

ABSTRACT
Cu ion (Cu) entry into human cells is mediated by CTR1 (also known as SLC31A1), the high-affinity Cu transporter. When extracellular Cu is raised, the cell is protected against excess accumulation by rapid internalization of the transporter. When Cu is lowered, the transporter returns to the membrane. We show in HEK293 cells overexpressing CTR1 that expression of either the C-terminal domain of AP180 (also known as SNAP91), a clathrin-coat assembly protein that sequesters clathrin, or a dominant-negative mutant of dynamin, decreases Cu-induced endocytosis of CTR1, as does a dynamin inhibitor and clathrin knockdown using siRNA. Utilizing imaging, siRNA techniques and a new high-throughput assay for endocytosis employing CLIP-tag methodology, we show that internalized CTR1 accumulates in early sorting endosomes and recycling compartments (containing Rab5 and EEA1), but not in late endosomes or lysosomal pathways. Using live cell fluorescence, we find that upon extracellular Cu removal CTR1 recycles to the cell surface through the slower-recycling Rab11-mediated pathway. These processes enable cells to dynamically alter transporter levels at the plasma membrane and acutely modulate entry as a safeguard against excess cellular Cu.
KEY WORDS: CTR1, Regulatory endocytosis, Metal ion homeostasis, Cu transporter recycling
Summary: Regulatory endocytosis and recycling processes of CTR1 via clathrin-mediated and Rab11 pathways enable cells to dynamically alter transporter levels at the plasma membrane and acutely modulate Cu entry.
INTRODUCTION
The plasma membrane content of a transporter determines the total flux of its substrate into (or out of) the cell. There is a need to regulate cellular composition to either protect against the toxic effects of certain substrates, as with elevation of P-glycoprotein and drug resistance (Bradley et al., 1989), or to ensure substrate is absorbed under low substrate conditions, as in the amino acid permease in yeast (Ghaddar et al., 2014). In these cases, transporter synthesis or degradation is regulated. There are many hormone-regulated transporters, among them the action of insulin on the trafficking of glucose transporter GLUT4 (Klip et al., 2014). In the present work, we provide evidence on the uptake mechanism and recycling pathway utilized by mammalian cells to regulate the uptake of Cu ions (hereafter referred to as Cu), an essential but toxic micronutrient. The regulation is unique among transporters in that it is controlled by the extracellular level of the substrate and acts directly on the transporter in a dynamic, acutely reversible mechanism that depends upon medium Cu and not alterations in synthesis or degradation of the transporter.
Cu is essential for normal mammalian cell physiology. It is utilized as a co-factor in many enzymatic processes including: protection against reactive oxygen species as a co-factor in superoxide dismutase (SOD), as a co-factor for cytochrome c oxidase, in neurons for neurotransmitter and brain peptide biosynthesis, and in the extracellular matrix as a co-factor for lysyl oxidase in collagen cross-linking (Iakovidis et al., 2011). However, because of its ready non-specific reaction with many amino acid side chains and ability to undergo Fenton chemistry and generate reactive oxygen species, Cu can be damaging at excessive levels. The cellular homeostasis of Cu is accomplished by a Cu proteome consisting of uptake transporter(s), specific metallochaperones, and glutathione (GSH) that deliver cellular Cu to its target proteins and exporters that protect against excess accumulation (Harrison et al., 2000; Maryon et al., 2013b). The main high-affinity Cu uptake protein, CTR1, delivers Cu to the intracellular milieu, whereas under low (normal) cellular Cu the export proteins ATP7A and ATP7B, Cu-activated P-type ATPases, are responsible for delivering Cu to the proteins in the secretory pathway. When intracellular Cu builds up, these trans-Golgi-resident proteins bud off in the membranes of vesicles into which the excess Cu has been pumped and fuse with the plasma membrane to expel excess cellular Cu (Hung et al., 1997; Petris et al., 1996). This is of clinical significance as Menkes disease and Wilson disease are associated with inherited dysfunction of ATP7A and ATP7B, respectively (Daniel et al., 2004). Elevated Cu levels have been implicated in tumor growth and have made Cu systems promising targets for anticancer therapy (Gupte and Mumper, 2009; Safi et al., 2014).
Cellular Cu homeostasis and protection against the potential toxic effects of excess intracellular Cu are achieved by relocation of the Cu-ATPases. Just as the Cu exporters ATP7A and ATP7B respond to elevated intracellular Cu, the Cu uptake protein CTR1 (also known as SLC31A1) can respond to elevated extracellular Cu. We showed previously that excess extracellular Cu caused a loss of plasma membrane CTR1, and proportional lowering of Cu entry rate. This internalization protects against Cu toxicity (Molloy and Kaplan, 2009). When extracellular Cu was returned to normal levels, CTR1 recycled to the plasma membrane. We termed this process regulatory endocytosis and proposed that it was a defense against acute elevations of extracellular Cu. The process could be repeated several times, was seen in a wide range of cells, and was an acute regulatory response to elevated medium Cu (Molloy and Kaplan, 2009). CTR1 is a small protein of 190 amino acid residues with three transmembrane segments, a variable extracellular amino terminus and a highly conserved intracellular carboxyl terminus of about 15 residues (Kaplan and Maryon, 2016). During a recent functional mutational analysis, we observed that mutations in the final three highly conserved residues (His-Cys-His) resulted in high rates of transport and the mutant CTR1 did not undergo regulatory endocytosis in elevated Cu (Maryon et al., 2013a). The basis for this loss of regulatory endocytosis is not understood. In order to adequately describe cellular Cu regulation it was necessary to learn more about the internalization process, and the intracellular pathway for internalization and recycling of CTR1.
In the present work, we have investigated the Cu-dependent CTR1 internalization and recycling process, and sought to identify the trafficking pathway in this acute regulatory endocytosis and recycling. We employed an expression system in which HEK cells overexpress CTR1 under the control of a tetracycline-sensitive promoter to minimize effects of long-term growth with excessive Cu uptake. CLIP-tagged CTR1 was also utilized, providing the ability to view cells in real-time using live cell imaging or to utilize a novel fluorescent plate assay technique for quantitating endocytosis. We find that CTR1 is internalized in a clathrin-mediated endocytic (CME) pathway when cells are exposed to excess Cu. Vesicles carrying CTR1 fuse with the early endosome, where CTR1 is then sorted into recycling compartments and returned to the cell surface upon Cu removal. Depletion of Rab11 GTPases, known to regulate the slow recycling pathway, prevents the return of endocytosed CTR1, establishing its role in CTR1 recycling. This substrate-dependent regulatory recycling is unique among metal ion transporters and provides new information on the cell biology of Cu transporters and their roles in the regulation of Cu homeostasis.
RESULTS
Internalized CTR1 co-localizes with early sorting endosomes and endocytosed transferrin
In elevated extracellular Cu, cell surface CTR1 is removed from the plasma membrane (Molloy and Kaplan, 2009; Petris et al., 2003). To visualize CTR1 trafficking, we labeled CTR1 proteins in HEK-CLIP–CTR1 cells using CLIP-Surface fluorescent antibodies and then visualized the localization of CTR1 after 30 min with or without 30 µM excess Cu. The CLIP tag is a 20 kDa mutant of the human DNA repair protein O6-alkylguanosine-DNA alkyltransferase (AGT) that reacts specifically and rapidly with benzylcytosine derivatives covalently labeling the CLIP-tag with a synthetic probe. For live cell imaging (Fig. 1A), HEK-CLIP–CTR1 cells were loaded with fluorescent transferrin (Tf), which binds to the transferrin receptor and enters the cell via clathrin-dependent endocytosis (Mayle et al., 2012), and CLIP-Surface label that only binds to CTR1 proteins at the plasma membrane. Cells were then treated with or without excess Cu (30 min) before imaging. Without added Cu, CLIP-labeled CTR1 (green) was at the plasma membrane, whereas Tf (red) was internalized into vesicular structures within the cell (Fig. 1A, left panel). After Cu treatment (Fig. 1A, right panel), CLIP-labeled CTR1 was internalized into punctate structures throughout the cytoplasm. Tf was internalized in both low and excess Cu-treated cells (Fig. 1A, left and right panels). Cu-treated cells showed internalized CTR1 in vesicle structures also containing Tf (merged images as yellow). Tf binds to transferrin receptor (TfR) at the surface and the ligand–receptor complex is endocytosed into clathrin-coated vesicles. To determine if the effect of Cu treatment is specific to CTR1, or whether other plasma membrane proteins are also internalized, we compared the localization of CTR1 with that of either TfR (Fig. 1B) or β-catenin (Fig. 1C). HEK-FLAG–CTR1 cells were treated with transferrin with or without 30 µM Cu (30 min), fixed, and probed for anti-FLAG (for CTR1) and TfR (Fig. 1B) or β-catenin (Fig. 1C). Confocal imaging shows that CTR1 follows a pathway like that of TfR when cells are treated with Cu. DAPI staining was used for cell counts, and DAPI stain intensity varied somewhat because of reagent incubation duration, manufacturer product lot, and length of time prior to imaging slides. Quantification of CTR1 co-localization with TfR was determined using ImageJ software (NIH) with the Coloc2 plug-in, and displayed in Table 1 as Manders' coefficients. Similar co-localization patterns of CTR1 and TfR after rapid internalization suggest a common pathway of cargo sorting and/or internalization. The effect of Cu on internalization is specific to CTR1 and not a result of internalization of whole membrane sheets, as β-catenin is not internalized in response to Cu (Fig. 1C). Co-localization of CTR1 with β-catenin is more prominent before Cu treatment, when CTR1 is mainly at the plasma membrane. Upon Cu addition, CTR1 internalizes and co-localization with β-catenin is significantly decreased (Table 1).
Fig. 1.

CTR1 co-localization with membrane or endosome markers after Cu treatment. (A) HEK-CLIP–CTR1 cells were incubated with CLIP-Surface–488 (green), followed by treatment with or without 30 µM Cu and with fluorescent transferrin (red) (30 min), and live cell imaging was performed. (B–D) For fixed cell immunofluorescence, HEK-FLAG–CTR1 cells were treated with or without Cu, fixed cells were labeled with anti-FLAG for CTR1 and either anti-TfR (B), anti-β-catenin (C), or antibody against early endosome marker EEA1 (D). (E) MDCK-FLAG–CTR1 cells treated with or without Cu were double-labeled with anti-FLAG for CTR1 and anti-EEA1. (F) SKCO15 cells transiently transfected to express FLAG-tagged CTR1 were treated with or without Cu (30 min), fixed, probed with anti-FLAG (CTR1). (G) HEK-CLIP–CTR1 cells treated with or without 30 µM Cu (30 min), fixed, and labeled with anti-TfR, anti-EEA1 and DAPI. Stained nuclei are visible in blue and merged fluorescence is displayed in yellow. Scale bars: 10 µm.
Table 1.
Quantification of co-localization between CTR1 and specified proteins during no-excess-Cu treatment or with 30 µM Cu treatment for 30 min
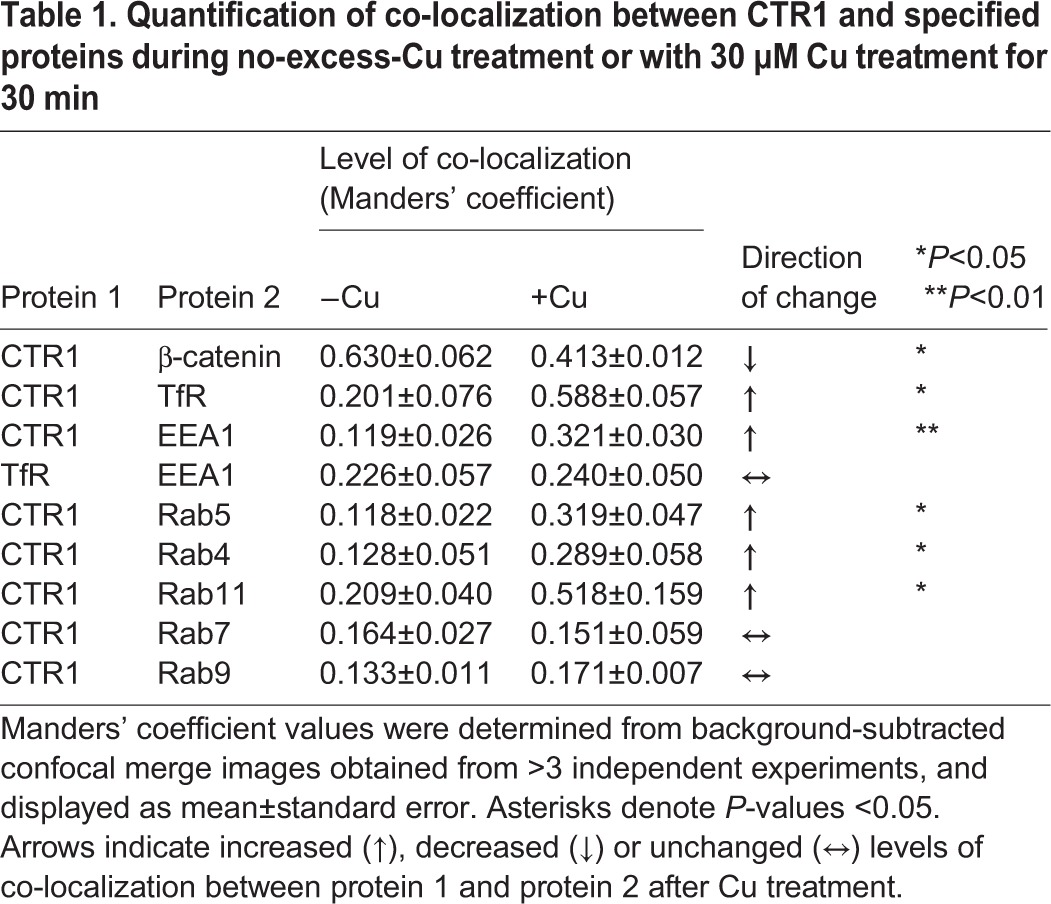
To determine the nature of the vesicles containing internalized CTR1, we treated HEK-FLAG–CTR1 cells with or without Cu, fixed and double-labeled with antibodies against FLAG (CTR1, red) and early endosome marker EEA1 (green, Fig. 1D) (Mu et al., 1995). Much of the CTR1 was localized in the early endosome after 30 min Cu treatment (Fig. 1D, right panel). Quantification of CTR1 co-localization with EEA1 is displayed in Table 1. The early endosomes are distributed in the cytoplasm and serve a sorting function, as proteins accumulate in their tubular extensions and vesicular regions that bud off and proceed to either recycling or lysosomal pathways (Mellman, 1996). To determine if the internalization of CTR1 is specific to HEK cells or whether other cells types also display a similar pattern of localization, we employed two different cell types, MDCK and SKCO15 cells. MDCK cells are derived from canine kidney, and SKCO15 cells from a metastatic site of a human colon. Internalization of CTR1 during Cu treatment was observed in both MDCK (Fig. 1E) and SKCO15 cells (Fig. 1F). We also determined whether Cu affects the amount of TfR endocytosis. We treated HEK-CLIP–CTR1 cells with or without 30 µM Cu (30 min), and then fixed cells were probed with anti-TfR and anti-EEA1 (Fig. 1G). Merged images show co-localization in yellow. No significant change in TfR co-localization with EEA1 occurred with Cu treatment (Table 1).
To quantify CTR1 internalization during various Cu treatments, we devised a novel and flexible high-throughput CLIP assay that detects only surface CTR1. This assay requires constitutive expression of CLIP-epitope-tagged CTR1 and the use of a fluorescent CLIP-Surface label. The CLIP-Surface label is impermeable, and will only label CLIP-tagged CTR1 proteins that are exposed to the extracellular medium. After treatment with Cu to initiate endocytosis, the CLIP label is added and allowed to bind to CTR1 proteins at the cell surface. Endocytosed CTR1 is not labeled. As we observed in previous experiments using cell surface biotinylation (Molloy and Kaplan, 2009), endocytosis was dependent on extracellular Cu concentration, incubation time and temperature. Biotinylation assays used to pull down surface CTR1 proteins after HEK-FLAG–CTR1 cells were treated with excess Cu at either 25°C or 37°C demonstrate that CTR1 endocytosis is permissible at 37°C but is inhibited at room temperature (Fig. S1A,B). This temperature inhibition of CTR1 endocytosis enabled us to incubate HEK-CLIP–CTR1 cells for sufficient times with CLIP label during the CLIP plate assay without causing increased endocytosis. Using the CLIP plate assay, we determined that increasing treatment duration with 10 µM Cu had a profound effect on CTR1 internalization, with ∼30% of surface CTR1 internalized after 30 min and over 60% after one hour, as visualized in plate reader well scan images (Fig. S1C) and quantified (Fig. S1D). Cu concentrations greater than 20 µM resulted in nearly 90% CTR1 internalization (Fig. S1E).
Endocytosis is dependent on clathrin and dynamin
Excess Cu addition causes internalization of CTR1 (Fig. 1). The effect of Cu on CTR1 localization occurs in multiple cell types (Fig. 1E,F) and is specific to CTR1, as it does not cause increased endocytosis of surface proteins such as β-catenin or TfR (Fig. 1C,G). The endocytic route of TfR is well-studied as a model for CME. After Tf binds to the TfR, the ligand–receptor complex is endocytosed into vesicles that are transported to the early endosomes where TfR continues along the recycling pathway back to the cell surface (Mayle et al., 2012). To determine if the CTR1 that is endocytosed after Cu treatment follows a clathrin-dependent endocytic pathway similar to that of TfR, we expressed dominant-negative forms of proteins dynamin-1 or carboxyl-domain-truncated clathrin coat assembly protein AP180 (also known as SNAP91). These mutant proteins, designated DynK44A and AP180c, prevent endocytosis by disrupting clathrin vesicle formation and budding (Damke et al., 1994; Hill et al., 2001) or by sequestering clathrin (Zhao et al., 2001), respectively. In Fig. 2, HEK-CLIP–CTR1 cells were transiently transfected for three days with plasmids containing DNA for either empty control (Fig. 2A), AP180c (Fig. 2B), wild-type dynamin (Fig. 2C), or mutant DynK44A (Fig. 2D). Cells were then CLIP-Surface-labeled, and treated with 30 µM Cu and Tf (30 min) before cell fixation, permeabilization and immunofluorescence. Transfection efficiency was determined by probing with either anti-HA for dynamin expression or anti-FLAG for AP180c expression (Fig. 2, third column panels, shown in purple). Approximately 30% of all cells expressed the exogenous mutants, and imaging fields containing both transfected and non-transfected cells were selected for analysis. Cells expressing the exogenous dynamin or AP180c protein were identified and marked with asterisks for easy visualization in CTR1 and TfR panels. In empty vector control or wild-type-dynamin-expressing cells (Fig. 2A,C), TfR (red) was present mostly in perinuclear and cytosolic regions, as previously observed in Fig. 1B, and CLIP–CTR1 (green) was localized mainly in intracellular vesicles in cells transfected with either empty vector control or wild-type dynamin and treated with excess Cu. These cells show co-localization of CTR1 and TfR in cytosolic regions (Fig. 2A,C, fourth column panels). In Fig. 2B and D, AP180c- or DynK44A-expressing cells (asterisks) show TfR (red) located mainly at the surface membrane, demonstrating that dynamin- and clathrin-dependent endocytosis was impaired. Cells labeled for mutant expression (asterisks) also cause CTR1 to be retained at the plasma membrane (Fig. 2B,D, left panels). CTR1 did not endocytose during excess Cu treatment when either clathrin- or dynamin-dependent pathways were impaired. Zoomed images demonstrate that CTR1 in these mutant-expressing cells co-localized with TfR at the plasma membrane (Fig. 2B,D, far right panels). It should be noted that although mutant expression of DynK44A or AP180c impairs clathrin-mediated endocytosis, the presence of endogenous clathrin and/or dynamin might still provide a route, though limited, of clathrin-mediated endocytosis.
Fig. 2.

Inhibiting dynamin-1 or clathrin assembly prevents CTR1 endocytosis: imaging analysis. CTR1 endocytosis was prevented in HEK-CLIP–CTR1 cells expressing dominant-negative DynK44A–HA (D) or AP180c–FLAG (B), but not in cells transfected with control empty vector (A) or expressing wild-type dynamin-1 (DynWT, C). Surface CTR1 proteins were fluorescently labeled with CLIP-Surface label (CTR1), and then cells were treated with Tf and 30 µM Cu for 30 min. Cells were fixed and probed with anti-TfR and either anti-FLAG or anti-HA antibodies. Confocal images at 100× are displayed, and 4× zoomed sections are shown in far right panels. Asterisks indicate cells that contain exogenous expression of dynamin or AP180c. CTR1 and TfR merge is displayed in yellow. Scale bars: 20 µm in first four columns, 10 µm in far right column.
Using immunofluorescence imaging, we found CTR1 endocytosis can be prevented by disrupting clathrin-mediated pathways with mutant DynK44A or AP180c (Fig. 2). To quantify the inhibition, we employed a high-throughput CLIP assay. The CLIP assay was used to determine how much CTR1 was retained at the cell membrane after 0, 30 or 100 µM Cu treatment for 30 min. Fig. 3A displays an example of a CLIP assay well scan visual readout using dual colorimetric analysis. Threshold values for the colorimetric display are user-defined (Fig. 3A), whereas quantified fluorescence values of CTR1 internalization compared with untreated control cells in histograms (Fig. 3B) were calculated and displayed as mean percentage±standard error. Fig. 3B shows that ∼55% of CTR1 was endocytosed in control cells. This is comparable with experiments using cell surface biotinylation (Molloy and Kaplan, 2009). Cells that express mutant DynK44A or AP180c had 20–30% more CTR1 retained at the cell surface than controls. Because not all of the cells expressed DynK44A or AP180c (Fig. 2) and endogenous clathrin and/or dynamin might retain limited function, the extent of inhibition of endocytosis is somewhat underestimated in this assay.
Fig. 3.
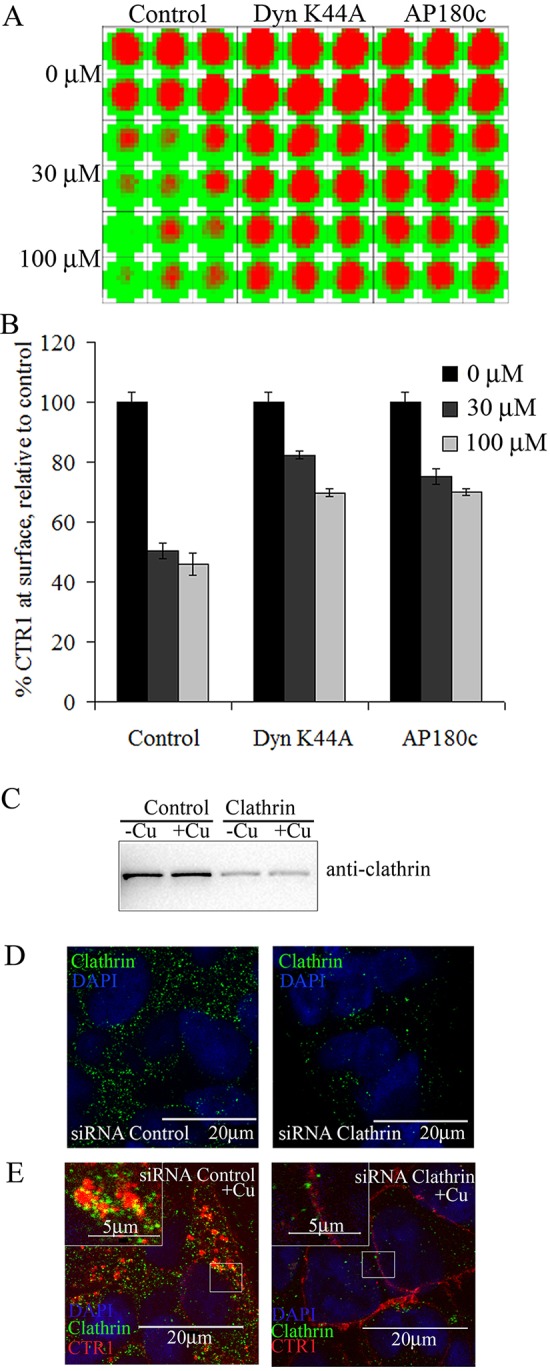
Cu-induced CTR1 internalization is mediated by clathrin and dynamin pathways: plate assay. HEK-CLIP–CTR1 cells transfected with empty vector control, DynK44A, or AP180c were treated with 30 or 100 µM Cu for 30 min, and surface CTR1 was labeled with fluorescent CLIP-Surface–547. (A) An example of a plate scan from the CLIP plate assay using a 10×10 scan matrix for each well. Red indicates CLIP fluorescence on surface CTR1, and green indicates lack of fluorescence. (B) Histogram showing the relative surface expression of CTR1 after Cu treatment compared with untreated control cells, mean±s.e.m., n=6. (C–E) HEK-CLIP–CTR1 cells were transiently transfected with siRNA against either scramble control or clathrin-targeted siRNA. (C) Lysates from siRNA-transfected cells treated with or without excess Cu were collected and subject to western blot analysis using anti-clathrin antibodies. (D) Cells were probed using anti-clathrin antibodies to show sufficient knockdown of clathrin (right panel) compared with control (left panel). (E) siRNA-transfected cells were labeled with Surface-CLIP (red) and then treated with or without 30 µM Cu for 30 min prior to fixation and anti-clathrin (green) immunofluorescent probing. Zoomed insets of selected regions are displayed. Scale bars: 20 µm, 5 µm in insets.
Selection of stable cells lines expressing DynK44A or AP180c is difficult because of reduced cell viability over time, as functioning clathrin and dynamin are vital for cell health (Damke et al., 1994). Thus, we had performed short-term transient transfection of DynK44A or AP180c over three days for studies described in Figs 2 and 3, reducing the negative effects of clathrin and/or dynamin depletion. The amount of mutant versus endogenous expression varied from cell to cell, making it difficult to accurately measure effects of CME inhibition. For more reliable quantification, we utilized a small molecule inhibitor that affects all of the cells, Dynasore. Dynasore inhibits dynamin-1, dynamin-2, and mitochondrial dynamin-like protein Drp1 (Macia et al., 2006). We tested whether Dynasore disrupts CTR1 endocytosis by treating HEK-FLAG–CTR1 cells with or without Dynasore for 24 h and then with or without 30 µM Cu (30 min). Using biotinylation, we determined that Dynasore inhibits essentially all Cu-induced CTR1 endocytosis, thus, Cu-activated endocytosis of CTR1 occurs through a dynamin- and clathrin-dependent pathway (Fig. S2).
We also employed siRNAs targeted for clathrin. Clathrin-targeted siRNA transfection decreases the expression of clathrin compared with control siRNA, as shown by a reduction in clathrin (Fig. 3C,D). To determine the effects of clathrin knockdown on CTR1 endocytosis, we employed cells transfected with control or clathrin siRNA, and then labeled CTR1 with CLIP-Surface reagent, treated with 30 µM Cu (30 min), fixed, and probed with anti-clathrin antibodies. Images of cells transfected with control siRNA show internalized CTR1, and zoomed inset shows partial co-localization with clathrin after Cu treatment (Fig. 3E, left panel). When clathrin levels were decreased by clathrin-targeted siRNA (Fig. 3E, right panel), most CTR1 proteins are restricted to the plasma membrane, suggesting that clathrin is required for Cu-induced CTR1 internalization.
CTR1 internalized after Cu treatment enters early, but not late, endosomes
During endocytosis, protein clusters formed in clathrin-coated pits are pinched off from the plasma membrane by dynamin. The resulting clathrin-coated vesicle is uncoated in the cytosol and fuses to early endosomes containing Rab5 (Mellman, 1996). Live cell imaging shows internalized CTR1 was present in Rab5-positive large early endosomes (Fig. 4A). Quantification of co-localization of CTR1 with Rab5 is displayed in Table 1. Early endosomes are the site for sorting cargo into one of two major pathways; lysosomal or recycling. Cargo trafficking through the lysosomal pathway is transported to the late endosome, and then to the lysosome. Rink et al. (2005) suggest that early endosomes can be converted into late endosomes by a process in which Rab7 replaces Rab5. Rab7 GTPases are known markers for late endosomes (Barbero et al., 2002). We double-labeled Cu-treated HEK-FLAG–CTR1 cells with anti-FLAG and anti-Rab7 antibodies to determine if co-localization occurs between CTR1 and Rab7. Co-localization of Rab7 with CTR1 was not seen in intracellular compartments after Cu treatment (Fig. 4B, left panel; quantification of co-localization in Table 1). Another GTPase, Rab9, is important for retrograde trafficking from endosomes to the trans-Golgi network (Barbero et al., 2002). Co-localization of Rab9 and CTR1 was present in few intracellular compartments but the majority of CTR1 was not co-localized with Rab9 (Fig. 4B, right panel; Table 1). This suggests that CTR1 does not enter lysosomal degradation pathways after short-term Cu-mediated endocytosis. Longer Cu (16 h) treatment times were not tested in this study, but have been previously reported to cause degradation (using 100 μM Cu) and some degradation after 3 h (Petris et al., 2003).
Fig. 4.
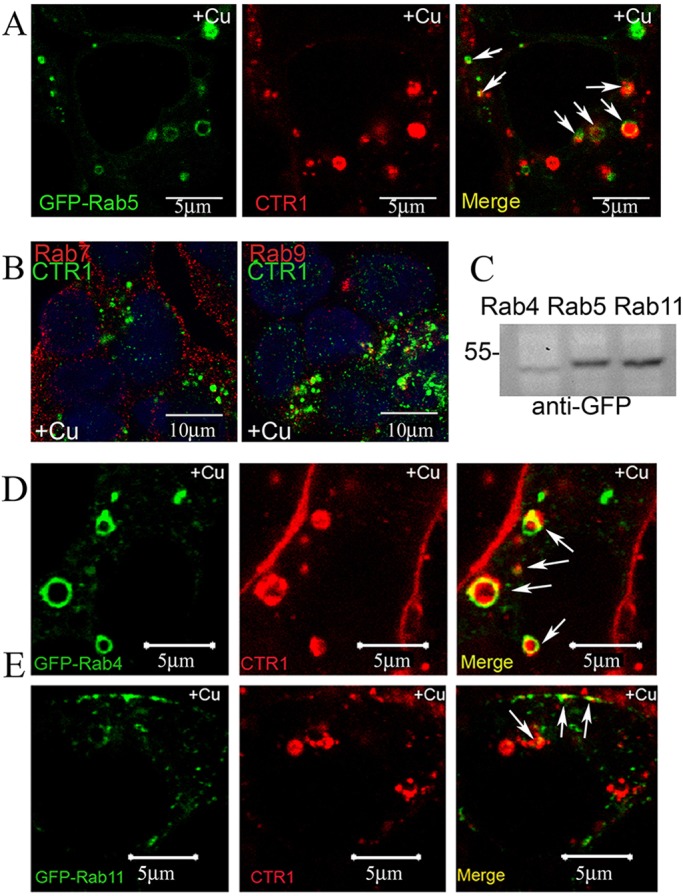
Internalized CTR1 co-localizes with early endosomes, but not with late endosomes. (A) HEK-CLIP–CTR1 cells expressing GFP–Rab5 were labeled with CLIP-Surface–547, treated with 30 μM Cu for 30 min, and subject to confocal microscopy. Intracellular compartments containing co-localized Rab5/CTR1 are visible in yellow (arrows). (B) HEK-FLAG–CTR1 cells treated with 30 μM Cu for 30 min were fixed and double-labeled with anti-FLAG (CTR1) and either anti-Rab7 or anti-Rab9 antibodies. (C) HEK-CLIP–CTR1 cells were transiently transfected with plasmids to express either GFP–Rab4, GFP–Rab5, or GFP–Rab11. Expression in total cell lysates was determined by western blot. (D,E) For live cell confocal fluorescence, transfected cells were labeled with CLIP-Surface and treated with 30 μM Cu for 30 min. CTR1 is internalized from the plasma membrane during Cu treatment and then co-localizes (arrows) with Rab4 (D) and Rab11 (E) GTPases. Scale bars: 5 μm in A,D,E; 10 μm in B.
Cu-induced CTR1 endocytosis is reversed via recycling pathways
Cu-induced CTR1 endocytosis is reversible upon Cu removal (Molloy and Kaplan, 2009). As early as 15 min after the removal of extracellular Cu, CTR1 reintegration into the surface membrane can be detected. The rapid re-insertion of CTR1 after Cu removal suggests that endocytosed CTR1 proteins are stored in intracellular endocytic compartments until Cu depletion occurs. This, in combination with a lack of evidence supporting late endosomal trafficking (Fig. 4B), suggests that CTR1 is transported from the early sorting endosome to a recycling pathway. There are two main routes for recycling proteins back to the cell surface after endocytosis; fast recycling is mediated by Rab4 GTPase and involves vesicle budding and tubule formation from the early endosome, whereas slower recycling is mediated by Rab11 GTPase. Proteins are trafficked from the early endosome to Rab11-positive compartments and then to the cell surface (Takahashi et al., 2012). Endocytosed TfR recycles to the cell surface via Rab11- (Mayle et al., 2012) or Rab4-positive compartments (Sönnichsen et al., 2000).
We utilized live cell fluorescence to visualize CTR1 localization with GTPase markers of recycling pathways. HEK-CLIP–CTR1 cells were transiently transfected with GFP–Rab4, GFP–Rab5, or GFP–Rab11, and Rab expression was assessed (Fig. 4C). Transfected cells were labeled with CLIP-Surface, and treated with 30 μM Cu (30 min). Live cell images show CTR1 was internalized into large sorting endosomes positive for Rab4 (Fig. 4D, white arrows). Rab11 GTPases are found (Fig. 4E) in subdomains of the early endosome and in small compartments consistent with recycling carriers (Sönnichsen et al., 2000). Several Rab11-positive compartments that contain CTR1 (Fig. 4E, white arrows) reside in punctate structures localized at or near the plasma membrane. Quantification of CTR1 co-localization with Rab GTPases is displayed in Table 1.
During Cu treatment, a large proportion of CTR1 aggregates in globular Rab4- and Rab5-positive endosomes, and some are visualized in small punctate compartments positive for Rab11 near the plasma membrane. Similar to the recycling pattern described earlier (Molloy and Kaplan, 2009), we find that removal of excess Cu causes internalized CTR1 to begin its return to the cell surface in as little as 5 min (Fig. 5A, middle panel). Upon Cu removal, CTR1 is co-localized less abundantly with globular endosomes, and a large portion of CTR1 is present with Rab11 near the plasma membrane (Fig. 5A), where Rab11 has been shown to regulate vesicle exocytosis (Takahashi et al., 2012). Time-resolved live cell fluorescence visualizes trafficking of CTR1/Rab11-containing compartments to the cell surface within 5 min of Cu removal from the extracellular growth media (Fig. 5B). Trafficking of CTR1/Rab11 recycling compartments to the plasma membrane occurred over ∼90 s from the time of vesicle arrival at the plasma membrane to apparent lateral dispersion of CTR1 cargo. Initial time-resolved images were collected at 5 min post-Cu-removal.
Fig. 5.
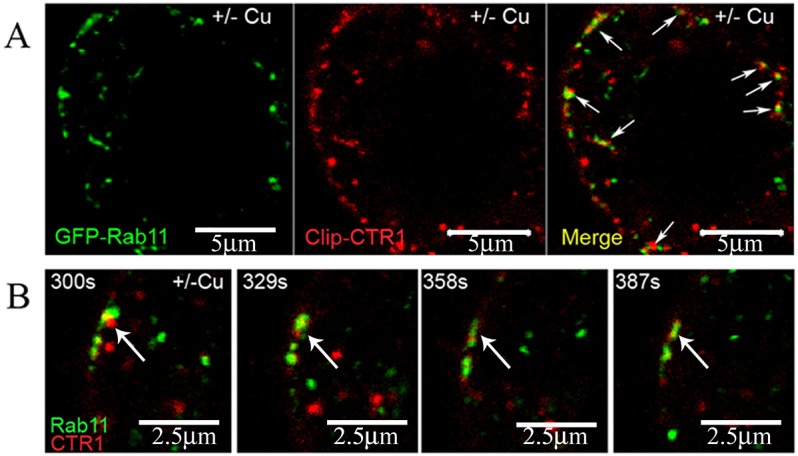
CTR1 is returned to the plasma membrane in Rab11-positive compartments. (A) Confocal images of cells treated with 30 μM Cu for 30 min and then Cu removed for 5 min show that endocytosed CTR1 is co-localized in small vesicles with Rab11(arrows) after Cu removal. (B) Compartments containing co-localized Rab11 and CTR1 (arrows) are dispersed near the surface membrane at 5 min post-Cu-removal. Scale bars: 5 μm in A, 2.5 μm in B.
To better visualize the return of CTR1 to the cell surface via Rab11 recycling pathways, we employed TIRF imaging. In Fig. 6A, control cells not treated with Cu showed CTR1 in a diffuse pattern near the cell surface with few small vesicular structures co-localizing with Rab11. When 30 μM Cu was added (30 min), CTR1 accumulated in larger punctate structures co-localized with Rab11 (Fig. 6B). At 15 min after Cu removal, CTR1 was retained in Rab11-positive endosomes near the cell surface (Fig. 6C) suggesting CTR1 recycles through a Rab11-dependent reinsertion pathway.
Fig. 6.
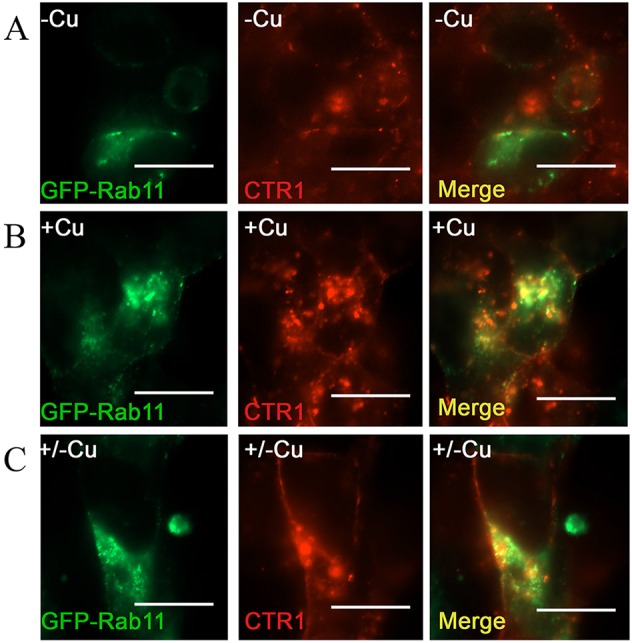
CTR1 is co-localized with Rab11 at the cell surface during exocytosis after Cu removal. TIRF fluorescence was used to visualize the surface localization of CTR1 and Rab11 after no excess Cu treatment (A), 30 μM Cu treatment for 30 min (B), and return of CTR1 to the cell surface in Rab11-positive recycling vesicles after excess Cu was removed for 15 min (C). Scale bars: 20 μm.
Rab4 is often found in early endosomes containing Rab5, a GTPase also located in sorting endosomes responsible for controlling endosomal fusion (Jopling et al., 2014; McCaffrey et al., 2001). Fusion is frequently accompanied by the formation of tubules that facilitate cargo recycling by developing into recycling vesicles that can enable either fast recycling directly back to the plasma membrane or can translocate to perinuclear regions in the cytoplasm and regulate the slower return to the plasma membrane via Rab11 (Mellman, 1996). The latter process has been shown to transport TfR back to the cell surface (Skjeldal et al., 2012). We observed a fusion event of homotypic endosomes within ∼1 min of initial contact (Fig. S3). Upon fusion, the formation of Rab4-positive tubules developed (Fig. S3, small arrow), although no CTR1 was detected in the tubule. Concurrently, a Rab4-negative subdomain of the fusing endosome contained CTR1, and vesicular budding was observed during homotypic endosome fusion (Fig. S3, arrowhead).
Based on immunofluorescence co-localization imaging, trafficking of endocytosed CTR1 occurs through Rab11 recycling pathways, but not through Rab4-positive tubules. To confirm this, we employed siRNAs for Rab4 or Rab11. Fig. S4A shows the extent of siRNA-mediated knockdown of Rab4 or Rab11 using quantitative PCR. Approximately 80% of Rab mRNA was knocked down compared with scramble control siRNA. We demonstrated that the return of internalized CTR1 to the cell surface after 1 h of Cu removal was inhibited by ∼35% when cells were transfected with Rab11 siRNAs, compared with control siRNA transfection (Fig. S4B). Silencing Rab4 did not have a significant effect on CTR1 return. This suggests that endocytosed CTR1 is recycled from intracellular compartments to the plasma membrane via the Rab11 recycling pathway.
DISCUSSION
The modulation of plasma membrane transporter activity is an essential aspect of cellular regulation. The varying requirements for substrates and metabolites during cell growth and differentiation and the fluctuations in substrate availability make this regulation essential for homeostasis. The internalization phenomena we describe here that acts acutely on CTR1, the human Cu transporter, is triggered rapidly by substrate in the extracellular media and is rapidly reversed by lowering of extracellular Cu, without the involvement of new transporter synthesis or degradation (Molloy and Kaplan, 2009). The internalization of CTR1 in response to excess Cu correlates with a decrease in Cu uptake into the cell.
Appropriate Cu homeostasis is vital in mammalian cells. Normal serum contains about 1–2 μM total Cu (McMillin et al., 2009) and internalization of CTR1 is caused by an addition as low as 2–5 μM Cu after exposure for 2 h, whereas the response to 20 μM Cu is complete in about 10 min (Molloy and Kaplan, 2009). Cu-concentration-dependence time courses show complex kinetics. The acute regulatory response described in the present work might be expected to play a role in the pulsatile appearance of the micronutrient in the diet. A balance is normally maintained between the uptake (predominantly by CTR1), the intracellular system responsible for its distribution to target proteins (through GSH and the metallochaperones), and the efflux transporters, Cu-activated ATPases ATP7A and ATP7B (Kaplan and Lutsenko, 2009). Internalization of CTR1 in the presence of excess extracellular Cu is reversible and CTR1 returns to the plasma membrane when extracellular Cu levels are reduced (Molloy and Kaplan, 2009). This regulatory endocytosis serves to protect the cell against excessive Cu accumulation by lowering the plasma membrane content of CTR1 and hence Cu entry. In the present work, we have utilized a range of imaging and siRNA approaches to characterize the trafficking process.
There are several forms of endocytosis, classified as either clathrin-mediated or clathrin-independent endocytosis pathways (CIE) (Doherty and McMahon, 2009). In CME, cargo is recruited by adaptor proteins, such as the AP2 complex, which also bind clathrin and enable clathrin pit formation at the membrane. Scission of clathrin pits is performed by dynamin, and clathrin vesicles are then transported to distinct endocytic compartments (Hill et al., 2001). TfR is a well-studied membrane protein that follows CME, a process enhanced by the addition of transferrin to extracellular media (Mayle et al., 2012). TfR is often used as a marker for CME and in recycling to the plasma membrane. CIE includes a multitude of pathways that do not require the involvement of clathrin (Doherty and McMahon, 2009).
CME requires several steps for vesicle formation and cargo internalization; initiation of pit formation, plasma membrane protein cargo selection, clathrin coat assembly, dynamin-mediated scission, and clathrin uncoating (Mousavi et al., 2004). TfR binds diferric transferrin at the cell surface and the ligand–receptor complex is then internalized into clathrin vesicles (Mayle et al., 2012). These vesicles fuse to sorting endosomes, where iron is released from the Tf–TfR complex. Tf–TfR then enters Rab4-positive (van Dam and Stoorvogel, 2002) or Rab11-positive (Takahashi et al., 2012) recycling compartments and recycles to the plasma membrane. We have shown that impairing the function of clathrin at the plasma membrane inhibits both TfR and CTR1 endocytosis (Figs 2,3). In addition to clathrin, dynamin is necessary for CME as it allows scission of the clathrin-coated pit from the plasma membrane (Hill et al., 2001). Expressing a non-functioning mutant DynK44A or employing Dynasore also prevents CTR1 endocytosis (see Fig. S2). Thus, endocytosis of CTR1, like TfR, is clathrin- and dynamin-1-dependent. CTR1 internalization occurs in several cell types (Fig. 1E,F), and based on our previous work, we hypothesize that the route of CTR1 endocytosis in those cell types is also clathrin-mediated.
Clathrin vesicle formation and selection of cargo occurs by AP2 recruitment to the plasma membrane (Collins et al., 2002). The μ2 and/or β2 subunits of AP2 recognize YXXØ or dileucine motifs on the cytoplasmic tail of transmembrane cargo proteins. CTR1 contains a potential μ2-binding motif, YNSM, in its cytoplasmic loop. Mutations in this motif of CTR1 showed decreased CTR1 internalization (Tsai et al., 2014). It seems likely that the YNSM motif in CTR1 might be the site for μ2 binding. The affinity of μ2 to bind YXXØ motifs is regulated by phosphorylation of the Thr156 residue of μ2. Binding sites of μ2 are blocked when the subunit is not phosphorylated preventing cargo protein recognition, but its phosphorylation results in a conformational change that allows binding to motifs on ligands (Jackson et al., 2010). The potential interactions between CTR1 and μ2, and the regulation of such interactions by AP2 phosphorylation are currently being investigated. In initial experiments we have attempted (unsuccessfully) to detect direct interactions between CTR1 and clathrin utilizing biotinylation. This might be because the interactions are transient or require greater sensitivity in their detection.
Once cargo proteins are endocytosed, they are transported through a series of vesicles and tubules, regulated by Rab GTPases, which are involved in nearly every step of endocytic trafficking and govern the destination of cargo-carrying compartments (Stenmark, 2009). Rab5 GTPases are involved in the initial stages of endocytosis and are localized primarily in the large early sorting endosome. Cargo located in endocytosed CIE or CME vesicles enters the early endosome, where it is sorted into compartments targeted to either the degradation or recycling pathway. The late endosome is involved in the degradation pathway and contains Rab7 and Rab9 GTPases (Rink et al., 2005). Cargo in Rab7- or Rab9-enriched domains is typically degraded in the lysosome. Cargo that enters the Rab4- or Rab11-enriched compartments is recycled to the plasma membrane (McCaffrey et al., 2001; Takahashi et al., 2012).
Upon internalization, CTR1 resides in specific Rab-GTPase-enriched compartments. Rab5 and Rab4 are enriched in early endosomes (Stenmark, 2009), and CTR1 co-localizes with Rab5 and Rab4 in these large intracellular compartments during Cu treatment (Fig. 4A,D). Rab4 is involved in rapid cargo recycling (McCaffrey et al., 2001), which occurs by cargo-filled tubule and vesicle formation from the early endosome and transport directly to the plasma membrane. CTR1 was found in large Rab4-positive endosomes (Fig. 4D), but Rab4-postive tubules extending from the early endosome did not contain CTR1 (Fig. S3). This, along with our data showing that Rab4 knockdown does not impair CTR1 recycling (Fig. S4), suggests that Rab4 is likely not involved in the direct trafficking and insertion of recycled CTR1 to the cell surface. Instead, a slowly recycling pool of CTR1 proteins is sorted from early endosomes to recycling endosomes, and is returned near the membrane by Rab11. Rab11 accompanies CTR1 in recycling endosomes located in perinuclear regions, and in compartments destined for the cell surface (Fig. 5). CTR1/Rab11-positive compartments nearing the plasma membrane can be observed by confocal and TIRF microscopy (Fig. 4E, Figs 5,6). When Rab11 function is impaired using siRNAs, the return of CTR1 to the plasma membrane is largely inhibited (Fig. S4B). Thus, we propose a model in which Rab11 mediates transport of the CTR1 proteins via a slower recycling pathway (Fig. 7).
Fig. 7.
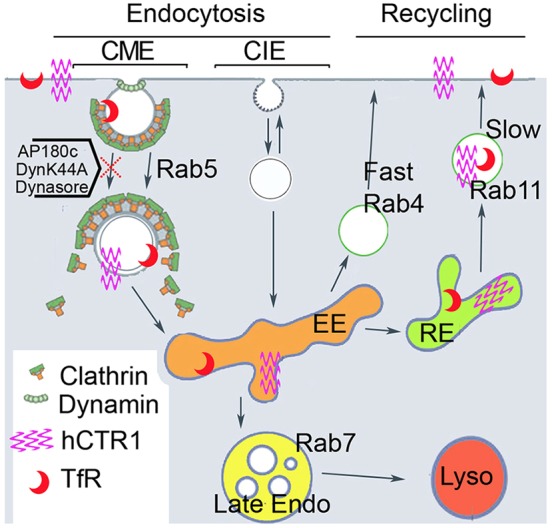
Proposed model of endocytosis and recycling pathways of CTR1. Under conditions of elevated extracellular Cu, CTR1 is endocytosed from the plasma membrane by a clathrin- and dynamin-mediated (CME) process. Internalized CTR1 is then localized in early endosomes and, when Cu is removed from the external media, it is transported back to the plasma membrane in a Rab11-dependent recycling pathway. CIE, clathrin-independent pathway; EE, early endosome; Late endo, late endosome; Lyso, lysosome; RE, recycling endosome.
The regulation of a metal ion transporter by means of alterations in its abundance in the plasma membrane has been reported previously in yeast. However, the endocytosis of the Mn transporter in response to the presence of Cd is a stress response, resulting in degradation of the transporter (Nikko et al., 2008). Similarly, endocytosis of the boron transporter in Arabidopsis thaliana, to avoid excess boron accumulation, occurs through transfer from the membranes to the vacuoles for degradation (Takano et al., 2005). The general amino acid permease, Gap1, of yeast also undergoes endocytosis in response to elevated levels of substrates via a ubiquitylation pathway (Ghaddar et al., 2014). It was concluded that the transporter engaged a conformation (caused by substrate binding) that would interact more efficiently with the endocytic machinery (Cain and Kaiser, 2011). In later work, the endocytosis was assumed to occur by means of a transient permease conformation that allowed recognition by arrestin-like adaptors. This was interpreted as requiring that this conformation be stable enough to increase its probability to interact with the adaptors. The model found support in the observation that a mutant of the permease that had a higher intrinsic Vmax than the wild-type transporter is largely resistant to substrate-induced endocytosis. This effect and its interpretation show a remarkable similarity to the substrate-induced endocytosis of CTR1, the human Cu transporter (Molloy and Kaplan, 2009). The distribution of CTR1 between the plasma membrane and intracellular sites can be modeled by a simple equilibrium where the rate constant governing the inward transition from the plasma membrane is stimulated by elevated extracellular Cu and the return steps from intracellular sites are Cu-independent. This simple model and the distributions at normal basal Cu (about 8% CTR1 is detected internally; Molloy and Kaplan, 2009) and at 100 μM Cu (about 80% is internal), suggests that there is an ∼40-fold increase in the rate of internalization at elevated Cu. The quantitative aspects of this model are under investigation.
We have previously reported that in a series of CTR1 mutants, truncations of the C-terminus or mutations in the terminal His-Cys-His triad have a higher Vmax for transport than the wild-type transporter. Furthermore, these mutants fail to undergo Cu-dependent endocytosis (Maryon et al., 2013a). This recapitulates the observations on the yeast amino acid permease system, but with CTR1 the substrate-dependent internalization is a dynamic reversible process involving endocytosis and recycling without transporter degradation or ubiquitylation. Our recent mutation studies on CTR1 have begun to define structural elements that are important for the regulatory endocytosis. It is clear from our own and earlier work (Guo et al., 2004) that mutations (M to L) in the important methionine triads (M150, 154) at the entry to the CTR1 pore, which render such CTR1 mutants unable to transport Cu, also are not internalized by excess extracellular Cu. Finally, the removal by substitution (188HCH to AAA) or truncation of the last seven or nine residues also produce CTR1 proteins that transport Cu more rapidly and fail to internalize in the face of excess Cu (Maryon et al., 2013a). This led us to propose that either structural elements at the cytoplasmic face of the protein must interact with the internalization machinery (identified in the present work) in a Cu-dependent fashion, or alternatively a specific Cu-dependent conformation of the protein that is a part of the normal transport cycle interacts with the internalization machinery and, in mutants with elevated transport rates, the crucial protein conformation has an insufficient dwell-time for productive interaction.
The current study provides a model for the endocytosis of CTR1 and regulation of CTR1 abundance at the cell surface (Fig. 7). CTR1 is internalized in clathrin-coated pits, and fission occurs by means of dynamin-1. Disruption of either of these processes results in the retention of CTR1 at the membrane. CTR1 proteins that are internalized during Cu treatment are trafficked to compartments in the cytosol that are mediated by Rab GTPases. CTR1 endocytosis is reversible and the removal of extracellular Cu enables CTR1 to relocalize at the plasma membrane via Rab11-mediated recycling pathways to once again mediate Cu entry. This work characterizes the pathways used in regulatory endocytosis of CTR1. The mechanism by which CTR1 internalization is triggered (by Cu acting at sites of lower affinity than transport sites) and the structural aspects of CTR1 essential to this process now need to be determined.
MATERIALS AND METHODS
Cell lines and constructs
HEK293 and MDCK Flp-In T-REx cells overexpressing CTR1 are isogenic and a tetracycline-regulated promoter allows CTR1 expression to be induced in the presence of tetracycline in growth media (Maryon et al., 2007). Growth in maximal tetracycline results in a maximally ∼20-fold increase in CTR1 expression per cell (Maryon et al., 2013a), and over the course of several days growth in basal media does not result in re-distribution of ATP7A or ATP7B or evidence of cytotoxicity. CTR1 sequences contain either a CLIP or FLAG epitope tag at their amino terminus, referred to as HEK-CLIP–CTR1, HEK-FLAG–CTR1, MDCK-FLAG–CTR1, or SKCO15-FLAG–CTR1. All cell lines were grown in Dulbecco's Minimal Essential Medium (DMEM) (Mediatech, Manassas, VA, USA) supplemented with 25 mM HEPES buffer (Mediatech) and 10% tetracycline-free fetal bovine serum (Atlanta Biologicals, Flowery Branch, GA, USA). HEK293- and MDCK-Flp-In-T-REx-derived cell lines were also supplemented with 400 μg/ml Hygromycin B (Mediatech) and 6 μg/ml Blasticidin (Research Products International Corp, Mt. Prospect, IL, USA). To induce expression of CTR1 in Flp-In T-REx cells, 1 μg/ml of tetracycline (Sigma, St. Louis, MO, USA) was added to the growth media for 48 h. Cell lines were maintained by washing twice with phosphate-buffered saline (PBS) and incubating with 0.05% trypsin-EDTA (GE Healthcare Life Sciences, Logan, UT, USA) until cell detachment, then cells were re-plated with fresh media at a 1:5–1:10 dilution. Cells were maintained in a humidified incubator with 5% CO2 at 37°C. pEGFP-C1-Rab4, pEGFP-C1-Rab5, and pEGFP-C1-Rab11 plasmids were a kind gift from Dr B. Yue (Park et al., 2010). pcDNA3.1/Dynamin1K44A-HA was a gift from Dr S. Schmid (Damke et al., 1994), and pFlag-CMV-2/AP180c construct was a kind gift from Dr J. Donaldson (National Institutes of Health, Bethesda, MD, USA). The pcDNA4/TO plasmid (Invitrogen, Grand Island, NY, USA) containing the FLAG-tagged CTR1 gene insert was used to transiently transfect SKCO15 cells, as recommended by the manufacturer's protocols. For transfection of plasmids, Lipofectamine 2000 Reagent (Invitrogen) was used according to the manufacturer's recommendations. Cells were incubated with transfection reagent mixture for 24 h and then media was replaced with fresh growth media for 48 h prior to selection, antibiotic addition for stable cell line creation, or experimental usage for transient transfections. Cell lines transfected with pEGFP plasmids were selected using G418 antibiotic (Thermo Fisher, Waltham, MA, USA). siRNAs against negative control, clathrin, Rab4A, or Rab11A (Qiagen, Valencia, CA, USA) were transiently transfected into HEK-FLAG–CTR1 cells using RNAiMAX Reagent (Invitrogen) according to the manufacturer's procedures, and then cells were incubated in fresh growth media for 72 h prior to assay.
Antibodies
Antibody dilutions used for immunoblotting were as follows: 1:10,000 of mouse anti-β-catenin (610153; BD Transduction Laboratories, San Jose, CA, USA), 1:5000 of mouse anti-GFP (11814460001; Roche, Indianapolis, IN, USA), 1:3000 rabbit anti-flag (A00170-40; Genscript, Piscaraway, NJ, USA). Peroxidase AffiniPure goat anti-mouse (115-035-146; Jackson Immunoresearch, West Grove, PA, USA) or goat anti-rabbit (111-035-144; Jackson Immunoresearch) secondary antibodies were diluted 1:5000. Antibodies used for immunofluorescence were diluted at 1:1000; rabbit anti-EEA1 (ab2900; Abcam, Cambridge, MA, USA), mouse anti-transferrin-receptor (TfR; ab84036; Abcam), mouse anti-HA (sc-7392; Santa Cruz, Dallas, TX, USA), mouse anti-FLAG (A00013; Genscript), mouse anti-Rab9 (MA3-067; Affinity Bioreagents, Golden, CO, USA), mouse anti-Rab7 (ab50533; Abcam), mouse anti-Rab11 (ab170134; Abcam), rabbit anti-Rab4 (PA3-912; Thermo Scientific, Rockford, IL, USA), and Surface-CLIP–547 or –488 (S9233S, S9232S; New England Biolabs, Ipswich, MA, USA).
Western blot analysis
Protein samples were subject to SDS-PAGE and western blot analysis (Clifford and Kaplan, 2008). To obtain total cell lysates, cells were washed twice with PBS, harvested by scraping, and whole cells pelleted at 1000 g for 5 min. Cell pellets were resuspended in cold lysis buffer (1% Triton X-100, 150 mM NaCl, 5 mM EDTA, 50 mM Tris pH 7.5). Suspensions were incubated end-over-end at 4°C for 1 h with protease inhibitors, and cell extracts were cleared by centrifugation at 10,000 g for 10 min. Protein contents of lysates were determined by the Bradford Protein Assay (BioRad, Hercules, CA, USA). Lysates were incubated at room temperature with 2× Laemmli sample buffer containing 10% β-mercaptoethanol (1 h). Samples were separated by 12% SDS-PAGE, transferred to polyvinylidene difluoride membranes and blocked with 5% milk in PBS (1 h). Antibodies were diluted in PBS containing 1% milk+0.1% Tween-20 and used for immunoprobing overnight (4°C). Following immunoprobing with antibodies, blots were washed 3 times with PBS+0.1% Tween-20. Chemiluminescent Western Blotting Substrate (Thermo Scientific) was used for peroxidase detection and signal intensity was quantified on BioRad Chemidoc XRS using BioRad Quantity One Version 4.6.2 software and corrected for protein loading using anti-β-catenin antibodies when appropriate.
Microscopy
Cells were trypsinized and re-plated on either glass coverslips for fixed cell immunofluorescence, or on glass-bottom plates for live cell imaging. CTR1 expression was induced with tetracycline (48 h). For fixed cell imaging, cells were fixed at room temperature (10 min) using 4% paraformaldehyde, then washed with PBS, permeabilized and blocked (1% bovine albumin, 0.1% Triton X-100 in PBS). Cells were double-labeled by probing with primary antibodies, consecutively, followed by washing with PBS, and fluorescent secondary antibody incubation. Goat anti-mouse and goat anti-rabbit fluorescent antibodies were obtained as FITC and Cy3 conjugates (Jackson Immunoresearch). Slides were covered with Vectashield mounting medium with DAPI (Vector Laboratories, Burlingame, CA, USA), and sealed with a glass coverslip. Images were detected and analyzed using Zeiss LSM 5 Pascal microscope (Carl Zeiss Microscopy, Thornwood, NY, USA) and quantification of co-localization was determined using ImageJ software (National Institutes of Health) with Coloc2 plug-ins. Fluorescent live cell imaging was performed using CLIP-Surface–488 or –547 (New England Biolabs) diluted 1:1000 in growth media to covalently label CTR1 at the plasma membrane, as described in the manufacturer's recommendations. Cells were washed three times in growth media and then treated with or without Cu and transferrin (Sigma) as indicated. Cell nuclei were stained with Hoescht (Thermo Scientific) for 5 min and cells were imaged with either standard confocal microscopy or total internal reflection fluorescence (TIRF). TIRF imaging was performed using the Zeiss Laser TIRF imaging system fitted with alpha Plan-Fluar 100×/1.45 objective and Pecon XL TIRF S incubation system, and images analyzed with Zeiss AxioVision software.
CLIP plate assay
HEK-CLIP–CTR1 cells were treated with tetracycline (1 μg/ml, 48 h) and used in the CLIP assay when 80% confluent. Cells are treated with Cu (0–100 μM) for the specified time at 37°C. CLIP-Surface–547 was added to the cells at a 1:500 dilution in growth media (30 min at room temperature), and nuclei stained with Hoescht 33342 (Thermo Fisher). Cells were washed three times with growth media and fluorescence was measured using a FluoStar Omega Plate Reader (BMG Labtech, Cary, NC, USA). Individual well scans of cells were performed using a 10×10 scan matrix. Relative fluorescence was determined using a standard curve of CLIP fluorescence from known cell numbers and individual wells were corrected for cell loss using Hoescht fluorescence. CLIP-Surface–547 is cell impermeable. Thus, CTR1 that remains on the cell surface after Cu treatment fluoresces, whereas internalized CTR1 proteins do not.
qPCR
To measure knockdown of endogenous Rab4 and Rab11 mRNA, total RNA from HEK-CLIP–CTR1 cells transfected with siRNA oligos against Rab4a, Rab11a, or control scramble were prepared. Cell pellets from 60-mm tissue culture dishes were used for RNA isolation using RNeasy/QIAshredder (Qiagen, Valencia, CA, USA) as instructed by the manufacturer. First-strand cDNA was made with SuperScript® III Reverse Transcriptase (Thermo Scientific), and qPCR was performed with human Rab4a, Rab11a, or GAPDH primers purchased from Qiagen and SYBR Green Mastermix (Qiagen), using a BioRad CFX96 Touch™. The Livak method for relative gene expression was used to quantify the expression of Rab4a and Rab11a in cells treated with siRNA or control [ΔΔCT=ΔCT (rab)-ΔCT(control)]. Reactions and calculations were performed according to the manufacturer's recommendations.
Acknowledgements
We would like to thank Dr N. Segev (UIC) and J. Donaldson (NIH) for helpful discussions and our anonymous referees for their constructive input.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
R.J.C. conducted most of the experiments and evaluated the data. E.B.M. designed the new endocytosis assay and advised on the manuscript. J.H.K. conceived and directed the project and evaluated data. R.J.C. and J.H.K. wrote and revised the manuscript.
Funding
This work was partially supported by the National Institutes of Health [grant 5P01 GM067166]. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.173351/-/DC1
References
- Barbero P., Bittova L. and Pfeffer S. R. (2002). Visualization of Rab9-mediated vesicle transport from endosomes to the trans-Golgi in living cells. J. Cell Biol. 156, 511-518. 10.1083/jcb.200109030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley G., Naik M. and Ling V. (1989). P-glycoprotein expression in multidrug-resistant human ovarian carcinoma cell lines. Cancer Res. 49, 2790-2796. [PubMed] [Google Scholar]
- Cain N. E. and Kaiser C. A. (2011). Transport activity-dependent intracellular sorting of the yeast general amino acid permease. Mol. Biol. Cell 22, 1919-1929. 10.1091/mbc.E10-10-0800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clifford R. J. and Kaplan J. H. (2008). {Beta}-subunit overexpression alters the stoicheometry of assembled na-K-ATPase subunits in MDCK cells. Am. J. Physiol. Renal Physiol. 295, F1314-F1323. 10.1152/ajprenal.90406.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins B. M., McCoy A. J., Kent H. M., Evans P. R. and Owen D. J. (2002). Molecular architecture and functional model of the endocytic AP2 complex. Cell 109, 523-535. 10.1016/S0092-8674(02)00735-3 [DOI] [PubMed] [Google Scholar]
- Damke H., Baba T., Warnock D. E. and Schmid S. L. (1994). Induction of mutant dynamin specifically blocks endocytic coated vesicle formation. J. Cell Biol. 127, 915-934. 10.1083/jcb.127.4.915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel K. G., Harbach R. H., Guida W. C. and Dou Q. P. (2004). Copper storage diseases: Menkes, Wilson's, and cancer. Front. Biosci. 9, 2652-2662. 10.2741/1424 [DOI] [PubMed] [Google Scholar]
- Doherty G. J. and McMahon H. T. (2009). Mechanisms of endocytosis. Annu. Rev. Biochem. 78, 857-902. 10.1146/annurev.biochem.78.081307.110540 [DOI] [PubMed] [Google Scholar]
- Ghaddar K., Merhi A., Saliba E., Krammer E. M., Prevost M. and Andre B. (2014). Substrate-induced ubiquitylation and endocytosis of yeast amino acid permeases. Mol. Cell. Biol. 34, 4447-4463. 10.1128/MCB.00699-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y., Smith K., Lee J., Thiele D. J. and Petris M. J. (2004). Identification of methionine-rich clusters that regulate copper-stimulated endocytosis of the human Ctr1 copper transporter. J. Biol. Chem. 279, 17428-17433. 10.1074/jbc.M401493200 [DOI] [PubMed] [Google Scholar]
- Gupte A. and Mumper R. J. (2009). Elevated copper and oxidative stress in cancer cells as a target for cancer treatment. Cancer Treat. Rev. 35, 32-46. 10.1016/j.ctrv.2008.07.004 [DOI] [PubMed] [Google Scholar]
- Harrison M. D., Jones C. E., Solioz M. and Dameron C. T. (2000). Intracellular copper routing: the role of copper chaperones. Trends Biochem. Sci. 25, 29-32. 10.1016/S0968-0004(99)01492-9 [DOI] [PubMed] [Google Scholar]
- Hill E., van der Kaay J., Downes C. P. and Smythe E. (2001). The role of dynamin and its binding partners in coated pit invagination and scission. J. Cell Biol. 152, 309-324. 10.1083/jcb.152.2.309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung I. H., Suzuki M., Yamaguchi Y., Yuan D. S., Klausner R. D. and Gitlin J. D. (1997). Biochemical characterization of the wilson disease protein and functional expression in the yeast saccharomyces cerevisiae. J. Biol. Chem. 272, 21461-21466. 10.1074/jbc.272.34.21461 [DOI] [PubMed] [Google Scholar]
- Iakovidis I., Delimaris I. and Piperakis S. M. (2011). Copper and its complexes in medicine: a biochemical approach. Mol. Biol. Int. 2011, 594529 10.4061/2011/594529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson L. P., Kelly B. T., McCoy A. J., Gaffry T., James L. C., Collins B. M., Höning S., Evans P. R. and Owen D. J. (2010). A large-scale conformational change couples membrane recruitment to cargo binding in the AP2 clathrin adaptor complex. Cell 141, 1220-1229. 10.1016/j.cell.2010.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jopling H. M., Odell A. F., Pellet-Many C., Latham A. M., Frankel P., Sivaprasadarao A., Walker J. H., Zachary I. C. and Ponnambalam S. (2014). Endosome-to-plasma membrane recycling of VEGFR2 receptor tyrosine kinase regulates endothelial function and blood vessel formation. Cells 3, 363-385. 10.3390/cells3020363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan J. H. and Lutsenko S. (2009). Copper transport in mammalian cells: special care for a metal with special needs. J. Biol. Chem. 284, 25461-25465. 10.1074/jbc.R109.031286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan J. H. and Maryon E. B. (2016). How mammalian cells acquire copper: an essential but potentially toxic metal. Biophys. J. 110, 7-13. 10.1016/j.bpj.2015.11.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klip A., Sun Y., Chiu T. T. and Foley K. P. (2014). Signal transduction meets vesicle traffic: the software and hardware of GLUT4 translocation. Am. J. Physiol. Cell Physiol. 306, C879-C886. 10.1152/ajpcell.00069.2014 [DOI] [PubMed] [Google Scholar]
- Macia E., Ehrlich M., Massol R., Boucrot E., Brunner C. and Kirchhausen T. (2006). Dynasore, a cell-permeable inhibitor of dynamin. Dev. Cell 10, 839-850. 10.1016/j.devcel.2006.04.002 [DOI] [PubMed] [Google Scholar]
- Maryon E. B., Molloy S. A. and Kaplan J. H. (2007). O-linked glycosylation at threonine 27 protects the copper transporter hCTR1 from proteolytic cleavage in mammalian cells. J. Biol. Chem. 282, 20376-20387. 10.1074/jbc.M701806200 [DOI] [PubMed] [Google Scholar]
- Maryon E. B., Molloy S. A., Ivy K., Yu H. and Kaplan J. H. (2013a). Rate and regulation of copper transport by human copper transporter 1 (hCTR1). J. Biol. Chem. 288, 18035-18046. 10.1074/jbc.M112.442426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maryon E. B., Molloy S. A. and Kaplan J. H. (2013b). Cellular glutathione plays a key role in copper uptake mediated by human copper transporter 1. Am. J. Physiol. Cell Physiol. 304, C768-C779. 10.1152/ajpcell.00417.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayle K. M., Le A. M. and Kamei D. T. (2012). The intracellular trafficking pathway of transferrin. Biochim. Biophys. Acta 1820, 264-281. 10.1016/j.bbagen.2011.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCaffrey M. W., Bielli A., Cantalupo G., Mora S., Roberti V., Santillo M., Drummond F. and Bucci C. (2001). Rab4 affects both recycling and degradative endosomal trafficking. FEBS Lett. 495, 21-30. 10.1016/S0014-5793(01)02359-6 [DOI] [PubMed] [Google Scholar]
- McMillin G. A., Travis J. J. and Hunt J. W. (2009). Direct measurement of free copper in serum or plasma ultrafiltrate. Am. J. Clin. Pathol. 131, 160-165. 10.1309/AJCP7Z9KBFINVGYF [DOI] [PubMed] [Google Scholar]
- Mellman I. (1996). Endocytosis and molecular sorting. Annu. Rev. Cell Dev. Biol. 12, 575-625. 10.1146/annurev.cellbio.12.1.575 [DOI] [PubMed] [Google Scholar]
- Molloy S. A. and Kaplan J. H. (2009). Copper-dependent recycling of hCTR1, the human high affinity copper transporter. J. Biol. Chem. 284, 29704-29713. 10.1074/jbc.M109.000166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mousavi S. A., Malerød L., Berg T. and Kjeken R. (2004). Clathrin-dependent endocytosis. Biochem. J. 377, 1-16. 10.1042/bj20031000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu F.-T., Callaghan J. M., Steele-Mortimer O., Stenmark H., Parton R. G., Campbell P. L., McCluskey J., Yeo J.-P., Tock E. P. C. and Toh B.-H. (1995). EEA1, an early endosome-associated protein. EEA1 is a conserved alpha-helical peripheral membrane protein flanked by cysteine “fingers” and contains a calmodulin-binding IQ motif. J. Biol. Chem. 270, 13503-13511. 10.1074/jbc.270.22.13503 [DOI] [PubMed] [Google Scholar]
- Nikko E., Sullivan J. A. and Pelham H. R. (2008). Arrestin-like proteins mediate ubiquitination and endocytosis of the yeast metal transporter Smf1. EMBO Rep. 9, 1216-1221. 10.1038/embor.2008.199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park B., Ying H., Shen X., Park J.-S., Qiu Y., Shyam R. and Yue B. Y. J. T. (2010). Impairment of protein trafficking upon overexpression and mutation of optineurin. PLoS ONE 5, e11547 10.1371/journal.pone.0011547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petris M. J., Mercer J. F., Culvenor J. G., Lockhart P., Gleeson P. A. and Camakaris J. (1996). Ligand-regulated transport of the menkes copper P-type ATPase efflux pump from the Golgi apparatus to the plasma membrane: a novel mechanism of regulated trafficking. EMBO J. 15, 6084-6095. [PMC free article] [PubMed] [Google Scholar]
- Petris M. J., Smith K., Lee J. and Thiele D. J. (2003). Copper-stimulated endocytosis and degradation of the human copper transporter, hCtr1. J. Biol. Chem. 278, 9639-9646. 10.1074/jbc.M209455200 [DOI] [PubMed] [Google Scholar]
- Rink J., Ghigo E., Kalaidzidis Y. and Zerial M. (2005). Rab conversion as a mechanism of progression from early to late endosomes. Cell 122, 735-749. 10.1016/j.cell.2005.06.043 [DOI] [PubMed] [Google Scholar]
- Safi R., Nelson E. R., Chitneni S. K., Franz K. J., George D. J., Zalutsky M. R. and McDonnell D. P. (2014). Copper signaling axis as a target for prostate cancer therapeutics. Cancer Res. 74, 5819-5831. 10.1158/0008-5472.CAN-13-3527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skjeldal F. M., Strunze S., Bergeland T., Walseng E., Gregers T. F. and Bakke O. (2012). The fusion of early endosomes induces molecular-motor-driven tubule formation and fission. J. Cell Sci. 125, 1910-1919. 10.1242/jcs.092569 [DOI] [PubMed] [Google Scholar]
- Sönnichsen B., De Renzis S., Nielsen E., Rietdorf J. and Zerial M. (2000). Distinct membrane domains on endosomes in the recycling pathway visualized by multicolor imaging of Rab4, Rab5, and Rab11. J. Cell Biol. 149, 901-914. 10.1083/jcb.149.4.901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenmark H. (2009). Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol. 10, 513-525. 10.1038/nrm2728 [DOI] [PubMed] [Google Scholar]
- Takahashi S., Kubo K., Waguri S., Yabashi A., Shin H.-W., Katoh Y. and Nakayama K. (2012). Rab11 regulates exocytosis of recycling vesicles at the plasma membrane. J. Cell Sci. 125, 4049-4057. 10.1242/jcs.102913 [DOI] [PubMed] [Google Scholar]
- Takano J., Miwa K., Yuan L., von Wiren N. and Fujiwara T. (2005). Endocytosis and degradation of BOR1, a boron transporter of arabidopsis thaliana, regulated by boron availability. Proc. Natl. Acad. Sci. USA 102, 12276-12281. 10.1073/pnas.0502060102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai C.-Y., Larson C. A., Safaei R. and Howell S. B. (2014). Molecular modulation of the copper and cisplatin transport function of CTR1 and its interaction with IRS-4. Biochem. Pharmacol. 90, 379-387. 10.1016/j.bcp.2014.06.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dam E. M. and Stoorvogel W. (2002). Dynamin-dependent transferrin receptor recycling by endosome-derived clathrin-coated vesicles. Mol. Biol. Cell 13, 169-182. 10.1091/mbc.01-07-0380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X., Greener T., Al-Hasani H., Cushman S. W., Eisenberg E. and Greene L. E. (2001). Expression of auxilin or AP180 inhibits endocytosis by mislocalizing clathrin: Evidence for formation of nascent pits containing AP1 or AP2 but not clathrin. J. Cell Sci. 114, 353-365. [DOI] [PubMed] [Google Scholar]