

Abstract
Nod-like receptor protein 3 (NLRP3) inflammasome has been implicated in alcoholic liver disease. Chronic alcohol consumption enhances gut permeability and causes microbial translocation. The present study explored the activation of the NLRP3 inflammasome by Escherichia coli RNA in hepatic stellate cells (HSCs), and the potential role of NLRP3 inflammasome in hepatic fibrosis. E. coli RNA transfection induced HSC-T6 cells to secrete and express mature interleukin-1 beta (IL-1β), which was abolished by NLRP3 siRNA pretreatment. In addition, E. coli RNA transfection enhanced caspase-1 expression, whereas reduced caspase-1 precursor (pro-caspase-1) expression. E. coli RNA-stimulated transforming growth factor beta 1 (TGF-β1) overproduction in HSC-T6 cells, which was blocked by recombinant IL-1 receptor antagonist (rIL-1Ra) or nuclear factor κB inhibitor BAY 11-7082. Furthermore, E. coli RNA-induced overexpression of pro-fibrogenic factors was suppressed by rIL-1Ra or TGF-β receptor inhibitor A83-01. These results demonstrate that E. coli RNA can stimulate NLRP3 inflammasome activation, which leads to excessive production of pro-fibrogenic factors, suggesting that NLRP3 inflammasome activation in HSCs may play a role in hepatic fibrosis.
KEY WORDS: Nod-like receptor protein 3, inflammasome, hepatic stellate cells, transforming growth factor β1
INTRODUCTION
Several members in nod-like receptor (NLR) family are related to inflammasomes [1]. By far, NLR protein 3 (NLRP3) inflammasome is the most extensively studied inflammasome, consisting of NLRP3, adaptor protein apoptosis associated speck-like protein (ASC) and serine protease caspase-1 precursor (procaspase-1) [2]. A variety of microbial pathogens can activate NLPR3 inflammasome, including fungi, bacteria and viruses, probably by their products, such as toxins, RNAs and DNAs [3,4]. On NLRP3 inflammasome activation, procaspase-1 precursor is turned into active caspase-1 by auto-cleavage. Subsequently, caspase-1 cleaves pro-IL-1β to form active IL-1β.
Chronic alcohol consumption is a common cause of liver injury. One of the most important underlying mechanisms is the gut-liver axis [5]. Studies have demonstrated that alcohol can lead to intestinal bacterial outgrowth and enteric dysbiosis [6]. More importantly, alcohol increases gut permeability, causing microbial translocation [7]. It has been revealed that both ethanol and its metabolic product acetaldehyde can disrupt epithelial tight junctions [7]. In addition, dysbiosis and inflammation also attribute to the disruption of intestinal epithelial integrity. As a result, bacterial products, such as endotoxin lipopolysaccharide, and bacterial RNAs and DNAs, translocate from intestinal lumen to liver.
Recently, NLRP3 inflammasome has been demonstrated to be implicated in alcoholic steatohepatitis [8]. It was revealed that increased IL-1β is required for the development of alcohol-induced liver disease, and due to the activation of NLRP3 inflammasome [8]. They identified Kupffer cells as sources of IL-1β [8]. In addition to Kupffer cells, NLRP3 inflammasome was also found to be activated in hepatic stellate cells (HSCs) [9], which may play a role in hepatic fibrosis. As bacterial RNA has been shown to activate NLRP3 inflammasome in macrophage, the present study explored the activation of the NLRP3 inflammasome by Escherichia coli RNA in HSCs and the role of NLRP3 inflammasome in alcoholic hepatic fibrosis.
MATERIALS AND METHODS
HSC cells and E. coli RNA transfection
Rat HSC-T6 cells (Procell, Wuhan, China) were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Gibco, USA), supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 µg/ml streptomycin, at 37°C with 5% CO2. E. coli (ATCC 25922) RNA (10 mg/ml) was used to transfect HSC-T6 cells via lipofectamine 2000 (Invitrogen) at a radio of 1 µl lipofectamine 2000 per 1 µg RNA. All experiments were done at least 3 times.
E. coli RNA extraction and RNase digestion
E. coli were grown in Luria-Bertani medium. Total E. coli RNA was extracted and purified using RNeasy Plus Mini kit (Qiagen, Shenzhen, China) according to the manufacturer’s protocol. In some experiments, RNase A (Sigma) was used to digest E. coli RNA at a concentration of 1 µg RNase A per 1 µg RNA for 60 minutes at 37°C.
Enzyme-linked immunosorbent assay (ELISA)
Secretion of IL-1β and transforming growth factor beta 1 (TGF-β1) was determined by examining the concentrations of IL-1β and TGF-β1 in cell supernatants via ELISA kits (R&D SYSTEMS, Shanghai, China), according to the manufacturer’s protocols.
Western blot
Equal amounts of total protein from each sample was subjected to 12% sulfate polyacrylamide gel electrophoresis, and transferred onto a nitrocellucose-ECL membrane. The membrane was probed with primary antibody for IL-1β (1:1000, Abcam), caspase-1 (1:1000, Santa Cruz) or NLRP3 (1:500, Santa Cruz), and then incubated with the peroxidase-conjugated secondary antibody (1:3000, Santa Cruz). Protein bands were detected by ECL (Pierce) and visualized by gel imaging system (Bio-Rad). β-actin was used as an internal control.
RNA interference
HSC-T6 cells were seeded into a 6-well plate at a density of 2 × 105, and transfected with NLRP3 siRNA and control siRNA (Santa Cruz, Texas, America), according to the manufacturer’s protocol. In brief, Solution A and Solution B were prepared, mixed and incubated for 30 minutes in room temperature. Solution A: 1 µg siRNA duplex was added into 100 µl siRNA transfection medium. Solution B: 8 µl transfection reagent was added into 100 µl siRNA transfection medium.
Real-time polymerase chain reaction (PCR)
RNA was isolated and purified from cells using RNeasy Plus Mini kit (Qiagen), according to the manufacturer’s protocol. 1 µg RNA was transcribed into cDNA using Superscript III reverse transcriptase (Invitrogen Life Technologies). Gene mRNA expression was determined by real-time PCR in a LightCycler system (Roche Diagnostics, Shanghai, China) with LightCycler DNA Master SYBR Green I Kit (Roche Diagnostics). Comparative CT method was used to quantify mRNA expression, normalizing CT values to β-actin which was used as an internal control. Primers for α-smooth muscle actin (α-SMA), collagen Type I α1 (COL1A1), tissue inhibitor of metalloproteinases 1 (TIMP-1), and β-actin were described by Son et al. [10].
Immunofluorescence
After incubation of 12 hours, HSC-T6 cells were fixed by 4% (w/v) formaldehyde solution for 15 minutes and washed with PBS at room temperature, and then lysed with 0.2% Triton X-100 (Biochemicals) for 5 minutes and blocked with 5% bovine serum albumin for 40 minutes. Sequentially, HSC-T6 cells were incubated with primary antibody for TGF-β1 (R&D SYSTEMS), and then with the rhodamine-conjugated secondary antibody (Santa Cruz). After washing with 4’,6-diamidino-2-phenylindole (DAPI) (Biochemicals), the cells were stained with DAPI, and observed by fluorescent confocal microscopy.
Statistical analysis
Statistical analysis was performed using SPSS version 13.0 (Chicago, IL, USA). Differences among groups were analyzed by one-way ANOVA and considered significant when p < 0.05.
RESULTS
E. coli RNA stimulates IL-1β secretion by HSCs
To explore whether E. coli RNA could activate NLRP3 inflammasome, we first examined IL-1β secretion by HSCs exposed to E. coli RNA for 12 hours. As shown by ELISA detection, E. coli RNA-induced HSC-T6 cells to secrete more IL-1β into cell supernatant (Figure 1A). However, the induction was abrogated after E. coli RNA was digested by RNase A (Figure 1A). We next examined the expression of IL-1β and IL-1β precursor (pro-IL-1β) in HSC-T6 cells by Western blot. It was found that IL-1β expression was elevated, whereas pro-IL-1β expression was reduced after E. coli RNA transfection (Figure 1B). Taken together, these results indicate that E. coli RNA promotes IL-1β maturation and subsequent secretion.
FIGURE 1.
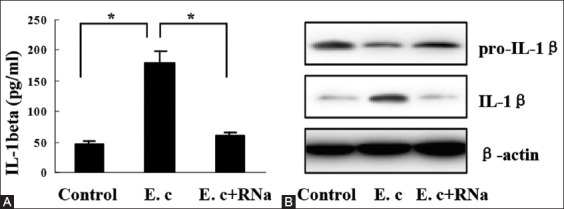
Interleukin-1 beta (IL-1β) induction by Escherichia coli RNA, (A) E. coli RNA stimulated IL-1β secretion by hepatic stellate cells (HSC)-T6 cells, which was abrogated upon RNase A digestion, (B) IL-1β expression was elevated, whereas pro-IL-1β expression was reduced in HSC-T6 cells after E. coli RNA transfection. *p < 0.01. Data are expressed as mean ± standard deviation. E. c: Escherichia coli RNA; RNa: RNase A.
IL-1β induction by E. coli RNA is NLRP3 inflammasome-dependent
To evaluate the role of the NLRP3 inflammasome in E. coli RNA-induced IL-1β secretion, we detected caspase-1 and NLRP3 expression in HSC-T6 cells. After transfection with E. coli RNA for 12 hours, HSC-T6 cells expressed more caspase-1, whereas less caspase-1 precursor (Pro-caspase-1), indicating E. coli RNA transfection activated caspase-1 (Figure 2A). NLRP3 expression was also found in HSC-T6 cells, but not affected by E. coli RNA (Figure 2B). When NLRP3 expression was silenced by NLRP3 siRNA, IL-1β overproduction by E. coli RNA were almost abolished (Figure 2B and C). These results suggest that NLRP3 inflammasome mediates IL-1β induction by E. coli RNA.
FIGURE 2.

Nod-like receptor protein 3 (NLRP3) inflammasome-dependent interleukin-1 beta (IL-1β) induction, (A) Escherichia coli RNA transfection increased caspase-1 expression, whereas decreased pro-caspase-1 expression in hepatic stellate cells (HSC)-T6 cells, (B) NLRP3 expression was observed in HSC-T6 cells, which was not affected by Escherichia coli RNA. NLRP3 expression was silenced after HSC-T6 cells were transfected with NLRP3 siRNA, (C) IL-1β induction by E. coli RNA was suppressed remarkably by NLRP3 siRNA transfection. *p < 0.01. Data are expressed as mean ± standard deviation. E.c: Escherichia coli RNA; RNa: RNase A; ConsiR: Control siRNA; NLsiR: Nod-like receptor protein 3 siRNA.
E. coli RNA induces TGF-β1 secretion by HSCs
To assess the involvement of NLRP3 inflammasome in hepatic fibrosis, we measured TGF-β1 levels in the supernatant of HSCs. It was found that E. coli RNA-transfected HSC-T6 cells secreted more TGF-β1, compared with untransfected control cells (Figure 3A). The induction was also abolished after E. coli RNA was digested by RNase A (Figure 3A). We further examined the expression of some pro-fibrogenic factors in HSC-T6 cells. As shown by real-time PCR detection, E. coli RNA transfection enhanced the expression of α-SMA, COL1A1, and TIMP-1 (Figure 3B-D).
FIGURE 3.
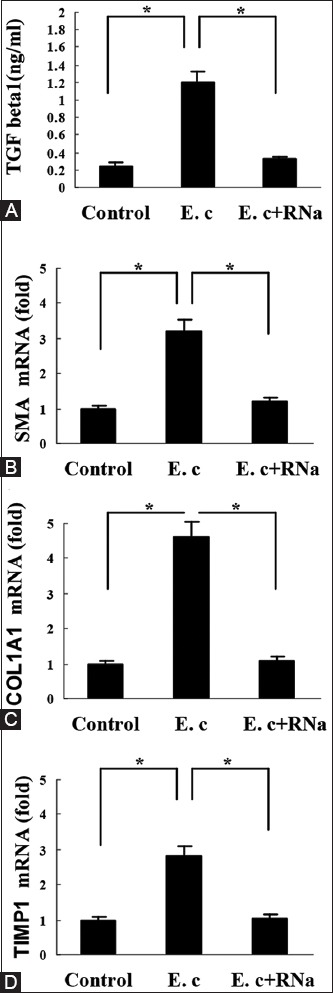
Induction of transforming growth factor beta 1 (TGF-β1) and other pro-fibrogenic factors by Escherichia coli RNA, (A) E. coli RNA stimulated TGF-β1 secretion by hepatic stellate cells (HSC)-T6 cells, which was abolished upon RNase A digestion, (B-D) E. coli RNA induced expression of for α-smooth muscle actin, collagen Type I α1, tissue inhibitor of metalloproteinases 1 in HSC-T6 cells, respectively. *p < 0.01. Data are expressed as mean ± standard deviation. E. c: Escherichia coli RNA; RNa: RNase A.
IL-1β-nuclear factor κB (NFκB) signaling mediates TGF-β1 induction by E. coli RNA
To determine the role of NLRP3 inflammasome in TGF-β1 induction, we pretreated HSC-T6 cells with recombinant IL-1 receptor antagonist (rIL-1Ra, 0.5 µg/ml, Sigma) for 8 hours. As shown by ELISA, TGF-β1 secretion induced by E. coli RNA was inhibited significantly (Figure 4A). We next analyzed the involvement of NFκB in TGF-β1 induction via pretreating HSC-T6 cells with NFκB inhibitor BAY 11-7082 (10 µmol/L, Sigma) for 6 hours. It was found that TGF-β1 induction was suppressed as well (Figure 4A). Consistent with TGF-β1 secretion, TGF-β1 content in HSC-T6 cells was elevated by E. coli RNA transfection, however, the upregulation was also blocked by rIL-1Ra or BAY 11-7082, as shown by Immunofluorescence (Figure 4B).
FIGURE 4.

Mediation of interleukin-1 beta (IL-1β)-nuclear factor κB (NFκB) signaling in transforming growth factor beta 1 (TGF-β1) induction by Escherichia coli RNA, (A) E. coli RNA-induced TGF-β1 secretion by hepatic stellate cells (HSC)-T6 cells was inhibited significantly by recombinant IL-1 receptor antagonist (rIL-1Ra) and NFκB inhibitor BAY 11-7082, respectively, (B) E. coli RNA-induced TGF-β1 production in HSC-T6 cells was suppressed by rIL-1Ra and BAY 11-7082, respectively. *p < 0.01. Data are expressed as mean ± standard deviation. E. c: Escherichia coli RNA.
IL-1β and TGF-β1 are involved in the induction of pro-fibrogenic factors by E. coli RNA
To confirm the involvement of NLRP3 inflammasome in the induction of pro-fibrogenic factors by E. coli RNA, we pretreated HSC-T6 cells with rIL-1Ra for 8 hours. Real-time PCR detection showed that E. coli RNA-induced expression of α-SMA, COL1A1, and TIMP-1 was inhibited remarkably (Figure 5A-C). Moreover, the increased expression of these factors was also suppressed by TGF-β receptor inhibitor A83-01 (1 µmol/L, Sigma), suggesting that the induction was TGF-β-dependent (Figure 5A-C).
FIGURE 5.
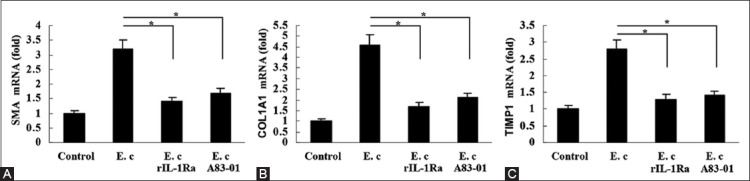
Involvement of interleukin-1 beta (IL-1β) and transforming growth factor beta 1 (TGF-β1) in the induction of pro-fibrogenic factors by Escherichia coli RNA, (A) E. coli RNA-induced α-smooth muscle actin expression in HSC-T6 cells was inhibited by recombinant IL-1 receptor antagonist (rIL-1Ra) and TGF-β receptor inhibitor A83-01, respectively, (B) E. coli RNA-induced collagen Type I α1 expression in hepatic stellate cells (HSC)-T6 cells was inhibited by rIL-1Ra and TGF-β receptor inhibitor A83-01, respectively, (C) E. coli RNA-induced tissue inhibitor of metalloproteinases 1 expression in HSC-T6 cells was inhibited by rIL-1Ra and TGF-β receptor inhibitor A83-01, respectively. *p < 0.01. Data are expressed as mean ± standard deviation. E. c: Escherichia coli RNA.
DISCUSSION
This study demonstrates E. coli RNA as an activator of NLRP3 inflammasome in HSCs, consistent with several studies on macrophages. Initially, infection of E. coli has been shown to induce caspase-1 activation in macrophage-dependent on adenosine triphosphate (ATP) [11]. However, in the absence of ATP stimulation, direct cytosolic delivery of bacterial products could also induce caspase-1 activation [12]. Kanneganti et al. observed that E. coli RNA-induced rapid activation of caspase-1 and secretion of IL-1β and IL-18 in macrophages [13]. Furthermore, RNA from E. coli was found to be able to activate the NLRP3 inflammasome not only in macrophages but also in unprimed dendritic cells [14]. It is largely unknown how bacterial RNA-induced NLRP3 inflammasome activation in these cells.
It has been demonstrated that NLRP3 inflammasome activation is involved in a series of liver diseases and injuries [15]. Recently, hepatitis C virus was reported to activate NLRP3 inflammasome in chronic hepatitis C patients [16]. Via different animal models, Petrasek et al. identified that alcoholic NLRP3 inflammasome activation and IL-1β overproduction were crucial in the pathogenesis of alcoholic liver diseases [8]. The mechanisms of NLRP3 inflammasome-mediated liver damage may be related with hepatocyte pyroptosis, liver inflammation, and fibrosis [17]. Importantly, NLRP3 inflammasome-associated damage can be attenuated by IL-1β antagonist, suggesting a potential role of IL-1Ra in the treatment of these liver diseases [8,17].
Increasing evidence indicates that NLRP3 inflammasome plays an important role in fibrosis [18]. Gasse et al. found that NLRP3 inflammasome was activated and essential in bleomycin-induced pulmonary fibrosis [19]. Moreover, asbestos and silica were identified to activate NLRP3 inflammasome [20], and silica-induced pulmonary fibrosis was dependent on NLRP3 inflammasome [21]. The NLRP3 inflammasome mediates fibrosis in systemic sclerosis [22] and myocardial infarction [23]. NLRP3 inflammasome was also involved in liver fibrosis. Study using NLRP3 knock-in mice has revealed that NLRP3 inflammasome activation induced liver fibrosis [17]. Recently, Wree et al. revealed a crucial role for the NLRP3 inflammasome in the development of fibrosis in non-alcoholic fatty liver disease [24].
The mechanisms underlying liver fibrosis induced by NLRP3 inflammasome activation are not complete understood. Given the crucial role of HSCs in liver fibrosis, it is reasonable to explore the expression and activation of the NLRP3 inflammasome in HSCs. Watanabe et al. identified that NLRP3 was expressed in primary mouse stellate cells and LX-2 HSCs, and activation of NLRP3 inflammasome with monosodium urate crystals upregulated TGF-β and collagen-1 expression [9]. Furthermore, animal studies showed that NLRP3-/- and ASC-/- mice were resistant to liver fibrosis induced by carbon tetrachloride or thioacetamide, with reduced expression of TGF-β and collagen-1 [9]. On the other side, NLRP3 knock in mice demonstrated HSC activation with collagen deposition in the liver [17].
CONCLUSION
E. coli RNA can induce the expression of TGF-β1 and some pro-fibrogenic factors dependent on NLRP3 inflammasome, suggesting that NLRP3 inflammasome activation in HSCs plays a role in liver fibrosis under chronic alcohol consumption.
DECLARATION OF INTERESTS
The authors state that there are no conflicts of interest.
REFERENCES
- [1].Franchi L, Eigenbrod T, Muñoz-Planillo R, Nuñez G. The inflammasome:a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol. 2009;10(3):241–7. doi: 10.1038/ni.1703. http://dx.doi.org/10.1038/ni.1703 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Cassel SL, Joly S, Sutterwala FS. The NLRP3 inflammasome:a sensor of immune danger signals. Semin Immunol. 2009;21(4):194–8. doi: 10.1016/j.smim.2009.05.002. http://dx.doi.org/10.1016/j.smim.2009.05.002 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Strowig T, Henao-Mejia J, Elinav E, Flavell R. Inflammasomes in health and disease. Nature. 2012;481(7381):278–86. doi: 10.1038/nature10759. http://dx.doi.org/10.1038/nature10759 . [DOI] [PubMed] [Google Scholar]
- [4].Franchi L, Muñoz-Planillo R, Núñez G. Sensing and reacting to microbes through the inflammasomes. Nat Immunol. 2012;13(4):325–32. doi: 10.1038/ni.2231. http://dx.doi.org/10.1038/ni.2231 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Szabo G, Bala S, Petrasek J, Gattu A. Gut-liver axis and sensing microbes. Dig Dis. 2010;28(6):737–44. doi: 10.1159/000324281. http://dx.doi.org/10.1159/000324281 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Schnabl B, Brenner DA. Interactions between the intestinal microbiome and liver diseases. Gastroenterology. 2014;146(6):1513–24. doi: 10.1053/j.gastro.2014.01.020. http://dx.doi.org/10.1053/j.gastro.2014.01.020 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Rao RK, Seth A, Sheth P. Recent advances in alcoholic liver disease I. Role of intestinal permeability and endotoxemia in alcoholic liver disease. Am J Physiol Gastrointest Liver Physiol. 2004;286(6):G881–4. doi: 10.1152/ajpgi.00006.2004. http://dx.doi.org/10.1152/ajpgi.00006.2004 . [DOI] [PubMed] [Google Scholar]
- [8].Petrasek J, Bala S, Csak T, Lippai D, Kodys K, Menashy V, et al. IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J Clin Invest. 2012;122(10):3476–89. doi: 10.1172/JCI60777. http://dx.doi.org/10.1172/JCI60777 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Watanabe A, Sohail MA, Gomes DA, Hashmi A, Nagata J, Sutterwala FS, et al. Inflammasome-mediated regulation of hepatic stellate cells. Am J Physiol Gastrointest Liver Physiol. 2009;296(6):G1248–57. doi: 10.1152/ajpgi.90223.2008. http://dx.doi.org/10.1371/journal.ppat.1003330 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Son G, Hines IN, Lindquist J, Schrum LW, Rippe RA. Inhibition of phosphatidylinositol 3-kinase signaling in hepatic stellate cells blocks the progression of hepatic fibrosis. Hepatology. 2009;50(5):1512–23. doi: 10.1002/hep.23186. http://dx.doi.org/10.1002/hep.23186 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Franchi L, Kanneganti TD, Dubyak GR, Núñez G. Differential requirement of P2X7 receptor and intracellular K+for caspase-1 activation induced by intracellular and extracellular bacteria. J Biol Chem. 2007;282(26):18810–8. doi: 10.1074/jbc.M610762200. http://dx.doi.org/10.1074/jbc.M610762200 . [DOI] [PubMed] [Google Scholar]
- [12].Kanneganti TD, Lamkanfi M, Kim YG, Chen G, Park JH, Franchi L, et al. Pannexin-1-mediated recognition of bacterial molecules activates the cryopyrin inflammasome independent of Toll-like receptor signaling. Immunity. 2007;26(4):433–43. doi: 10.1016/j.immuni.2007.03.008. http://dx.doi.org/10.1016/j.immuni.2007.03.008 . [DOI] [PubMed] [Google Scholar]
- [13].Kanneganti TD, Ozören N, Body-Malapel M, Amer A, Park JH, Franchi L, et al. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature. 2006;440(7081):233–6. doi: 10.1038/nature04517. http://dx.doi.org/10.1038/nature04517 . [DOI] [PubMed] [Google Scholar]
- [14].Eigenbrod T, Franchi L, Muñoz-Planillo R, Kirschning CJ, Freudenberg MA, Núñez G, et al. Bacterial RNA mediates activation of caspase-1 and IL-1ßrelease independently of TLRs 3, 7, 9 and TRIF but is dependent on UNC93B. J Immunol. 2012;189(1):328–36. doi: 10.4049/jimmunol.1103258. http://dx.doi.org/10.4049/jimmunol.1103258 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Szabo G, Csak T. Inflammasomes in liver diseases. J Hepatol. 2012;57(3):642–54. doi: 10.1016/j.jhep.2012.03.035. http://dx.doi.org/10.1016/j.jhep.2012.03.035 . [DOI] [PubMed] [Google Scholar]
- [16].Negash AA, Ramos HJ, Crochet N, Lau DT, Doehle B, Papic N, et al. IL-1ßproduction through the NLRP3 inflammasome by hepatic macrophages links hepatitis C virus infection with liver inflammation and disease. PLoS Pathog. 2013;9(4):e1003330. doi: 10.1371/journal.ppat.1003330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wree A, Eguchi A, McGeough MD, Pena CA, Johnson CD, Canbay A, et al. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology. 2014;59(3):898–910. doi: 10.1002/hep.26592. http://dx.doi.org/10.1002/hep.26592 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Artlett CM. Inflammasomes in wound healing and fibrosis. J Pathol. 2013;229(2):157–67. doi: 10.1002/path.4116. http://dx.doi.org/10.1002/path.4116 . [DOI] [PubMed] [Google Scholar]
- [19].Gasse P, Mary C, Guenon I, Noulin N, Charron S, Schnyder-Candrian S, et al. IL-1R1/MyD88 signaling and the inflammasome are essential in pulmonary inflammation and fibrosis in mice. J Clin Invest. 2007;117(12):3786–99. doi: 10.1172/JCI32285. http://dx.doi.org/10.1172/jci32285 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Dostert C, Pétrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320(5876):674–7. doi: 10.1126/science.1156995. http://dx.doi.org/10.1126/science.1156995 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Cassel SL, Eisenbarth SC, Iyer SS, Sadler JJ, Colegio OR, Tephly LA, et al. The Nalp3 inflammasome is essential for the development of silicosis. Proc Natl Acad Sci U S A. 2008;105(26):9035–40. doi: 10.1073/pnas.0803933105. http://dx.doi.org/10.1073/pnas.0803933105 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Artlett CM, Sassi-Gaha S, Rieger JL, Boesteanu AC, Feghali-Bostwick CA, Katsikis PD. The inflammasome activating caspase 1 mediates fibrosis and myofibroblast differentiation in systemic sclerosis. Arthritis Rheum. 2011;63(11):3563–74. doi: 10.1002/art.30568. http://dx.doi.org/10.1002/art.30568 . [DOI] [PubMed] [Google Scholar]
- [23].Shinde AV, Frangogiannis NG. Fibroblasts in myocardial infarction:a role in inflammation and repair. J Mol Cell Cardiol. 2014;70:74–82. doi: 10.1016/j.yjmcc.2013.11.015. http://dx.doi.org/10.1016/j.yjmcc.2013.11.015 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wree A, McGeough MD, Peña CA, Schlattjan M, Li H, Inzaugarat ME, et al. NLRP3 inflammasome activation is required for fibrosis development in NAFLD. J Mol Med (Berl) 2014;92(10):1069–82. doi: 10.1007/s00109-014-1170-1. http://dx.doi.org/10.1007/s00109-014-1170-1 . [DOI] [PMC free article] [PubMed] [Google Scholar]