

Abstract
The intermolecular addition of the α-C–H bonds of unactivated dialkylamines to olefins in the presence of the chloro amido complex [TaCl3(NEt2)2]2 (2) is described. This process forms the branched insertion products in high yields (up to 96%) and selectivities, and represents a rare example of an intermolecular amine-olefin coupling reaction that does not require pre-activation of either substrate. The reaction is shown to encompass the addition of linear- and branched-methylamines, as well as secondary C–H bonds. The related chloroanilido complex [TaCl3(NMePh)2]2 (4) is also shown to catalyze the addition of Nalkyl-arylamines to olefins at temperatures as low as 90 °C. 1H NMR spectroscopy, identification of the catalyst structure, and deuterium-labeling experiments all suggest that reactions catalyzed by 2 and 4 occur by turnover-limiting generation of an η2- imine complex is turnover-limiting and occurs by elimination of amine. These labeling studies also imply that more favorable partitioning of the η2-imine complex toward reaction with alkene versus regeneration of the starting bis-amido complex accounts for the higher reactivity of the mixed halide amido catalyst versus a homoleptic amido complex.
Graphical Abstract
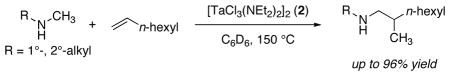
Intermolecular reactions between unstrained alkenes and amines are rare, and additions of unactivated alkylamines to alkenes are particularly unusual. In almost all reported cases occurring in substantial yields, these reactions proceed by addition of the N–H bond and are limited to processes in which the amine is first activated as an amide or sulfonamide,1 or to reactions involving strained olefins or ethylene.2 An exception is a single hydroamination of 1-pentene with propylamine reported by Marks.3
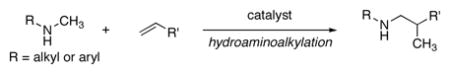
We recently described the addition of the α-C–H bonds of N-alkyl arylamines to alkenes (hydroaminoalkylation, eq 1).4 Complexes derived from the homoleptic amide Ta(NMe2)5 (1) catalyzed the hydroaminoalkylation in yields of up to 96%. However, the reactions required heating at 160–165 °C and an aryl substituent on nitrogen to modulate the properties of the amine. Until now, no analogous high-yielding additions of dialkylamines to alkenes have been reported.5 Here, we show that such additions occur in high yield in the presence of chlorotantalum amide catalysts. This process represents a rare intermolecular addition of an amine to an alkene that does not require activating substituents on either reaction partner.6 We also show that chlorotantalum anilide complexes catalyze the hydroaminoalkylation of alkenes with N-alkyl-arylamines under mild condition, and provide data that suggest the formation of an η2-imine complex is the turnover--limiting step of the catalytic cycle.
 |
(1) |
Our studies began with the syntheses of complexes in which a fraction of the five dimethylamido ligands of 1 were exchanged with chelating polyamines and polyols.7 Although complexes derived from a combination of 1 and biphenols (such as 2,2′-biphenol) exhibited some catalytic activity, complexes containing more electron-donating aliphatic alcohols and amines were inactive. These data led us to test complexes containing less electron-donating halide ligands, and these displayed the desired reactivity. The reaction of N-methyl-phenethylamine with 1-octene in the presence of 2 mol% of the chloroamido complex [TaCl3(NEt2)2]2 (2)8 generated the branched addition product in high yield for the first time (Table 1, entry 1). Under otherwise identical conditions but with 1 as precatalyst, this product was obtained in <5% yield.
Table 1.
Alkylation of Dialkylamines with 1-Octene.
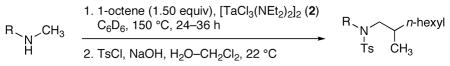
| |||||
|---|---|---|---|---|---|
| entry | alkylamine | mol% 2 | sulfonamide | yield aminea | yield sulfonamideb |
| 1 |
|
2 |
|
93%c | 85% |
| 2 |
|
2 |
|
91% | 81% |
| 3 |
|
2 |

|
96% | 91% |
| 4 |
|
3 |

|
94% | 89% |
| 5 |
|
4 |
|
47% | 41% |
| 6 |
|
3 |
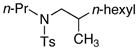
|
78% | 72% |
| 7 |
|
2 |

|
91% | 86%d |
Determined by integration of the 1H NMR spectrum of the crude reaction mixture containing added internal standard.
Isolated yield of sulfonamide after purification by flash-column chromatography.
In a separate experiment, the amine product was isolated in 77% yield by flash-column chromatography.
Single diastereomer (stereochemistry not assigned).
The data in Table 1 illustrate that complex 2 catalyzes the formation of the branched addition products from reaction of 1-octene with several types of dialkylamines in high yields and selectivities. Yields of the amine determined by 1H NMR spectroscopy are provided in the table. The product from the reaction of entry 1 was isolated by flash-column chromatography (77% yield), and the products of other reactions were isolated after conversion to the para-toluenesulfonamide derivative. The reactions of the unsymmetrical methylamines in entries 1–6 formed exclusively the product from addition of the methyl C–H bonds. Linear alkyl and branched alkyl methylamines added in high yield, although tert-alkyl methylamines, such as N-methyl-tertbutylamine, were unreactive. N-Methyl-phenethylamine and N-methyl-benzylamine both reacted selectively at the methyl group, despite the presence of benzylic hydrogens. The addition of secondary amine α-C–H bonds also occurred in high yield (entry 7).
A putative mechanism for the hydroaminoalkylation reaction (Scheme 1) consists of elimination of amine from a tantalum bis(amide) to form an η2-imine complex (3),9 insertion of the olefin into the tantalum–carbon bond of this intermediate, and protonolysis by the amine reagent to regenerate the starting bis(amide) and liberate the product. To determine the origins of the selectivity for alkylation at the methyl position in the amine (entries 1–6), a mixture of the catalyst precursor 2 and N-methylpropylamine-N-d were heated at 150 °C for 25 h. Analysis of the crude reaction mixture by 1H- and 2H-NMR spectroscopy revealed the presence of deuterium in the methyl group of the amine (0.56 D-atom), but none could be detected in the propyl substituent. These results imply that the observed regioselectivity derives from selective metalation at a methyl group in the presence of a methylene group.
Scheme 1.

Proposed mechanism for olefin hydroaminoalkylation.
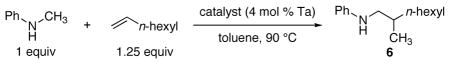
Hydroaminoalkylations of α-olefins with N-alkyl-arylamines occur when catalyzed by the closely related chlorotantalum anilide 4, prepared by the two-step sequence shown in equation 2.7 Use of the anilide 4 rather than the chlorotantalum amide 2 in this addition eliminates complications arising from exchange of the arylamine with the alkylamido groups of catalyst 2. Complex 4 catalyzed the addition of N-alkyl-arylamines to alkenes under milder conditions than the homoleptic amido complex 1. Additions catalyzed by 4 generated product 6 at temperatures as low as 90 °C (Table 2, entry 1), whereas the same reaction catalyzed by 1 required heating to 160–165 °C.4 This reaction catalyzed by 1 did not generate detectable amounts of product at 90 °C (entry 2).
Table 2.
Coupling of N-methylaniline and 1-octene at 90 °C by mixed chloroanilido complexes.
| ||||
|---|---|---|---|---|
| Entry | Catalyst Precursor | % Yield 6a
|
||
| 2.3 h | 5.1 h | 24 h | ||
| 1 | [Cl3Ta(NMePh)2]2 (4) | 34b | 53b | 72b |
| 2 | Ta(NMe2)5 (1) | 0c | 0c | 0c |
| 3 | [Cl4Ta(NMePh)]·OEt2 (5) | 2.1 | 3.8 | 14 |
Determined by GC using dodecane as an internal standard.
Yield corrected for additional substrate introduced by catalyst.
None was observed under conditions where >0.05% could be detected.
| (2) |
In contrast to reactions catalyzed by bis(anilide) 4, little product was obtained when the mono(anilide) 5 was used instead (Table 2, entry 3). This result suggests that the key η2-imine intermediate 3 does not form as readily by elimination of HCl·amine (eq 3) as by elimination of amine.
 |
(3) |
To gain further information on the relative rates of different steps of the catalytic cycle, we conducted NMR and deuterium-labeling studies of reactions mediated by 2 and 4. First, the catalytic alkylation of N-methylaniline with 1-octene was conducted with 15 mol% 4 (30 mol% Ta) and monitored by 1H NMR spectroscopy. Although several tantalum complexes derived from binding and exchange of the reactant and product amines were identified, no signals that we could assign to a metalacyclopentane complex were observed. Moreover, reactions conducted with 15 mol% 4 and terminated after 69% conversion by addition of excess DCl in D2O did not lead to detectable amounts of product containing deuterium at the methyl substituent, as determined by 1H NMR and GC-mass spectrometry. The analogous experiment conducted using 15 mol% 2 with diethylamine as substrate also did not lead to detectable amounts of deuterium incorporation at the methyl substituent of the product. Next, to probe the equilibrium between a bis(amide) complex and an η2-imine complex and free amine, we monitored the thermolysis of isolated complexes 2 and 4 by 1H NMR spectroscopy at 150 °C and 90 °C, respectively. Under these conditions with either complex, no free amine or η2-imine complex was observed. Taken together, these data imply that the proposed metallayclopentane intermediate is consumed faster than it is formed, that the equilibrium constant for elimination of amine from 2 and 4 is much less than unity, and that formation of the putative η2-imine intermediate is turnover-limiting under the reaction conditions.
We also conducted deuterium-labeling studies to provide information on the origin of the relative reactivities of Ta(NR2)5 and the chlorotantalum amido catalysts. We previously showed that the hydroaminoalkylation of 1-octene with N-(methyl-d3)-aniline using homoleptic amide 1 as precatalyst formed product 6-dn, in which only 45% of the deuterium was retained at the methylene position of the product (eq 4).4 The loss of deuterium implies that formation of the η2-imine intermediate during reactions catalyzed by 1 is reversible. In contrast, the reaction of N-(methyl-d3)-aniline with 1-octene catalyzed by 4 formed product 6-dn that retained almost all of the expected deuterium on the α-carbon (eq 4).10 This result suggests that reversion to a bis(amide) by protonlysis of the Ta–C bond of 3 by the N–H bond of the reagent does not occur to a significant extent in reactions catalyzed by 4 and that the partitioning of the η2-imine complex toward insertion of alkene vs reversion to a bis(anilide) is more favorable for complexes derived from 4 than from 1.
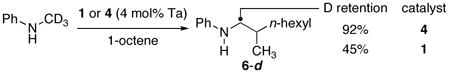 |
(4) |
In summary, we have shown that chlorotantalum amide and anilide complexes catalyze the hydroaminoalkylation of olefins with unprecedented efficiency. We attribute the enhanced activity to a more favorable partitioning of an η2-imine complex toward addition of the olefin, rather than reversion to the starting bis(amide), and we attribute the more favorable partitioning of this intermediate to the reduced steric hindrance and electron density on the chlorotantalum complex. Most generally, the alkylations of dialkylamines in this work constitute rare examples of intermolecular reactions between an alkylamine and an alkene without preactivation of either substrate.
Supplementary Material
Acknowledgments
Financial support from the National Institutes of Health (NRSA fellowship to S.B.H.) and the NSF (to J.F.H.) is gratefully acknowledged.
Footnotes
Supporting Information Available: Detailed experimental procedures and spectral data of all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Karshtedt D, Bell AT, Tilley TD. J Am Chem Soc. 2005;127:12640. doi: 10.1021/ja052836d. [DOI] [PubMed] [Google Scholar]; (b) Furstner A, Davies PW. Angew Chem, Int Ed Engl. 2007;46:3410. doi: 10.1002/anie.200604335. [DOI] [PubMed] [Google Scholar]; (c) Stephen A, Hashmi K. Chem Rev. 2007;107:3180. doi: 10.1021/cr000436x. [DOI] [PubMed] [Google Scholar]
- 2.Coulson DR. Tetrahedron Lett. 1971;12:429. [Google Scholar]
- 3.(a) Li Y, Marks TJ. Organometallics. 1996;15:3770. [Google Scholar]; (b) Ryu JS, Li GY, Marks TJ. J Am Chem Soc. 2003;125:12584. doi: 10.1021/ja035867m. [DOI] [PubMed] [Google Scholar]
- 4.Herzon SB, Hartwig JF. J Am Chem Soc. 2007;129:6690. doi: 10.1021/ja0718366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.For the first reported examples of hydroaminoalkylation, see: Clerici MG, Maspero F. Synthesis. 1980:305.Nugent WA, Ovenall DW, Holmes SJ. Organometallics. 1983;2:161.
- 6.For the addition of amine α-C–H bonds to olefins using activated amines substrates, see: Jun CH, Hwang DC, Na SJ. Chem Commun. 1998:1405.Chatani N, Asaumi T, Yorimitsu S, Ikeda T, Kakiuchi F, Murai S. J Am Chem Soc. 2001;123:10935. doi: 10.1021/ja011540e.
- 7.See the Supporting Information for details.
- 8.Chao YW, Polson S, Wigley DE. Polyhedron. 1990;9:2709. [Google Scholar]
- 9.(a) Takahashi Y, Onoyama N, Ishikawa Y, Motojima S, Sugiyama K. Chem Lett. 1978:525. [Google Scholar]; (b) Airoldi C, Bradley DC, Vuru G. Transition Met Chem. 1979;4:64. [Google Scholar]
- 10.The amount shown is corrected for the N-CH3 groups of the catalyst.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.