

Abstract
Chemical communication between cells ensures coordination of behavior. In prokaryotes, this chemical communication is usually referred to as quorum sensing, while eukaryotic cells signal through hormones. In the past years, a growing number of reports have shown that bacterial quorum sensing signals, called autoinducers, signal to eukaryotic cells, mimicking hormones. Conversely, host hormones can signal to bacterial cells through converging pathways to autoinducer signaling. This inter-kingdom signaling mediates symbiotic and pathogenic relationships between bacteria, mammalian and plant hosts.
Graphical Abstract
Introduction
Bacteria and host have co-evolved for billions of years, through which they have been exposed to many signaling molecules produced and released by each other. Hence, it is not surprising that microorganisms intercept eukaryotic signaling systems for successful colonization of the host.
Bacterial cells are able to communicate through the production, release and detection of autoinducers to engage in multi-cellular behavior and coordinate gene expression at a population level. This chemical communication was named quorum sensing (QS) [1]. QS regulates a multitude of traits, such as production of virulence factors, motility and symbiosis [2].
The recent discovery of the crosstalk between bacterial signaling molecules and host-derived hormones has inaugurated a new field for research, and contributed to a better understanding of the pathogen-host relationship [3]. This inter-kingdom signaling appears to be as ancient as the relationship between bacteria and host. In this review, we discuss recent reports concerning the chemical communication between bacteria and host, from both a prokaryotic and a eukaryotic point of view.
Quorum sensing molecules talking to the host
QS was first described in the regulation of bioluminescence in Vibrio fischeri [4]. The luciferase operon in V. fischeri is regulated by two proteins, LuxI, which is the acyl-homoserine-lactone (AHL) autoinducer synthase, and LuxR, which is activated by this autoinducer to increase transcription of the luciferase operon [5,6]. Since this initial description, homologues of LuxR-LuxI have been identified in other bacteria and regulate a variety of phenotypes [2,7,8]. One of the most studied AHL-based QS system is the P. aeruginosa’s. P. aeruginosa has two hierarchical AHL-QS systems, LasR/I and RhlR/I. LasI synthesizes oxo-C12-homoserine lactone (HSL), which activates LasR, and RhlI, synthesizes C4-HSL, which activates RhlR [9]. Although the involvement of AHLs in the bacterial pathogenesis has been extensively studied, less is known about the effects of AHLs on eukaryotic cells. However, there is increasing evidence that oxo-C12-HSL is able to modulate signal transduction and immune responses of the eukaryotic host cell, in vitro and in vivo [10].
P. aeruginosa forms a biofilm inside the lungs of cystic fibrosis (CF) patients, and oxo-C12-HSL molecules can be detected in sputum [11]. The CF lung is characterized by an intense inflammatory response that compromises the organ. In vitro studies indicate that oxo-C12-HSL triggers IL-8 production in bronchial epithelial cells, through induction of the transcription factors NF-kB and AP-2 [12].
In vivo studies using the adult mouse acute lung infection model indicate that a P. aeruginosa ΔlasI/rhlI mutant, defective for the production of oxo-C12-HSL and C4-HSL, is attenuated for disease in mice [13]. oxo-C12-HSL, but not C4-HSL, induces a potent inflammatory response in vivo. Expression analysis indicated increased expression of the chemokines MIP-2 (macrophage inflammatory protein), MCP-1 (monocyte chemotactic protein), MiP1-beta, IP-10, (TCA3) T-cell activation gene 3, IL-1α and IL-6 [13].
These data contrast with results obtained by Tateda et al [14], who observed a decrease in MCP-1. Activated T cells exposed to oxo-C12-HSL showed an increased production of IFN-γ, comparable to that induced by the control IL-12, but not IL-4, suggesting that oxo-C12-HSL induces a Th1 inflammatory response [12,13]. These data suggest that oxo-C12-HSL stimulates inflammation in host tissues.
Smith et al (2002) [13] also reported that oxo-C12-HSL increased the expression of Cyclooxygenase 2 (Cox-2), an enzyme involved in the metabolism of arachidonic acid, in lung fibroblasts, inducing the nuclear translocation of NF-κB. Hence, production of oxo-C12-HSL, and subsequent induction of Cox-2 and PGE2, could help P.aeruginosa to disseminate through the host’s body. In addition to immunodulatory effects, oxo-C12-HSL induces apoptosis in bone-marrow derived macrophages and neutrophiles [14].
Although there is increasing evidence, that oxo-C12-HSL manipulates host cell signaling, the mechanism of this regulation remains unknown. Williams et al.[15] showed that oxo-C12-HSL enters and function within mammalian cells. High concentrations of oxo-C12-HSL induced apoptosis due to calcium mobilization from the endoplasmic reticulum (ER). In contrast, the increase in intracellular calcium did not correlate to the immunomodulatory effects, suggesting the existence of two distinct mammalian receptors for oxo-C12-HSL, one mediating apoptosis via Ca increase, and the other mediating a proinflammatory response [16]. Jahoor et al (2008) [17] hypothesized that the eukaryotic receptor for AHLs is a nuclear hormone receptor (NHR). These authors examined the profile of NHRs expressed in cells upon exposure to oxo-C12-HSL. The two members of the peroxissome proliferators-activated receptor (PPAR), PPARbeta-delta and PPARgamma were expressed and responsive to oxo-C12-HSL in a dose-dependent manner. PPARs are transcription factors that inhibit expression of proinflammatory genes. Transcriptional activity of PPAR-gamma was inhibited by oxo-C12-HSL even in the presence of the PPAR-gamma agonist rosiglitazone, while PPAR-beta-delta had its activity increased. These experiments indicated that oxo-C12-HSL is a ligand for PPAR-beta-delta, reducing its binding to target promoters. In vitro experiments have further shown that oxo-C12-HSL and rosiglitazone modulate PPAR-gamma activity, where oxo-C12-HSL act as antagonist.
Host response to bacterial autoinducers
Many studies have shown that successful colonization of respiratory tract by P. aeruginosa involves expression of genes coordinately regulated by QS. These data have raised the possibility that mammalian cells could have developed mechanisms to disrupt QS signaling as a defense against bacterial infection. Chun et al. [18] reported that oxo-C12-HSL was selectively inactivated by human airway epithelia, and the degrading activity was mediated by heat-sensitive factor associated with the cell rather than a secreted factor. One mechanism that seems to play a major role in circumventing QS-regulated virulence by bacterial pathogens is the production of paraoxonases (PONs) by mammalian cells [19]. PONs constitute a family of conserved enzymes (PON1, PON2 and PON3). PON1 and PON3 are found in the blood, while PON2 is ubiquitously expressed in many tissues such as lung and kidney and brain. PONs were initially described to hydrolyze organophosphates, but also present a calcium-dependent esterase activity over a wide range of substrates, including lactones. Although their biological function is still not well defined, accumulating evidence suggests that its lactonase activity could represent an innate mechanism of defense against bacterial autoinducers [19].
Yang et al [20] have provided evidence of a lactonase activity towards the P. aeruginosa AHLs oxo-C12-HSL, which was conserved in the serum of mammalian species. These authors suggested that PONs were responsible for the AHL inactivation, which is supported by a previous reported from Chun et al [18]. Ozer et al [21] were the first to provide evidence of the inhibitory effect of PONs over P. aeruginosa virulence. Analysis of biofilm formation by P. aeruginosa upon exposure to mouse serum and serum containing human PON1 showed that PON1 blocked biofilm formation. In contrast, in vivo studies using the murine peritonitis/sepsis model of P. aeruginosa infection showed that pon1- knockout mice were protected from P. aeruginosa infection. Speculations about putative compensatory mechanisms operating in the pon1- knockout mice are supported by a recent study conducted by Teiber [19].
Teiber et al [19] used biochemical and genetic approaches in an attempt to identify other mammalian enzymes that could target oxo-C12-HSL in addition to PONs [19]. This study showed that the calcium-dependent esterase activity was mediated by an esterase, which is irreversibly inactivated by EDTA or depletion of metal ions, consistent with activity of PON. PON1 lactonase activity is present in mouse and human sera, while PON2 activity is found in cell lysates [21].
In vitro studies performed by Stoltz et al [22] seem to agree with the previous data. The authors evaluated the ability of murine tracheal epithelial cells collected from PON1, PON2 or PON3 knockout mice to degrade oxo-C12-HSL. PON2-knockout cell lysates presented diminished ability to inactivate oxo-C12-HSL, while intact cells derived from each one of the knockout mice retained their inactivating ability. In addition, it was found that bacteria exposed to sera from PON1 knockout mice had increased QS activity. When the same experiment was performed using murine tracheal epithelial cells, PON2 knockout cells increased QS in P. aeruginosa.
Host hormones talking to bacteria
Eukaryotic organisms utilize several hormones to regulate different aspects of their physiology and maintain homeostasis. Prokaryotic cells signal through autoinducers (AIs), which are hormone-like compounds that allow microorganisms to coordinate behaviors in a population level.
In the gastrointestinal (GI) tract, the number of commensal bacteria exceeds the number of eukaryotic cells by one order of magnitude. Co-evolution of prokaryotic species and their respective eukaryotic host have exposed bacteria to host hormones and eukaryotic cells to the bacterial AIs during host colonization. Therefore, it is not surprising that some pathogenic species have high-jacked these signaling systems to promote disease.
The knowledge host hormones such as catecholamines can promote bacterial growth is not recent [23]. Conflicting reports sugges . In the past few years, there increasing experimental evidence have shown that some pathogenic bacteria are able to recognize host signaling molecules to turn on production of virulence factors.
The Epi/NE/AI-3 quorum sensing system was discovered by Sperandio et al (1999) during an investigation of the regulation of virulence gene expression in EHEC. Cell-to-cell communication involved the detection of a new autoinducer, named AI-3 (Sperandio et al, 2003). Synthesis of AI-3 was initially associated with the gene luxS, since luxS mutants were unable to produce the autoinducer (Sperandio et al, 2003). However, further studies demonstrated that AI-3 synthesis was not dependent on luxS. Actually, metabolic changes caused by the lack of luxS left the bacteria unable to produce AI-3 (Walters et al, 2006).
Studies performed by Sperandio et al (2003) have shown that the enteric pathogen EHEC senses AI-3 produced by the microbial gastrointestinal flora to activate the expression of virulence genes, encoded by locus of enterocyte effacement (LEE). The pathogenicity island LEE encodes for a type III secretion system, which injects bacterial effectors inside the eukaryotic host cells. Expression of the LEE genes lead to formation of attaching and effacing (AE) lesions. AE lesions are characterized by destruction of the microvilli and rearrengment of the actin cytoskeleton, leading to the formation of pedestals that cups the bacteria individually.
EHEC hijacks eukaryotic hormones present in the gastrointestinal tract to activate expression of the virulence genes and promote colonization of the human colon mucosa. Once have reached the human colon, EHEC is able to recognize epinephrine and norepinephrine and activates transcription of the LEE genes and flagella regulon. These catecholamines were able to substitute AI-3 to activate the expression of virulence genes in EHEC. These results provide, for the first time, evidence of crosstalk between prokaryotic and eukaryotic signaling systems (Sperandio et al, 2003). Further work has shown that AI-3/epi/nore crosstalk is mediated by a sensor histidine kinase present at the bacterial surface, named QseC. This membrane-embebed sensor kinase can sense specifically the bacterial hormone-like compound AI-3 as well as the host hormones epinephrine and norepinephrine, binding directly to these signals [25]. The chemical structure of AI-3 is still unknown, but it is known that AI-3 is an aromatic compound. AI-3 seems to bind to the same receptor as epinephrine and norepinephrine, since the use of adrenergic receptors can block the activation of LEE genes and flagellar genes mediated by AI-3.
Biochemical evidence shows that QseBC constitute a functional two-component signal transduction system. Upon sensing AI-3/epi/nore in the gastrointestinal tract, the sensor kinase QseC is activated and undergoes autophosphorylation; subsequently, QseC transduces the signal to its cognate response regulator QseB, which activates the transcription of the master regulator of the flagella regulon, flhDC. In addition, recognition of AI-3/epi/NE by QseC can be specifically blocked by the administration of the alfa-adrenergic antagonist phentolamine, showing that QseC is a bacterial adrenergic receptor. QseC was demonstrated to play an important role in pathogenesis in vivo. By using a rabbit infection model, qseC mutants were shown to be attenuated for virulence. These studies have started the novel field of research, focused on the adrenergic regulation of virulence [26]. Adding a little more complexity in the interkingdom-regulation of virulence in EHEC, two more players were discovered. Recently, a novel two-component system involved in regulation of AE lesion in EHEC has been described, the QseEF system [27]. QseE is the sensor kinase and QseF is the cognate response regulator. QseEF transcription is activated by epinephrine via QseC. Although the signal recognized by the sensor kinase QseE remains to be identified, the results of its activation could be assessed by phenotypic analysis of non-polar mutants of qseE and qseF. QseF activates expression of EspFu, a non-LEE encoded effector that plays a pivotal role in pedestal formation by EHEC [28]. EspFu is translocated by the LEE-encoded TTSS into the enterocytes, where it promotes actin nucleation and polymerization, leading to formation of the pedestal-like structures underneath the bacterium, characteristic of the AE lesions.
The AI-3/epi/NE signaling system doesn’t seem to be restricted to EHEC. In silico analysis have found homologous of QseC in other bacterial species such as Salmonella sp, Shigella flexneri, Francisella tularensis, Haemophilus influenzae, Erwinia carotovoa, Pasteurella multocida, Ralstonia eutropha, Chromobacterium violaceum and others [29]. In vivo studies provide evidence that QseC histidine sensor kinase plays a major role in Salmonella typhimurium [30] and Francisella tularensis [31], since qseC mutants of these strains are attenuated in animal models.
Salmonella
Recent investigations suggest that NE can play a role in the pathogenesis of Salmonella. Bearson and Bearson [30] have studied the gene expression profile of Salmonella enterica serovar typhimurium to 2mM NE. Microarray analysis showed up-regulation of flagellar genes fliA, fliY and fjB. These results were further confirmed by phenotypic analysis using motility plates. Enhanced motility caused by exposure to NE was blocked by phetolamine. In order to investigate the role of the qseC gene in Salmonella, a qseC mutant was constructed, and its motility phenotype was analyzed. Salmonella qseC mutant presented decreased motility compared to wt, even in the presence of NE, so the authors suggest that NE modulates Salmonella motility via the QseC sensor. In addition, the authors investigated the role of QseC in vivo during colonization of a swine model. Competition assays using WT and qseC mutant showed that the qseC mutant Salmonella was impaired in its ability to colonize the gastrointestinal tract of swines, so leading to speculations about the possible involvement of the AI-3/epi/NE interkingdom signaling during Salmonella colonization in vivo.
In a recent investigation of the effects of NE in the growth of Salmonella enterica, Methner et al[24] reported that . Surprinsingly, pre-treatment of bacteria with 50ug/mL of NE before inoculation resulted in increased intestinal colonization and systemic spread to the liver in chicken and mouse models. Mutations in the genes tonB and entS, involved in iron aquisition, didn’t have any impairment in their ability to colonize chickens or mice, reinforcing the idea that NE in more a signaling molecule than an iron source for Salmonella enterica in vivo.
Targeting bacterial cell-to-cell signaling mechanisms seems to be a clever strategy for development of new antimicrobial agents. Since QseC is not present in mammals and its importance for virulence in human pathogens, Rasko et al [29] performed a high-throughput screen in an attempt to identify inhibitors of QseC. This approach has successfully identified LED209, a small molecule able to inhibit QseC-mediated virulence gene expression activated by Epi/NE at a picomolar range. LED209 was able to inhibit Salmonella typhimurium and Francisella tularensis virulence in vivo. The same effect was observed for EHEC in vitro but not in vivo. Importantly, in vitro cell culture assays has demonstrated that LED209 is not harmful for eukaryotic cells, not being able to block adrenergic regulation in these cells.
Vibrio parahemolyticus
Catecholamines are not restricted to Enterobacteraceae. Nakano et al (2007) investigated the effect of NE in the modulation of virulence in emerging pathogen V. parahemolyticus, providing additional evidence of adrenergic regulation of bacterial virulence. Exposure of Vibrio parahemolyticus to 50uM of norepinephrine (NE) enhanced cytotoxicity towards Caco-2 cells, in a TTSS1- (but not TTSS2) dependent manner. Since this effect was blocked by administration of the adrenergic receptors phentolamine and propranolol, it suggests that NE-enhanced cytotoxicity would be mediated by adrenergic receptors displayed by Caco-2 cells. Analysis of gene expression upon exposure to NE showed increased expression of genes associated with TTSS1, such as vscQ and vscU. Using a rat ligated ileal loop model, it was demonstrated that 100uM of NE caused increased accumulation of fluid. Interestingly, the increased enterotoxic activity caused by NE was blocked by administration of alfa1-adrenergic antagonist prazosin but not the alfa-2 adrenergic antagonist yohimbine or the beta-adrenergic antagonist propranolol. It suggests that the NE can modulate virulence of V. parahemolyticus by interacting with alfa1-adrenergic receptors presented by the pathogen.
Dynorphin: opioids as a novel hormone hijacked by pathogenic bacteria
It is well established that in an organism undergoing, several hormones are released to restore homeostasis. Between the stress-related hormones are included the catecholamines adrenaline and noradrenaline; and opioids such as endorphin and dynorphin. Several reports show host-derived signaling molecules are able to activate bacterial virulence during stress responses. Given that opioids abundantly released during stress responses and at inflammatory sites, Zaborina et al [32] decided to investigate the influence of opioids in the quorum sensing system of P. aeruginosa. P. aeruginosa colonizes the lung of cystic fibrosis patients, where it forms a biofilm. Biofilm production is regulated by a complex quorum sensing mechanism involving 3 signaling molecules: HQNO, HHQ and PQS. Interestingly, the results showed that P. aeruginosa PAO1 could recognize k-opioid receptor agonists U-50,488 and dynorphin, and use it to enhance virulence by increasing the production of the three QS signals by up-regulating the operon pqsABCDE. There is evidence of opioids accumulation at inflammatory sites; so, this mechanism can be operating in the lung of CF patients, contributing to a persistent colonization by P. aeruginosa.
5-Rhomboid Proteins: Convergent Evolution
Rhomboid (RHO) proteins comprise a conserved family of serine proteases present in involved in the epidermal growth factor (EGF) signaling pathway. Although it has been more studied in Drosophila melanogaster development, RHO proteins are widespread, with homologous being found in eubacteria, archea, yeast and plants [33]. They present low level of sequence-similarity; in contrast, they present similarity at the structural level as well as conserved aminoacid residues, which constitute a RHO domain[33]. RHO proteins are polytopic 7-transmembrane domain proteins displaying serine protease activity, responsible for processing the inactive membrane-associated form of EGFr into a secreted and active form.
The first prokaryotic member of the RHO family to be genetically characterized was the product of the gene aarA from the Gram-negative pathogen Providencia stuartii. The gene aarA is involved in the production of an unknown quorum sensing molecule, which represses the expression of an acetyl-transferase that modifies peptidoglycan and aminoglycosides. The observation that a RHO-related protein is involved in cell-to-cell signaling in this bacterium raised the speculation about evolutive convergence of the RHO proteins [34]. In an attempt to elucidate the function of RHO, Gallio et al [33] constructed a mutant of Drosophila defective for the production of RHO and a aarA-mutant of P. stuartii. They further tried to rescue the phenotypes of these mutants by complementing each mutant with a plasmid containing the gene encoding RHO protein of the reciprocal organism. Using wing development as a RHO-dependent reporter phenotype in flies, this group reported that aarA from P. stuartii was able to rescue wing development in rho-1- D. melanogaster. In addition, Drosophila rho-1 was proved to substitute AarA from P. stuartii and rescue cell-to-cell communication activity in the aarA mutant bacteria. The authors speculate that RHO signaling could represent an ancient mechanism of cell communication, which could have been crucial for the evolution of multicellularity.
Recently, Stevenson et al [35] reported the identification of a natural substrate for the aarA-encoded RHO protein of P. stuartii. In addition, they provided in vitro and in vivo evidence that the P. stuartii RHO protein mediates cleavage of TatA protein, resulting in activation of this integrant of the twin-arginine translocase (Tat) protein secretion system.
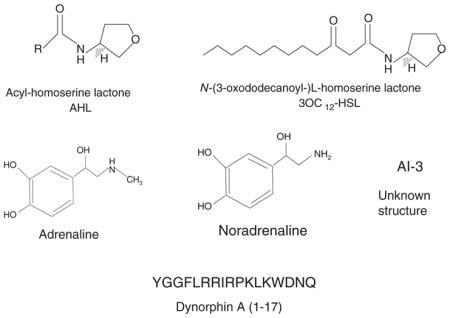
Figure 1.

Chemical signals used in cell-to-cell communication between bacteria and host.
Table 1.
Signals, receptors and effects involved in crosstalk between pathogenic bacteria and mammalian hosts
| Pathogen | Signal source | Bacterial receptor | Host receptor | Effects in bacteria | Effects in the host |
|---|---|---|---|---|---|
| Pseudomonas aeruginosa | 3OC12-HSL bacterial | LasR | PPARβ/δ PPARγ | Production of virulence factors (elastase, exotosin A), biofilm, regulation of rhl QS system | PPARβ/δ energy, homeostasis, cell proliferation and differentiation PPARγ: anti-inflammatory |
| EHEC Salmonella Francisella | AI-3 bacterial | QseC | ? | Activation of motility, T3SS, Shiga-toxin | ? |
| EHEC Salmonella Francisella | Adrenaline Noradrenaline Host | QseC | Adrenergic receptors | Activation of motility, T3SS, Shiga-toxin | Stress responses Electrolyte balance Intestinal motility |
| Pseudomonas aeruginosa | Dynorphin A Host | ? | Kappa-opiod receptors | Increased expression of piocyanin and quorum sensing molecules quinolone-based | Modulation of analgesic responses |
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fuqua WC, Winans SC, Greenberg EP. Quorum sensing in bacteria: the LuxR-LuxI family of cell density-responsive transcriptional regulators. J Bacteriol. 1994;176:269–275. doi: 10.1128/jb.176.2.269-275.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.de Kievit TR, Iglewski BH. Bacterial quorum sensing in pathogenic relationships. Infect Immun. 2000;68:4839–4849. doi: 10.1128/iai.68.9.4839-4849.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hughes DT, Sperandio V. Inter-kingdom signaling: communication between bacteria and host. Nature Reviews Microbiology. 2008;6:111–120. doi: 10.1038/nrmicro1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nealson KH, Platt T, Hastings JW. Cellular control of the synthesis and activity of the bacterial luminescent system. J Bacteriol. 1970;104:313–322. doi: 10.1128/jb.104.1.313-322.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Engebrecht J, Nealson K, Silverman M. Bacterial bioluminescence: isolation and genetic analysis of functions from Vibrio fischeri. Cell. 1983;32:773–781. doi: 10.1016/0092-8674(83)90063-6. [DOI] [PubMed] [Google Scholar]
- 6.Engebrecht J, Silverman M. Identification of genes and gene products necessary for bacterial bioluminescence. Proc Natl Acad Sci U S A. 1984;81:4154–4158. doi: 10.1073/pnas.81.13.4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parsek MR, Greenberg EP. Acyl-homoserine lactone quorum sensing in gram-negative bacteria: a signaling mechanism involved in associations with higher organisms. Proc Natl Acad Sci U S A. 2000;97:8789–8793. doi: 10.1073/pnas.97.16.8789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davies DG, Parsek MR, Pearson JP, et al. The involvement of cell-to-cell signals in the development of a bacterial biofilm. Science. 1998;280:295–298. doi: 10.1126/science.280.5361.295. [DOI] [PubMed] [Google Scholar]
- 9.Schuster M, Greenberg EP. A network of networks: quorum-sensing gene regulation in Pseudomonas aeruginosa. Int J Med Microbiol. 2006;296:73–81. doi: 10.1016/j.ijmm.2006.01.036. [DOI] [PubMed] [Google Scholar]
- 10.Kravchenko VV, Kaufmann GF, Mathison JC, et al. Modulation of gene expression via disruption of NF-kappaB signaling by a bacterial small molecule. Science. 2008;321:259–263. doi: 10.1126/science.1156499. [DOI] [PubMed] [Google Scholar]
- 11.Singh PK, Schaefer AL, Parsek MR, et al. Quorum-sensing signals indicate that cystic fibrosis lungs are infected with bacterial biofilms. Nature. 2000;407:762–764. doi: 10.1038/35037627. [DOI] [PubMed] [Google Scholar]
- 12.Smith RS, Fedyk ER, Springer TA, et al. IL-8 production in human lung fibroblasts and epithelial cells activated by the Pseudomonas autoinducer N-3-oxododecanoyl homoserine lactone is transcriptionally regulated by NF-kappa B and activator protein-2. J Immunol. 2001;167:366–374. doi: 10.4049/jimmunol.167.1.366. [DOI] [PubMed] [Google Scholar]
- 13.Smith RS, Harris SG, Phipps R, et al. The Pseudomonas aeruginosa quorum-sensing molecule N-(3-oxododecanoyl)homoserine lactone contributes to virulence and induces inflammation in vivo. J Bacteriol. 2002;184:1132–1139. doi: 10.1128/jb.184.4.1132-1139.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tateda K, Ishii Y, Horikawa M, et al. The Pseudomonas aeruginosa autoinducer N-3-oxododecanoyl homoserine lactone accelerates apoptosis in macrophages and neutrophils. Infect Immun. 2003;71:5785–5793. doi: 10.1128/IAI.71.10.5785-5793.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Williams SC, Patterson EK, Carty NL, et al. Pseudomonas aeruginosa autoinducer enters and functions in mammalian cells. J Bacteriol. 2004;186:2281–2287. doi: 10.1128/JB.186.8.2281-2287.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shiner EK, Terentyev D, Bryan A, et al. Pseudomonas aeruginosa autoinducer modulates host cell responses through calcium signalling. Cell Microbiol. 2006;8:1601–1610. doi: 10.1111/j.1462-5822.2006.00734.x. [DOI] [PubMed] [Google Scholar]
- 17.Jahoor A, Patel R, Bryan A, et al. Peroxisome proliferator-activated receptors mediate host cell proinflammatory responses to Pseudomonas aeruginosa autoinducer. J Bacteriol. 2008;190:4408–4415. doi: 10.1128/JB.01444-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chun CK, Ozer EA, Welsh MJ, et al. Inactivation of a Pseudomonas aeruginosa quorum-sensing signal by human airway epithelia. Proc Natl Acad Sci U S A. 2004;101:3587–3590. doi: 10.1073/pnas.0308750101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Teiber JF, Horke S, Haines DC, et al. Dominant role of paraoxonases in inactivation of the Pseudomonas aeruginosa quorum-sensing signal N-(3-oxododecanoyl)-L-homoserine lactone. Infect Immun. 2008;76:2512–2519. doi: 10.1128/IAI.01606-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang F, Wang LH, Wang J, et al. Quorum quenching enzyme activity is widely conserved in the sera of mammalian species. FEBS Lett. 2005;579:3713–3717. doi: 10.1016/j.febslet.2005.05.060. [DOI] [PubMed] [Google Scholar]
- 21.Ozer EA, Pezzulo A, Shih DM, et al. Human and murine paraoxonase 1 are host modulators of Pseudomonas aeruginosa quorum-sensing. FEMS Microbiol Lett. 2005;253:29–37. doi: 10.1016/j.femsle.2005.09.023. [DOI] [PubMed] [Google Scholar]
- 22.Stoltz DA, Ozer EA, Ng CJ, et al. Paraoxonase-2 deficiency enhances Pseudomonas aeruginosa quorum sensing in murine tracheal epithelia. Am J Physiol Lung Cell Mol Physiol. 2007;292: L852–860. doi: 10.1152/ajplung.00370.2006. [DOI] [PubMed] [Google Scholar]
- 23.Lyte M, Ernst S. Catecholamine induced growth of gram negative bacteria. Life Sci. 1992;50:203–212. doi: 10.1016/0024-3205(92)90273-r. [DOI] [PubMed] [Google Scholar]
- 24.Methner U, Rabsch W, Reissbrodt R, et al. Effect of norepinephrine on colonisation and systemic spread of Salmonella enterica in infected animals: role of catecholate siderophore precursors and degradation products. Int J Med Microbiol. 2008;298:429–439. doi: 10.1016/j.ijmm.2007.07.013. [DOI] [PubMed] [Google Scholar]
- 25.Clarke MB, Hughes DT, Zhu C, et al. The QseC sensor kinase: a bacterial adrenergic receptor. Proc Natl Acad Sci U S A. 2006;103:10420–10425. doi: 10.1073/pnas.0604343103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Waldor MK, Sperandio V. Adrenergic regulation of bacterial virulence. J Infect Dis. 2007;195:1248–1249. doi: 10.1086/513281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reading NC, Torres AG, Kendall MM, et al. A novel two-component signaling system that activates transcription of an enterohemorrhagic Escherichia coli effector involved in remodeling of host actin. J Bacteriol. 2007;189:2468–2476. doi: 10.1128/JB.01848-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Campellone KG, Robbins D, Leong JM. EspFU is a translocated EHEC effector that interacts with Tir and N-WASP and promotes Nck-independent actin assembly. Dev Cell. 2004;7:217–228. doi: 10.1016/j.devcel.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 29.Rasko DA, Moreira CG, Li de R, et al. Targeting QseC signaling and virulence for antibiotic development. Science. 2008;321:1078–1080. doi: 10.1126/science.1160354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spector MP, Garcia del Portillo F, Bearson SM, et al. The rpoS-dependent starvation-stress response locus stiA encodes a nitrate reductase (narZYWV) required for carbon-starvation-inducible thermotolerance and acid tolerance in Salmonella typhimurium. Microbiology. 1999;145(Pt 11): 3035–3045. doi: 10.1099/00221287-145-11-3035. [DOI] [PubMed] [Google Scholar]
- 31.Weiss DS, Brotcke A, Henry T, et al. In vivo negative selection screen identifies genes required for Francisella virulence. Proc Natl Acad Sci U S A. 2007;104:6037–6042. doi: 10.1073/pnas.0609675104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zaborina O, Lepine F, Xiao G, et al. Dynorphin activates quorum sensing quinolone signaling in Pseudomonas aeruginosa. PLoS Pathog. 2007;3:e35. doi: 10.1371/journal.ppat.0030035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gallio M, Sturgill G, Rather P, et al. A conserved mechanism for extracellular signaling in eukaryotes and prokaryotes. Proc Natl Acad Sci U S A. 2002;99:12208–12213. doi: 10.1073/pnas.192138799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baca-DeLancey RR, South MM, Ding X, et al. Escherichia coli genes regulated by cell-to-cell signaling. Proc Natl Acad Sci U S A. 1999;96:4610–4614. doi: 10.1073/pnas.96.8.4610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jacobs MR, Koornhof HJ, Robins-Browne RM, et al. Emergence of multiply resistant pneumococci. N Engl J Med. 1978;299:735–740. doi: 10.1056/NEJM197810052991402. [DOI] [PubMed] [Google Scholar]
- 36.Lopez MA, Bannenberg G, Castresana C. Controlling hormone signaling is a plant and pathogen challenge for growth and survival. Curr Opin Plant Biol. 2008;11:420–427. doi: 10.1016/j.pbi.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 37.Bryant JA, Wylie BR, Yuan FF, et al. Effect of blood donation on the establishment of normal ranges of lymphocyte subsets. Transfusion. 1996;36:559–566. doi: 10.1046/j.1537-2995.1996.36696269517.x. [DOI] [PubMed] [Google Scholar]
- 38.Chevrot R, Rosen R, Haudecoeur E, et al. GABA controls the level of quorum-sensing signal in Agrobacterium tumefaciens. Proc Natl Acad Sci U S A. 2006;103:7460–7464. doi: 10.1073/pnas.0600313103. [DOI] [PMC free article] [PubMed] [Google Scholar]