this study that the COX-2 selectivity of the molecule 8 is higher than the standard celecoxib. The rest of 34 molecules were examined and found to have more stability when compared to the standard.
Table 4.
Interaction energy values of tyrosine derivatives and celecoxib with COX-2 protein
| Name | C-Docker energya | –C-Docker interaction energya | Initial potential energya | Initial RMS gradient | Electrostatic energya | Potential energya | VDW energya | RMS gradient |
|---|---|---|---|---|---|---|---|---|
| Molecule_8 | −78.7003 | 4.96727 | −74.2658 | 16.3263 | −199.774 | −155.629 | 3.78158 | 0.09694 |
| Molecule_54 | −46.1094 | 3.80434 | 9.73689 | 40.9916 | −161.106 | −129.460 | 5.45173 | 0.09737 |
| Molecule_23 | −45.4158 | 1.08668 | −2.98987 | 44.0659 | −177.976 | −139.880 | 5.17214 | 0.09761 |
| Molecule_6 | −40.1233 | 1.50834 | 339.920 | 91.3010 | −131.124 | −106.360 | 1.62197 | 0.09667 |
| Molecule_14 | −38.0308 | 9.72515 | −93.3437 | 6.78104 | −128.448 | −98.3557 | 2.48238 | 0.08123 |
| Molecule_50 | −32.9449 | −3.8949 | 25.7593 | 47.7124 | −133.414 | −112.472 | −0.05903 | 0.08110 |
| Molecule_25 | −29.4798 | 14.5849 | 14.0717 | 43.5888 | −142.506 | −118.107 | 1.45489 | 0.07719 |
| Molecule_51 | −28.5191 | 0.90861 | 32.0508 | 46.2969 | −130.255 | −100.616 | 2.08610 | 0.08137 |
| Molecule_24 | −28.4505 | 16.3299 | 534.240 | 568.860 | −140.619 | −104.274 | 3.04089 | 0.09149 |
| Molecule_11 | −26.1386 | 6.71301 | 61.7373 | 44.7827 | −151.439 | −136.499 | −1.89433 | 0.09066 |
| Molecule_10 | −23.4787 | 21.0787 | 71.8921 | 63.5300 | −157.857 | −126.759 | 6.24669 | 0.09716 |
| Molecule_20 | −21.3714 | 17.7833 | −17.3987 | 40.1040 | −120.253 | −99.3152 | 4.00568 | 0.09156 |
| Molecule_21 | −20.4346 | 21.4014 | −55.1410 | 4.32287 | −79.2812 | −58.7042 | 1.69521 | 0.08615 |
| Molecule_57 | −15.0159 | 13.1286 | 28.7444 | 53.0095 | −162.501 | −128.173 | 8.40306 | 0.09806 |
| Molecule_58 | −12.0458 | 3.82902 | −56.6613 | 20.5821 | −152.031 | −129.655 | 3.88633 | 0.09311 |
| Molecule_7 | −5.28412 | 22.9306 | 55,568.4 | 75,666.6 | −150.99 | −121.105 | 1.02490 | 0.09610 |
| Molecule_67 | −3.39829 | 11.7573 | 26.7832 | 42.2915 | −123.539 | −103.737 | 2.94580 | 0.09285 |
| Molecule_59 | −1.19358 | 14.2210 | −89.0706 | 14.9038 | −132.811 | −104.716 | 3.23357 | 0.08664 |
| Molecule_13 | 0.274143 | 19.9477 | 73.5331 | 48.1590 | −130.355 | −111.012 | 0.14158 | 0.09153 |
| Molecule_17 | 0.957257 | 13.0669 | −13.8560 | 6.29743 | −42.2671 | −25.9434 | −1.71694 | 0.08943 |
| Molecule_15 | 0.961175 | 18.3100 | −37.7612 | 6.14656 | −75.5862 | −50.066 | 3.31430 | 0.08757 |
| Molecule_102 | 4.763580 | 38.6705 | −1.47768 | 6.52181 | −20.0264 | −13.5243 | −6.95490 | 0.08871 |
| Molecule_52 | 7.997040 | 16.8484 | 487.293 | 105.229 | −155.493 | −119.589 | −0.16776 | 0.09978 |
| Molecule_26 | 8.272020 | 21.8179 | 42.0749 | 38.5993 | −138.631 | −94.2976 | 2.46568 | 0.08678 |
| Molecule_146 | 8.494660 | 38.3012 | 13.8468 | 6.52572 | −12.4255 | −7.32723 | −7.01223 | 0.09203 |
| Molecule_103 | 9.218700 | 41.1444 | −0.67757 | 6.08668 | −21.515 | −13.0472 | −4.12813 | 0.09382 |
| Molecule_12 | 9.37307 | 23.8593 | 599.169 | 124.448 | −133.207 | −95.3196 | −0.06212 | 0.09771 |
| Molecule_99 | 10.0093 | 38.4689 | 10.9431 | 4.96463 | −5.95502 | 0.97673 | −4.40147 | 0.09965 |
| Molecule_154 | 10.8974 | 42.4735 | 10.9119 | 7.15111 | −16.5170 | −5.14193 | −5.71648 | 0.86146 |
| Molecule_9 | 11.5098 | 24.9470 | 65.3320 | 46.7416 | −120.920 | −71.2045 | 2.39190 | 0.09569 |
| Molecule_113 | 12.1402 | 41.6382 | 28,289.50 | 39,402.2 | −136.929 | −83.7555 | 2.70303 | 0.09185 |
| Molecule_60 | 12.5198 | 19.1621 | −13.2592 | 6.10851 | −41.6039 | −25.2553 | −0.89500 | 0.08769 |
| Molecule_115 | 12.5673 | 46.3928 | −11.8405 | 6.12586 | −44.0638 | −23.2031 | 1.68847 | 0.09377 |
| Molecule_141 | 12.8093 | 32.4320 | −5.41508 | 4.20617 | −29.7202 | −17.6498 | −0.35700 | 0.09705 |
| Molecule_117 | 17.0983 | 38.6338 | 2021.79 | 2299.10 | 59.3405 | 96.7284 | 3.50862 | 0.09529 |
| Celecoxib | 17.33395 | 33.9253 | 13.8933 | 42.5446 | −139.661 | −117.986 | 6.21732 | 0.09936 |
| Molecule_142 | 17.3898 | 38.1553 | −79.2244 | 15.0265 | −128.565 | −93.4084 | 2.66973 | 0.09101 |
| Molecule_100 | 17.9025 | 29.9299 | 6222.05 | 6373.98 | −160.241 | −112.801 | 1.91017 | 0.08234 |
| Molecule_105 | 17.9411 | 39.9044 | 3.14311 | 5.19254 | −19.9248 | −12.5248 | −3.32516 | 0.09908 |
| Molecule_110 | 18.7239 | 44.9821 | 6.43866 | 5.82078 | −15.4753 | −6.75722 | −1.88785 | 0.09904 |
| Molecule_107 | 20.4115 | 36.5229 | 18.9587 | 5.76753 | −11.5264 | 0.057840 | −2.43628 | 0.09055 |
| Molecule_98 | 20.7154 | 30.5548 | −1.67995 | 5.36936 | −21.3184 | −9.82350 | −1.23215 | 0.09869 |
| Molecule_104 | 23.4169 | 41.1073 | 6.86145 | 4.83158 | −23.4274 | −2.71648 | −5.74037 | 0.0969 |
| Molecule_114 | 24.2130 | 49.8248 | 5.38092 | 5.21023 | −12.5330 | −4.10493 | −4.12701 | 0.08961 |
| Molecule_101 | 24.4073 | 38.7888 | 9.88176 | 16.3253 | −17.7871 | −5.53674 | −3.82927 | 0.08234 |
| Molecule_143 | 25.1057 | 38.8955 | 4.73240 | 5.43985 | −23.7807 | −9.70408 | −2.10607 | 0.08908 |
| Molecule_159 | 25.8484 | 38.2027 | 6.37157 | 5.98037 | −34.4232 | −3.90721 | −5.13228 | 0.09613 |
| Molecule_122 | 26.6459 | 39.9598 | 2.21425 | 6.49133 | −34.9642 | −14.2581 | −6.04252 | 0.08389 |
| Molecule_111 | 29.4514 | 42.7783 | 38.9483 | 46.4718 | −127.864 | −82.6885 | 3.20745 | 0.08620 |
| Molecule_118 | 30.2871 | 43.3848 | 13.8351 | 7.06563 | −23.0209 | −1.49548 | −7.16945 | 0.08975 |
| Molecule_144 | 31.0438 | 41.6663 | 16.4609 | 6.42745 | −18.8412 | 4.78885 | −6.92729 | 0.09901 |
| Molecule_150 | 34.7730 | 46.4684 | 36.3206 | 7.23252 | −7.94199 | 11.2902 | −2.19893 | 0.08886 |
| Molecule_112 | 35.1376 | 46.2887 | 8.39953 | 6.33198 | −33.0042 | −4.69838 | −5.58198 | 0.09233 |
| Molecule_151 | 35.3649 | 45.4588 | 33.7593 | 7.10073 | −14.0113 | 18.3148 | −3.51091 | 0.08832 |
| Molecule_152 | 41.9392 | 45.3560 | 90.9266 | 30.9076 | −12.7073 | 16.6751 | −3.82475 | 0.09296 |
aThe energies of the molecules are indicated in kcal/mol unit
Fig. 4.
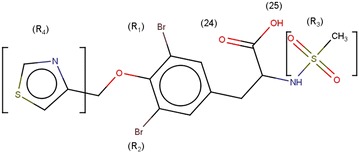
Structure of molecule 8
Fig. 5.
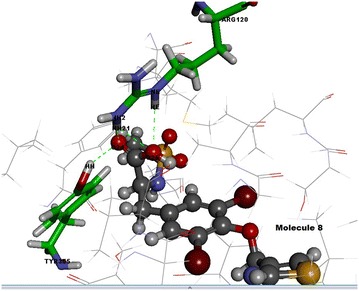
Interactions of molecule 8 with active site amino acids of COX-2 protein
Fig. 6.
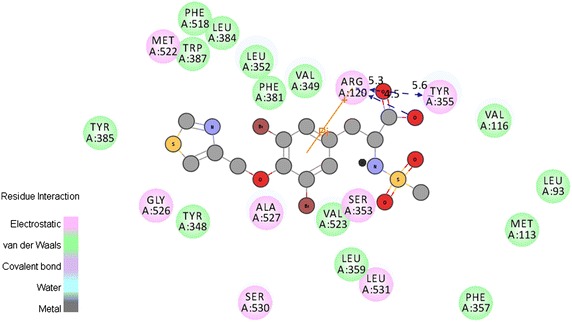
2D Interactions view of molecule 8 with active site amino acids of COX-2 protein
The COX-2 selectivity of the 55 tyrosine derivatives was compared with COX-1 enzyme. In this COX-1 docking study, the designed molecule had not created appropriate conformation inside the active site of COX-1 enzyme due to the bulky amino acid residue Ilu523 and non-polar moieties of the His513. The VDW space of the tyrosine molecules in COX-1 chemical space of the active site is in conflict with the receptor essential volume. This conflict creates steric repulsion between side chain amino acids of the COX-1 and designed molecules. It strongly evidenced that there is a large decrease in the affinity of the designed tyrosine derivatives with COX-1 when compared to the celecoxib. The above results proved that the tyrosine derivatives are more selective on COX-2 than COX-1.
Ulcerogenic interaction
The enzyme COX-1 played pivotal role in the maintenance of mucosal integrity in the gastrointestinal tract. It is believed that the ulcerogenic effects of non-steroidal anti-inflammatory drugs is owing to exclusive inhibition of COX-1 [18]. The interaction between the designed 55 tyrosine moiety and COX-1 protein aided to identify the ulcerogenicity level of designed molecules. The results of docking studies (C-Docker) revealed that the designed tyrosine derivatives exhibited more binding energy which was in contrast with the standard celecoxib (Table 5). The standard drug formed, one sigma-π, π-cationic and two hydrogen bond interaction with the Ile523, Arg120, Gln192 and Lue352 amino acids respectively (Fig. 7). These bonds support the celecoxib to fit into the cavity of COX-1 enzyme. On the other hand, the designed tyrosine derivatives formed hydrogen bonds with the Tyr385 and Ser530 (Fig. 8) and there is no other additional interaction with the active site amino acids of COX-1 receptor. Also, the electro negative groups (-Br, -I) of the designed molecules forms intermolecular bumps which disfavors the binding capability of the molecules. These unstable conformations of the designed molecule prove their negligible ulcerogenic side effect.
Table 5.
C-Docker values for the tyrosine derivatives with COX-1 and hERG protein
| Name of the molecule | COX-1 | hERG | ||
|---|---|---|---|---|
| C-Docker energy | –C-Docker interaction energy | C-Docker energy | –C-Docker interaction energy | |
| Molecule_11 | 11.1931 | 39.4964 | 25.6376 | 36.6014 |
| Molecule_7 | 15.4566 | 35.6748 | 25.7715 | 38.8912 |
| Molecule_102 | 18.612 | 45.7385 | 6.12591 | 32.7374 |
| Molecule_99 | 22.7057 | 45.0127 | 13.8034 | 33.4137 |
| Molecule_10 | 23.7368 | 49.2067 | 17.2666 | 37.8672 |
| Molecule_113 | 25.291 | 51.6406 | 11.4752 | 37.2134 |
| Molecule_14 | 25.5442 | 45.5213 | 27.0423 | 39.9337 |
| Molecule_50 | 27.2685 | 42.4608 | 37.1187 | 35.2034 |
| Molecule_54 | 27.9592 | 45.2948 | 30.1138 | 37.0948 |
| Molecule_154 | 28.0068 | 48.3108 | 22.5487 | 37.1437 |
| Molecule_103 | 28.5622 | 49.3350 | 12.6594 | 33.7937 |
| Molecule_23 | 28.9051 | 51.4072 | 27.8294 | 36.9222 |
| Molecule_146 | 29.7938 | 48.7906 | 12.7369 | 29.7554 |
| Molecule_117 | 32.24 | 50.9400 | 17.502 | 35.4744 |
| Molecule_122 | 32.4296 | 41.5757 | 23.1628 | 31.3608 |
| Molecule_21 | 32.7128 | 54.7328 | 26.6071 | 40.1819 |
| Molecule_8 | 33.0627 | 39.4668 | 36.4622 | 33.5932 |
| Molecule_115 | 33.5042 | 53.2589 | 16.3142 | 35.3354 |
| Molecule_105 | 33.9553 | 48.2822 | 20.7848 | 31.2565 |
| Molecule_114 | 34.2029 | 54.9272 | 19.9584 | 37.415 |
| Molecule_25 | 34.4117 | 51.3171 | 32.1083 | 41.2442 |
| Molecule_100 | 34.7976 | 46.4333 | 21.2323 | 30.1093 |
| Molecule_110 | 34.9249 | 53.1217 | 16.7253 | 34.8426 |
| Molecule_6 | 35.033 | 45.8393 | 41.2549 | 36.2332 |
| Molecule_26 | 35.2188 | 44.5275 | 43.3746 | 38.2527 |
| Molecule_51 | 35.9835 | 42.5881 | 41.6024 | 38.3912 |
| Molecule_107 | 36.0181 | 46.6087 | 21.6437 | 29.0359 |
| Molecule_159 | 36.4927 | 43.0306 | 25.964 | 32.5955 |
| Molecule_142 | 37.3162 | 49.1502 | 22.6982 | 30.9975 |
| Molecule_141 | 37.8732 | 49.0256 | 23.9516 | 32.7221 |
| Molecule_15 | 37.9714 | 43.6680 | 37.2577 | 34.7344 |
| Molecule_58 | 38.0398 | 42.8592 | 43.586 | 36.3985 |
| Molecule_13 | 39.4551 | 49.4952 | 35.9365 | 38.5156 |
| Molecule_52 | 41.2608 | 41.016 | 48.0603 | 37.6181 |
| Molecule_67 | 41.3861 | 43.4004 | 40.4953 | 37.1043 |
| Molecule_59 | 41.5117 | 49.588 | 51.4686 | 44.477 |
| Molecule_104 | 41.5637 | 48.3222 | 25.0942 | 32.4227 |
| Molecule_101 | 42.6379 | 48.1987 | 25.8003 | 32.6898 |
| Molecule_98 | 42.9202 | 48.7329 | 27.3695 | 33.4591 |
| Molecule_143 | 43.1506 | 48.7602 | 26.4997 | 32.3527 |
| Molecule_9 | 43.4413 | 49.4292 | 40.1671 | 35.6967 |
| Molecule_20 | 44.4891 | 48.6384 | 45.1393 | 37.1212 |
| Molecule_24 | 45.1278 | 54.9812 | 39.8631 | 37.5571 |
| Molecule_12 | 45.21 | 49.8787 | 41.6676 | 37.8414 |
| Molecule_111 | 45.9116 | 51.7703 | 27.1743 | 33.3161 |
| Molecule_144 | 46.9694 | 46.4174 | 34.4717 | 35.0657 |
| Molecule_112 | 47.1361 | 50.3979 | 32.8243 | 35.6348 |
| Molecule_151 | 48.0494 | 53.3779 | 35.1662 | 37.0065 |
| Molecule_150 | 48.0592 | 53.4856 | 29.3814 | 36.5024 |
| Molecule_118 | 48.319 | 52.7029 | 29.5539 | 33.4975 |
| Molecule_17 | 48.6628 | 48.8614 | 45.811 | 38.6698 |
| Molecule_60 | 49.6967 | 49.4645 | 56.8447 | 44.3316 |
| Molecule_152 | 52.4378 | 51.2126 | 40.7779 | 35.7427 |
| Celecoxib | 19.4457 | 51.7111 | −0.642396 | 30.7255 |
Fig. 7.
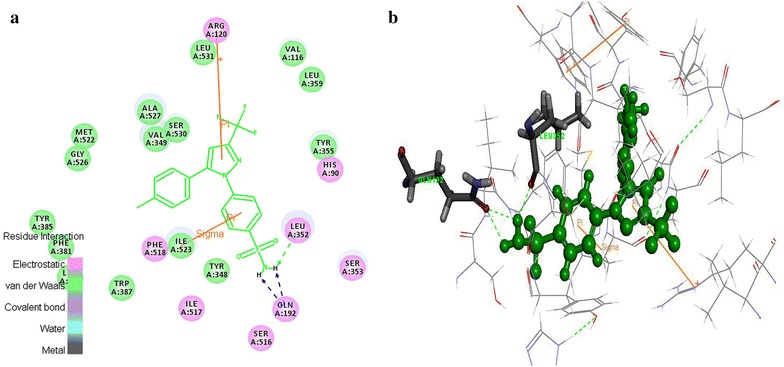
Celecoxib interaction map with the COX-1 protein a 2D view of non-bonded interactions b 3D interaction view
Fig. 8.
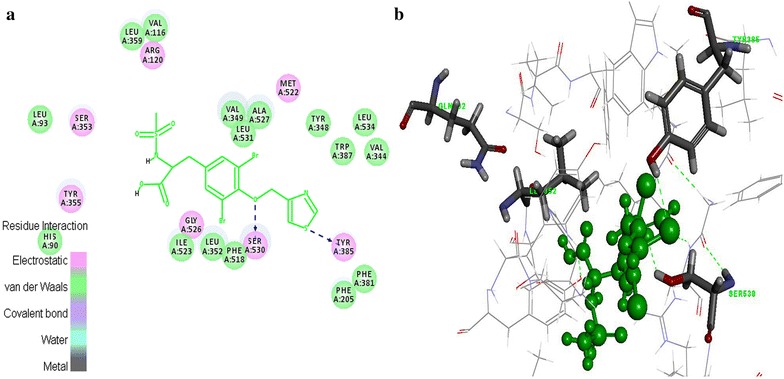
Tyrosine derivatives interaction map with the COX-1 protein a 2D view of non-bonded interactions b 3D interaction view of hERG protein interaction
hERG protein interaction studies
The hERG is the most critical channel involved in drug induced Torsade de Pointes (TdP) arrhythmias. Extra cellular application of celecoxib causes rapid suppression of hERG channels which induces the cardiac disturbances [19]. Evaluation of spatial orientation of the designed molecule interactions with the hERG protein recognizes the cardiotoxicity level of molecules [20]. The results of docking studies indicated that among the 55 designed molecules, 52 molecules possessed more interaction energy against the standard (Table 5). It revealed that these molecules are having less binding affinity to the active site residues of the hERG protein. In standard celecoxib, the benzyl ring creates π-π interaction with the Tyr652 (Fig. 9). This enables the celecoxib to fit well into the hydrophobic pocket of COX-2 protein. On contrary, tyrosine derivatives did not form any π-π interactions and the extra volume of the electronegative group substitutions in the R1 and R2 positions which repulse the molecules to bind in the active site (Fig. 10). Hence, the cardiotoxicity of the designed molecules were less when compared to the celecoxib. The selected 35 tyrosine molecules demonstrated high COX-2 selectivity, less COX-1 (ulcerogenic) and hERG (cardiotoxicity) binding affinity. Further, these molecules were examined by ADMET descriptors calculation and OSIRIS properties explorer.
Fig. 9.
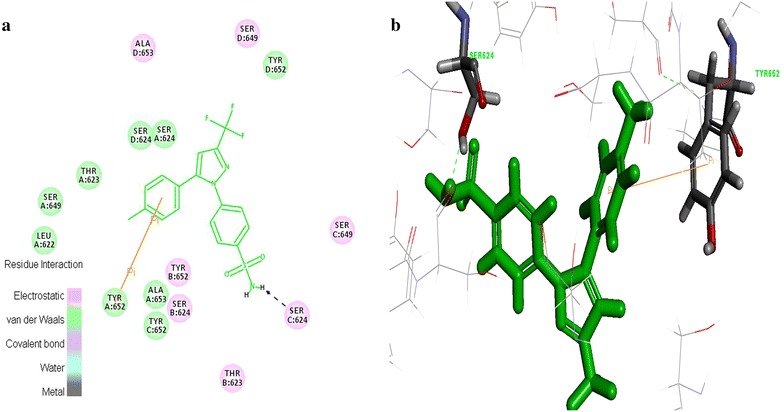
Standard celecoxib interaction map with the hERG protein a 2D view of non-bonded interactions b 3D interaction view
Fig. 10.
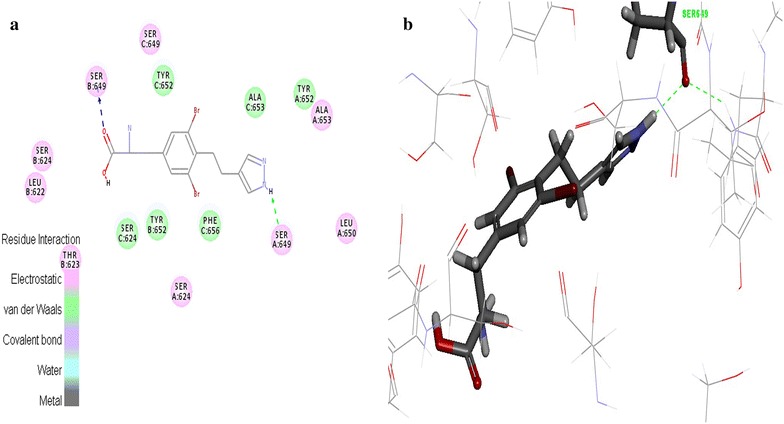
Tyrosine derivatives interaction map with the hERG protein a 2D view of non-bonded interactions b 3D interaction view
Docking protocol validation
The results of RMSD values of redocked native co-crystallized ligand of each PDB entry revealed that native ligand conformations including 3NT1 and best docked ligand conformation exactly binds in the experimental protein binding mode. In the docking study performed by first method, RMSD values of best docked conformations ranged from 0.8436 to 1.7674 Å. According to validation protocol, RMSD values of best docked conformation should be ≤2.0 Å [21]. It represents that this docking protocol is able to find an appropriate binding mode. The designed 55 molecules were redocked into the active site of the COX-2 (3NT1) receptor and confirms that these docked molecules followed the similar binding method as in native co-crystallised ligand (Table 6).
Table 6.
Native co-crystallised ligands and its respective PDB ID with its redocked RMSD values
| Co-crystallized ligand | PDB ID | RMSD (Ǻ) |
|---|---|---|
| CEL682 | 3LN1 | 1.7674 |
| NPS5 | 3NT1 | 1.3330 |
| DIF701 | 3N8Y | 0.8436 |
| IBP601 | 4PHA | 1.0834 |
| Molecule 8 | 3NT1 | 1.0810 |
In the second method, the selected docking protocol parameters accurately distinguished the selective and non-selective COX-2 inhibitors. It is illuminated by the docking results in which C-Docker energy of selective COX-2 inhibitors fall in the negative kcal/mol range and the non-selective inhibitors energies fall in the range of positive kcal/mol (Table 7). Additionally, the binding site (3NT1) analysis of the drug receptor complexes revealed that all the selective COX-2 inhibitors formed π interaction with the active site amino acids which are major force for molecular recognition and join with hydrophobic interaction [22]. But, non-selective COX inhibitors formed hydrogen bond, VDW and electrostatic interactions only (Fig. 11). It clearly proves that the selective COX-2 inhibitors and designed 55 molecules possessed more selectivity compared to the non-selective inhibitors. This proposed model predicted the correlation between C-Docker energy and the experimental IC50 value of the selective and non-selective inhibitors. The correlation coefficient was predicted to be 0.835 (r2) (Fig. 12). This correlation strongly indicates that the docking protocol of this study possessed good predicting ability as well as it distinguishes the selective and non-selective COX-2 inhibitors precisely.
Table 7.
C-Docker energy values of the selective and non-selective inhibitors
| Selective COX-2 inhibitors | C-Docker energy value (kcal/mol) | Non-selective COX-2 inhibitors | C-Docker energy value (kcal/mol) |
|---|---|---|---|
| Rofecoxib | −19.0343 | Diclofenac | 5.45905 |
| Valdecoxib | −9.2766 | Ketorolac | 12.2429 |
| Etoricoxib | −3.32262 | Aspirin | 29.113 |
| Naproxen | 32.0361 | ||
| Ibuprofen | 39.7383 |
Fig. 11.
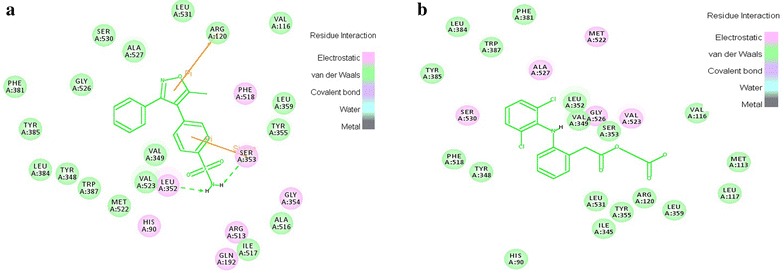
Interactions of selective and non-selective COX-2 inhibitors. a Rofecoxib b Aceclofenac
Fig. 12.
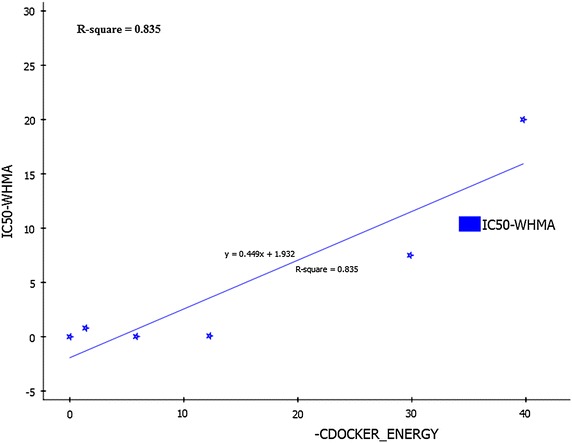
Correlation point plot of C-Docker energy and the experimental activity (IC50) of the nonselective COX-2 inhibitors
Toxicity
ADMET descriptors
In the present work, we have assessed ADMET (absorption, distribution, metabolism, excretion, and toxicity) properties of the 35 compounds which were selected from the docking report. ADMET descriptors were calculated to filter the poor tyrosine molecule with undesired pharmacokinetic and toxicity properties [23]. This step prevents wasting of time, chemicals as well as animal studies of tyrosine derivatives. The pharmacokinetic profile of all the molecules was predicted by means of six pre-calculated ADMET models provided by ADS 2.5 software. The ADMET plot shows the 95 and 99 % confidence ellipse for the HIA and BBB models (Fig. 13). The 95 % confidence ellipse represents the region of chemical space with molecules having excellent absorption through cell membrane. According to this model, for a designed molecule to have an optimal cell permeability, it should follow the criteria of PSA < 140 Å2 and AlogP98 < 5) [24]. The selected 35 molecules have shown PSA < 140 Å2 and AlogP98 < 5 which satisfied the criteria.
Fig. 13.
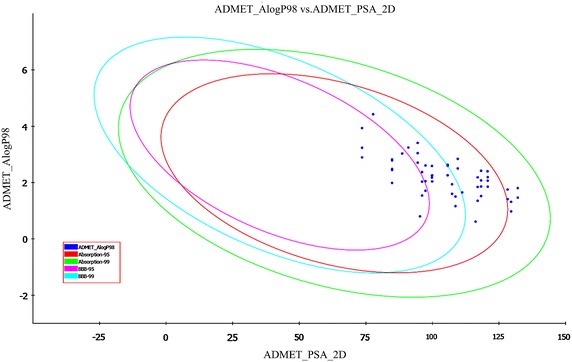
The 95 and 99 % confidence limit ellipses corresponding to the BBB and HIA models for tyrosine derivatives
These selected molecules as well as standard celecoxib fall in the 95 and 99 % confidence ellipse for both HIA and BBB (Fig. 13). The HIA of the tyrosine derivatives ranges from 0 (good absorption) to 1 (moderate absorption) (Table 8). It indicates the good bioavailability of designed molecules to produce desired therapeutic effect. BBB penetration of the designed molecules indicated undefined to low penetration, except the molecule 141. On the other hand, celecoxib exhibited moderate penetration to the BBB (Table 8). The aqueous solubility plays a vital role in the bioavailability of the drug. The designed tyrosine derivatives have solubility in the range of 2 (low soluble) to 3 (soluble) as referred in Table 9. Further, the hepatotoxicity level of all the molecules were calculated, the molecules with liver toxic nature were filtered out. Similarly, all the molecules were found to be satisfactory with respect to CYP 450 2D6 liver enzyme, suggesting that the tyrosine derivatives were non inhibitors of the metabolic enzyme. Finally, the PPB prediction denotes that all the designed molecules have binding ≤90 % clearly revealing that the molecules have good bioavailability and are not likely to be highly bound to carrier proteins in the blood [25].
Table 8.
ADMET predictions of 35 tyrosine molecules and celecoxib
| Name of the molecule | Absorption level | AlogP98 | PSA 2D | BBB level | Solubility | Solubility level | Hepatotoxicity level | CYP 2D6 | PPB level |
|---|---|---|---|---|---|---|---|---|---|
| Molecule_6 | 0 | 2.843 | 109.513 | 4 | −4.495 | 2 | 0 | 0 | 0 |
| Molecule_8 | 0 | 2.634 | 105.719 | 4 | −4.197 | 2 | 0 | 0 | 0 |
| Molecule_9 | 0 | 1.862 | 118.273 | 4 | −3.523 | 3 | 1 | 0 | 0 |
| Molecule_10 | 1 | 1.317 | 129.534 | 4 | −3.217 | 3 | 0 | 0 | 0 |
| Molecule_11 | 0 | 2.402 | 120.689 | 4 | −4.027 | 2 | 0 | 0 | 0 |
| Molecule_12 | 0 | 1.852 | 120.774 | 4 | −3.883 | 3 | 1 | 0 | 0 |
| Molecule_13 | 0 | 2.419 | 118.273 | 4 | −4.009 | 2 | 1 | 0 | 0 |
| Molecule_14 | 1 | 1.804 | 132.035 | 4 | −4.096 | 2 | 0 | 0 | 0 |
| Molecule_15 | 0 | 3.047 | 94.458 | 3 | −4.388 | 2 | 1 | 0 | 0 |
| Molecule_17 | 0 | 2.503 | 109.513 | 4 | −4.197 | 2 | 1 | 0 | 0 |
| Molecule_20 | 0 | 1.522 | 118.273 | 4 | −3.225 | 3 | 0 | 0 | 0 |
| Molecule_21 | 1 | 0.976 | 129.534 | 4 | −2.919 | 3 | 0 | 0 | 0 |
| Molecule_23 | 0 | 2.068 | 120.774 | 4 | −4.071 | 2 | 0 | 0 | 0 |
| Molecule_24 | 0 | 2.078 | 118.273 | 4 | −3.711 | 3 | 0 | 0 | 0 |
| Molecule_25 | 1 | 1.464 | 132.035 | 4 | −3.798 | 3 | 0 | 0 | 0 |
| Molecule_26 | 0 | 2.707 | 94.458 | 3 | −4.09 | 2 | 1 | 0 | 0 |
| Molecule_50 | 0 | 2.599 | 105.719 | 4 | −3.984 | 3 | 0 | 0 | 0 |
| Molecule_51 | 0 | 2.186 | 116.98 | 4 | −3.793 | 3 | 0 | 0 | 0 |
| Molecule_54 | 0 | 0.613 | 116.198 | 4 | −2.789 | 3 | 0 | 0 | 0 |
| Molecule_58 | 0 | 2.259 | 105.719 | 3 | −3.686 | 3 | 0 | 0 | 0 |
| Molecule_67 | 0 | 1.354 | 116.98 | 4 | −2.83 | 3 | 0 | 0 | 0 |
| Molecule_99 | 0 | 3.245 | 90.972 | 3 | −4.639 | 2 | 1 | 0 | 0 |
| Molecule_102 | 0 | 1.505 | 108.662 | 3 | −3.598 | 3 | 1 | 0 | 0 |
| Molecule_103 | 0 | 2.59 | 99.817 | 3 | −4.351 | 2 | 1 | 0 | 0 |
| Molecule_113 | 0 | 1.164 | 108.662 | 3 | −3.3 | 3 | 1 | 0 | 0 |
| Molecule_115 | 0 | 2.256 | 99.902 | 3 | −4.114 | 2 | 1 | 0 | 0 |
| Molecule_117 | 0 | 1.652 | 111.163 | 4 | −3.925 | 3 | 1 | 0 | 0 |
| Molecule_141 | 0 | 3.937 | 73.586 | 2 | −4.973 | 2 | 1 | 1 | 0 |
| Molecule_146 | 0 | 0.801 | 95.326 | 3 | −2.859 | 3 | 1 | 0 | 0 |
| Molecule_154 | 0 | 2.18 | 99.817 | 3 | −3.996 | 3 | 1 | 0 | 0 |
| Molecule_7 | 0 | 2.193 | 120.774 | 4 | −4.182 | 2 | 0 | 0 | 0 |
| Molecule_52 | 1 | 1.753 | 128.241 | 4 | −3.584 | 3 | 1 | 0 | 0 |
| Molecule_57 | 0 | 3.409 | 94.458 | 3 | −4.521 | 2 | 0 | 0 | 0 |
| Molecule_59 | 0 | 1.846 | 116.98 | 4 | −3.495 | 3 | 0 | 0 | 0 |
| Molecule_60 | 1 | 1.413 | 128.241 | 4 | −3.286 | 3 | 1 | 0 | 0 |
| Celecoxib | 0 | 4.428 | 77.75 | 2 | −6.603 | 1 | 1 | 0 | 1 |
Table 9.
ADMET descriptor models
| Name of the ADMET model | Prediction levels |
|---|---|
| Human intestinal absorption | 0 (Good absorption) |
| 1 (Moderate absorption) | |
| 2 (Low absorption) | |
| 3 (Very low absorption) | |
| Aqueous solubility | 0 (Extremely low) |
| 1 (No, very low, but possible) | |
| 2 (Yes, low) | |
| 3 (Yes, good) | |
| 4 (Yes, optimal) | |
| 5 (Too soluble) | |
| Blood brain barrier (BBB) | 0 (Very high penetration) |
| 1 (High penetration) | |
| 2 (Medium penetration) | |
| 3 (Low penetration) | |
| 4 (Undefined penetration) | |
| Cytochrome P450 2D6 (CYP 2D6) | 0 (Non−inhibitor) |
| 1 (Inhibitor) | |
| Hepatotoxicity | 0 (Nontoxic) |
| 1 (Toxic) | |
| Plasma protein binding (PBB) | 0 (Binding is <90 %) |
| 1 (Binding is >90 %) | |
| 2 (Binding is >95 % |
Osiris property explorer
The result of toxicity analysis of designed molecules showed low toxicity tendency except the molecules 103 and 113. The drug-likeness value of standard and designed molecule exhibited the fragment content of the drug. If the drug-likeness value of designed molecules is increasing, then it has the same fragment content with existing drugs. Table 10 shows that the drug-likeness value of the tyrosine derivatives were higher than the standard celecoxib (−8.11), with the exception of 102, 103, 117, 141, 146 and 154 (−10.82 to −11.92). This results predict that among 35, 29 molecules exhibited same fragment content of the drugs. It confirms the drug likeness properties of these compounds.
Table 10.
Toxicity of tyrosine derivatives and standard drug based on OSIRIS property explorer
| Molecule | Mutagenicity | Tumorigenic | Irritant | Reproductive effect | Drug likeness | Drug score |
|---|---|---|---|---|---|---|
| Molecule_6 | Green | Green | Green | Green | 1.88 | 0.63 |
| Molecule_8 | Green | Green | Green | Green | 2.25 | 0.62 |
| Molecule_9 | Green | Green | Green | Green | 1.83 | 0.60 |
| Molecule_10 | Green | Green | Green | Green | 2.46 | 0.66 |
| Molecule_11 | Green | Green | Green | Green | 0.87 | 0.53 |
| Molecule_12 | Green | Green | Green | Green | 2.46 | 0.67 |
| Molecule_13 | Green | Green | Green | Green | 2.61 | 0.65 |
| Molecule_14 | Green | Green | Green | Green | −2.08 | 0.39 |
| Molecule_15 | Green | Green | Green | Green | 2.03 | 0.54 |
| Molecule_17 | Green | Green | Green | Green | 4.74 | 0.54 |
| Molecule_20 | Green | Green | Green | Green | 4.69 | 0.50 |
| Molecule_21 | Green | Green | Green | Green | 5.29 | 0.55 |
| Molecule_23 | Green | Green | Green | Green | 5.54 | 0.57 |
| Molecule_24 | Green | Green | Green | Green | 5.43 | 0.53 |
| Molecule_25 | Green | Green | Green | Green | 0.74 | 0.48 |
| Molecule_26 | Green | Green | Green | Green | 4.88 | 0.45 |
| Molecule_50 | Green | Green | Green | Green | 2.34 | 0.45 |
| Molecule_51 | Green | Green | Green | Green | 1.46 | 0.60 |
| Molecule_54 | Green | Green | Green | Green | 1.77 | 0.59 |
| Molecule_58 | Green | Green | Green | Green | 4.31 | 0.51 |
| Molecule_67 | Green | Green | Green | Green | 2.39 | 0.46 |
| Molecule_99 | Green | Green | Green | Green | −0.06 | 0.46 |
| Molecule_102 | Green | Green | Green | Green | −10.82 | 0.39 |
| Molecule_103 | Green | Yellow | Red | Green | −15.1 | 0.18 |
| Molecule_113 | Green | Green | Red | Green | −7.79 | 0.32 |
| Molecule_115 | Green | Green | Green | Green | −7.28 | 0.33 |
| Molecule_117 | Green | Green | Green | Green | −11.92 | 0.33 |
| Molecule_141 | Green | Green | Green | Green | −17.18 | 0.34 |
| Molecule_146 | Green | Green | Green | Green | −11.29 | 0.35 |
| Molecule_154 | Green | Green | Green | Green | −8.91 | 0.31 |
| Molecule_7 | Green | Green | Green | Green | 3.47 | 0.70 |
| Molecule_52 | Green | Green | Green | Green | −2.79 | 0.21 |
| Molecule_57 | Green | Green | Green | Green | −0.83 | 0.49 |
| Molecule_59 | Green | Green | Green | Green | 4.36 | 0.52 |
| Molecule_60 | Green | Green | Green | Green | 2.39 | 0.29 |
| Celecoxib | Green | Green | Green | Green | −8.11 | 0.37 |
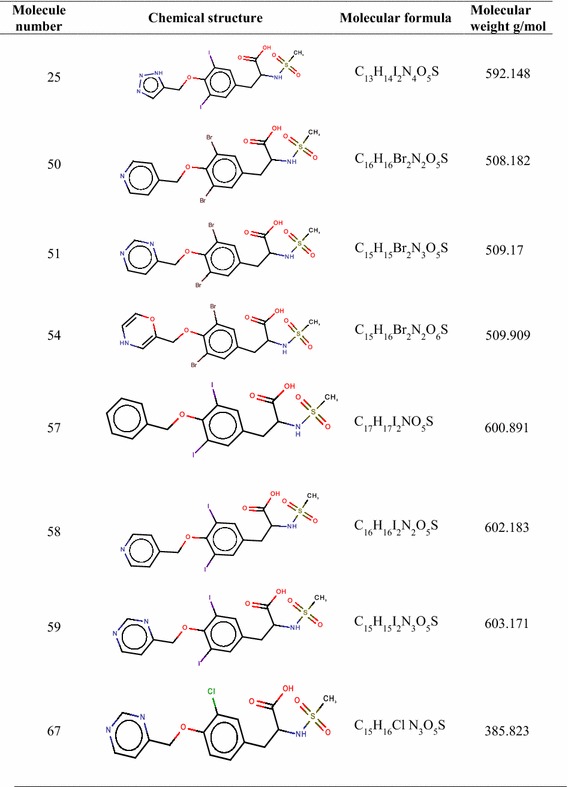
The drug score value is the combination of solubility, molecular weight, logP, drug likeness and toxicity risk. It is used for evaluating the potential of the drug candidate. When the drug score is better, then the compound is predictive to be a drug candidate [26]. The drug score value of standard celecoxib is found to contain 0.37. Finally 19 compounds which possessed drug score greater than the standard were shortlisted for further studies (Tables 11, 12).
Table 11.
Details of shortlisted potent COX-2 inhibitors
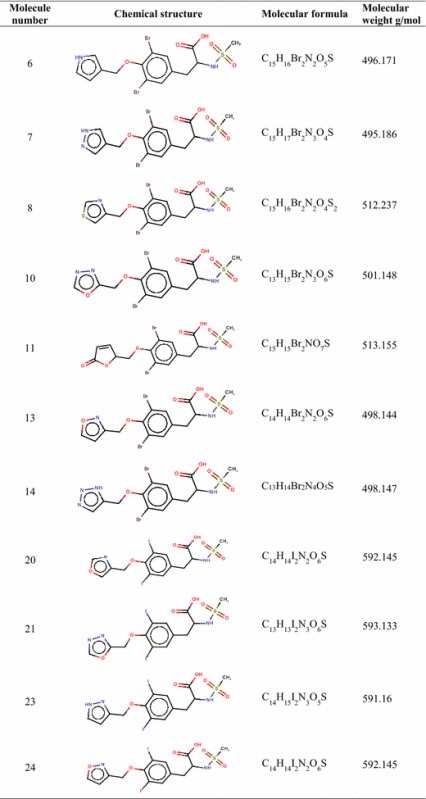
Table 12.
Details of shortlisted potent COX-2 inhibitors
Conclusion
In the current work, 55 tyrosine structural analogues on docking with COX-2, COX-1 and hERG revealed that 35 molecules have more affinity at active site residues of COX-2 enzyme and less interaction with the other two proteins (COX-1, hERG) than standard celecoxib. This information proved to exhibit potential of high selective, less ulcerogenic and cardiotoxicity of the designed novel anti-inflammatory molecules. Further, the result of ADMET and Osiris property explorer helped to eliminate 16 unwanted toxic fragments contained tyrosine molecules. Finally, 19 hits with good pharmacokinetic parameter and negligible toxicity was proceeded for synthesis. Hence, it is concluded that the predicted parameters are exclusively used as a basis for the further design of tyrosine derivatives and understand the mechanism of COX-2 related enzymatic inhibition reactions. The next step of the potent safe anti-inflammatory drug identification involves the synthesis and biological evaluation of the selected molecules which are in progress.
Authors’ contributions
It is certified that all authors have participated sufficiently in the work to take public responsibility for the content, including participation in the concept, design, analysis, writing, or revision of the manuscript. Furthermore, each manuscript author certified that this material or similar material has not submitted to or published in any other publication. Dr. AP: A Conception, design of study and approval of final version of manuscript. Dr. DS: Participated in computational studies. Ms. AU: Contributed to design the study and Drafting of manuscript. Mr. NI: Carried out the computational studies and participated in the Data analysis. All authors read and approved the final manuscript.
Acknowledgements
The authors are thankful to the Department of Science and Technology (DST-SERB), New Delhi for their financial assistance provided for this research (SR/S1/OC-48/2011 Dt: 14-052013).
Competing interests
The authors declare that they have no competing interests.
Funding
The present project was supported by grants from the Department of Science and Technology (DST-SERB), Government of India, New Delhi (SR/S1/OC-48/2011 Dt: 14-05-2013).
Abbreviations
- ADS
accelyrs discovery studio
- Arg
arginine
- BBB
blood brain barrier
- COX-1
cyclooxygenase-1
- COX-2
cyclooxygenase-2
- CYP 2D6
cytochrome P450 2D6
- Gln
glutamine
- HIA
human intestinal absorption
- HM
homology modeling
- His
histidine
- Ile
isoleucine
- Lue
leucine
- PDB
Protein Data Bank
- Phe
phenylalanine
- PPB
plasma protein binding
- PSA_2D
2D polar surface area
- RMS
root mean square
- SAR
structure activity relationship
- Ser
serine
- TdP
torsade de pointes
- Tyr
tyrosine
- Val
valine
- VDW
van der Waals
Contributor Information
Ayarivan Puratchikody, Phone: 0431-2407995, Email: puratchipharma@gmail.com.
Dharmaraj Sriram, Email: drdsriram1@gmail.com.
Appavoo Umamaheswari, Email: umapharmaaut@gmail.com.
Navabshan Irfan, Email: nirfanahamed1985@live.com.
References
- 1.Amaravani M, Nirmal KP, Vadde R. COX-2 structural analysis and docking studies with gallic acid structural analogues. Springer Plus. 2012;1:58. doi: 10.1186/2193-1801-1-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lipsky PE, Abramson SB, Crofford L, DuBios RN, Simon L, Van de Putte LBA. The classification of cyclooxygenase inhibitors. J Rheumatol. 1998;25:2298–2303. [PubMed] [Google Scholar]
- 3.Subbaramaiah K, Telang N, Ramonetti JT, Araki R, Devito B, Weksker BB, Dannenberg AJ. Transcription of cyclooxygenase-2 is enhanced in transformed mammary epithelial cells. Cancer Res. 1996;56:4424–4429. [PubMed] [Google Scholar]
- 4.Prasit P, Wang Z, Brideau C, Chan CC, Charleson S, Lish cro W, Ethier D, Evans JF, FordHutchinson AW, Gautheir JY, Gordon R, Guay J, Gresser M, Kargman S, Kennedy B, Leblanc Y, Leger S, Mancini J, O’neil GP, Ouellet M, Percival MD, Perrier H, Riendeau D, Rodger I, Zamboni R. The discovery of rofecoxib, [MK966, Vioxx, 4-(4 V-methylsulfonylphenyl)-3-phenyl-2(5H)-furanone], an orally active cyclooxygenase-2 inhibitor. Bioorg Med Chem Lett. 1999;9:1773–1778. doi: 10.1016/S0960-894X(99)00288-7. [DOI] [PubMed] [Google Scholar]
- 5.Zargi A, Arfaei S. Selective COX-2 inhibitors: a review of their structural—activity relationships. Iran J Pharm Res. 2011;4:655–683. [PMC free article] [PubMed] [Google Scholar]
- 6.Mukherjee D, Nissen SE, Topol EJ. Risk of cardiovascular events associated with selective COX-2 inhibitors. J Amer Med Assoc. 2001;8:954–959. doi: 10.1001/jama.286.8.954. [DOI] [PubMed] [Google Scholar]
- 7.Liu S, Fu X, Schmitz FJ, Kelly-Borges M, Psammaplysin F. A new Bromotyrosine derivative from a sponge, Aplysinella sp. J Nat Prod. 1997;60:614–615. doi: 10.1021/np970070s. [DOI] [PubMed] [Google Scholar]
- 8.Tilvi S, Roadringues C, Naik CG, Parameswaran PS, Wahidhulla S. New bromotyrosine alkaloids from the marine sponge Psammaplysilla purpurea. Tetraheadron. 2004;60:10207–10215. doi: 10.1016/j.tet.2004.09.009. [DOI] [Google Scholar]
- 9.Leone-Stumpf D (2001) Synthesis and chromatography of [RuCp]+-labelled diaryl ether peptoids as precursors of the bastadins from the marine sponge lanthella basta. PhD Thesis. Combined faculties for the natural sciences and for mathematics of the Ruperto-Carola University, Heidelberg
- 10.Wagner AB. SciFinder Scholar 2006, an empirical analysis of research topic query processing. J Chem Inf Model. 2006;46:767–774. doi: 10.1021/ci050481b. [DOI] [PubMed] [Google Scholar]
- 11.Wang Y, Bolton E, Dracheva S, Karapetyan K, Shoemaker BA, Suzek TO, Wang J, Xiao J, Zhang J, Bryant SH. An overview of the PubChem BioAssay resource. Nucl Acids Res. 2010;38:D255–D266. doi: 10.1093/nar/gkp965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Islam MA, Pillay TS. Structural requirements for potential HIV-integrase inhibitors identified using pharmacophore-based virtual screening and molecular dynmics studies. Mol BioSyst. 2016;12:982–993. doi: 10.1039/C5MB00767D. [DOI] [PubMed] [Google Scholar]
- 13.Cook D, Brown D, Alexander R, March R, Morgan P, Satterthwaite G, Pangalos MN. Lessons learned from the fate of AstraZeneca’s drug pipeline: a five-dimensional framework. Nat Rev Drug Discov. 2014;13:419–431. doi: 10.1038/nrd4309. [DOI] [PubMed] [Google Scholar]
- 14.Osiris property explorer [http://www.organicchemistry.Org/prog/peo/druglikeness.html]
- 15.Osiris property explorer [http://www.organicchemistry.Org/prog/peo/drugscore.html]
- 16.Picot D, Loll PJ, Garavito RM. The X-ray crystal structure of the membrane protein prostaglandin H2 synthase-1. Nature. 1994;367:243–249. doi: 10.1038/367243a0. [DOI] [PubMed] [Google Scholar]
- 17.Kurumbail RG, Kiefer JR, Marnett LJ. Cyclooxygenase enzymes: catalysis and inhibition. Curr Opin Struct Biol. 2001;11:752–760. doi: 10.1016/S0959-440X(01)00277-9. [DOI] [PubMed] [Google Scholar]
- 18.Honn VK, Marnett JL, Santosh N, Jones LR, Wong PY-K. Eicosanoids and other Bioactive Lipids in Cancer, Inflammation, and Radiation Injury. Newyork: Plenum Press; 1997. [Google Scholar]
- 19.Rolov RV, Ignatova II, Singh S. Inhibition of hERG potassium channels by celecoxib and its mechanism. PLoS ONE. 2011 doi: 10.1371/journal.pone.0026344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang B, Liu Z, Ma Z, Li M, Du L. Astemizole derivatives as fluorescent probes for hERG potassium channel imaging. Med Chem Lett. 2016 doi: 10.1021/acsmedchemlett.5b00360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Azam F, Abugrain IM, Sanalla MH, Fatahalla R, Abdassalam I, Rajab I. In Silico investigation of the structural requirements for the AMPA receptor antagonism by quinoxaline derivatives. Bioinformation. 2013;9:2–7. doi: 10.6026/97320630009864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dougherty AD. The cation-π interaction. Acc Chem Res. 2013;46:885–893. doi: 10.1021/ar300265y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cumming JG, Davis AM, Muresan S, Haeberlein M, Chen H. Chemical predictive modelling to improve compound quality. Nat Rev Drug Discov. 2013;12:948–962. doi: 10.1038/nrd4128. [DOI] [PubMed] [Google Scholar]
- 24.Singh S, Srivastava P. Molecular docking studies of myricetin and its analogues against human PDK-1 kinase as candidate drugs for cancer. Comput Mol Biosci. 2015;5:20–33. doi: 10.4236/cmb.2015.52004. [DOI] [Google Scholar]
- 25.Ali F, Zahrani A, Arshad MN, Asiri AM, Mahmood T, Gilani MA, El RM. Synthesis and structural properties methylene malononitrile derivatives; a combined experimental and theoretical insight. Chem Cent J. 2016;10:1–15. doi: 10.1186/s13065-016-0148-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pedrosa LF, Furtado ACR, Pinheiro LCS, Vaz MGF, Macedo WP, Guedes GP, Resende JALC, Bernardino AMR, Souza MC. Synthesis, crystal structures, and in silico toxicity prediction of thienopyridine phosphoramidates. Synth Commun. 2013;43:3373–3386. doi: 10.1080/00397911.2013.786092. [DOI] [Google Scholar]