

ABSTRACT
Transcriptional activation of telomerase reverse transcriptase (TERT) gene is a rate-limiting determinant in the reactivation of telomerase expression in cancers. TERT promoter mutations represent one of the fundamental mechanisms of TERT reactivation in cancer development. We review recent studies that elucidate the molecular mechanisms underscoring activation of mutant TERT promoters.
Reactivation of telomerase occurs in the majority (more than 90%) of human malignancies and is an essential pre-requisite for the immortalization of most transformed cells.1,2 This is primarily due to the key biological functions of telomerase ribonucleoprotein complex in telomeric DNA extension and maintenance of telomere homeostasis, which confer cancer cells with a limitless proliferative capacity.3,4 While telomerase expression is high in embryonic and adult stem cells, telomerase activity is absent in differentiated somatic cells due to transcriptional silencing of the telomerase reverse transcriptase (TERT) gene, which encodes its catalytic subunit that synthesizes TTAGGG nucleotide repeats at chromosome ends.5 Reconstitution of telomerase activity occurs through transcriptional de-repression of TERT and represents one of the hallmark events in cancer progression that enables tumor cells to divide and proliferate indefinitely without encountering the adverse effects of progressive telomere attrition.
Multiple transcription factors, including Myc, β-catenin and NF-κB have been documented to control human TERT promoter activity and several studies have suggested that the chromatin environment plays a critical role in the regulation of TERT transcription.6-10 However, the molecular events leading to the TERT promoter being constitutively switched on during cancer development remains an intriguing field to be resolved. Recently, two highly recurrent point mutations in the TERT promoter were identified in a multitude of cancer types including melanoma (where these mutations were initially described), glioblastoma multiforme (GBM), urothelial cancer, thyroid carcinoma and hepatocellular carcinoma (HCC).11-15 Occurring at nucleotide residues -124 and -146 upstream from the ATG start site (commonly referred to as C228T and C250T, respectively) in a mutually exclusive manner, the two somatic mutations resulted in the de novo generation of consensus binding motifs for E-twenty-six (ETS) transcription factors.11,12 The high prevalence of TERT promoter mutations in various advanced cancers and their direct correlation with augmented TERT transcription, increased telomere length and telomerase activity in primary tumors as well as poor patient outcome suggest that they represent a fundamental mechanism of telomerase reactivation in human cancers.14,16,1717 Indeed, recent cumulative evidence from a number of laboratories supports the notion that TERT promoter mutations are gain of function, driver events in cancer progression.
It is known that the new CCGGAA/T binding sites created by both C228T and C250T hotspot mutations can be recognized by the ETS family of 28 different transcription factors.18 While most ETS factors are ubiquitously expressed in untransformed human tissues, an ETS gene that is usually present in low abundance in normal tissues may be selectively overexpressed in certain tumors or cancer cell lines.18 Thus Bell and colleagues analyzed the expression pattern of ETS genes in GBM tumors and performed a siRNA screen in GBM cell lines harboring mutant TERT promoters to identify which ETS factor was specifically recruited to the mutation site.19 Their screen revealed at least three ETS factors (ETS1, GABPA and ETV3) whose knock-down prominently reduced TERT expression in mutant GBM cells.19 However, in their analysis, Bell et al. found that GABPA knockdown resulted in a more rapid and severe reduction of TERT expression than ETS1 down-regulation.19 In addition, ETV3 is known to function as a transcriptional repressor and was thus ruled out as a potential activator of mutant TERT promoter.19 Subsequent investigation of published chromatin immunoprecipitation-sequencing (ChIP-seq) data from various ENCODE cancer cell lines using bioinformatics prediction tools and experimental validation via biochemical assays led the group to suggest that GABP (GA-binding protein) is specifically recruited to mutant TERT promoters during telomerase reactivation.19
GABP is the only ETS family transcription factor that exists as an obligate multimeric complex comprising of GABPα, which contains a DNA-binding ETS domain, and its dimerization partner, GABPβ, which possesses transactivation properties.20,21 Previous studies have suggested that oncogenic and non-oncogenic ETS factors occupy distinct transcriptional targets genome-wide.18,22 The wild-type TERT promoter, for example, contains a number of native ETS binding sites with which certain ETS transcription factors are associated for telomerase activation.17 However, in the event of TERT promoter mutations, the mechanisms by which GABP is selectively recruited to mutant promoter to reactivate TERT have not been characterized. It is not clear, for instance, which mechanisms determine the specificity of GABP among the remaining ETS factors that are upregulated during oncogenesis for the mutant TERT promoter. Moreover, which chromatin factors or environment regulate the recruitment and stabilization of GABP at the mutant allele remains to be defined. Although Bell et al. have suggested that adjacent pre-existing ETS motifs cooperatively facilitate GABP heterotetramer recruitment on TERT promoter mutations to activate TERT transcription, the group has relied on site-directed mutagenesis assays of artificial mutant TERT promoter reporter constructs, which are unlikely to adequately reflect the in vivo chromatin context.19
The human TERT promoter harbors several CpG nucleotide clusters and its hypermethylation has been associated with transcriptional silencing of TERT gene.10 Epigenetic deregulation has been long considered to be one of the mechanisms of TERT reactivation in cancers.10,17 However, the epigenetic mechanisms leading to activation of highly prevalent TERT promoter mutations in cancers have not been well established. In a recent study, Stern and co-authors examined the chromatin status of wild-type and C228T TERT promoters in HCC cell lines and found that in contrast to wild-type TERT promoter, C228T-mutant promoters harbored an epigenetic signature of active chromatin.23 Sanger sequencing of PCR products from ChIP assays revealed that histone marks associated with active gene transcription were preferentially located on the mutant relative to wild-type allele.23 In contrast, the wild-type allele was enriched for a repressive histone mark that was absent on mutant allele.23 Similar to earlier observations by Bell et al., the researchers also detected the specific binding of GABP to the C228T-mutant allele in HCC cell lines.23
Clearly, several central questions are prompted from the two independent studies and need to be addressed. It is currently undefined which are the epigenetic factors that mediate the epigenetic switch on the mutant allele and whether additional chromatin regulators are required to recruit GABP to mutant TERT promoter. While both TERT promoter mutations create identical ETS binding motifs de novo, it remains to be established if both sites are exclusively bound by GABP or whether they are similarly regulated, at the molecular level, during TERT reactivation.
Recent work from our lab suggests that the C228T and C250T mutant TERT promoters are functionally distinct and operate in a context-dependent manner.24,25 In particular, we found that non-canonical NF-κB subunit, p52, dimerizes with ETS1/2 factors selectively on the C250T-mutant TERT promoter to activate TERT transcription.24 De novo motif analysis of publicly available ChIP-seq data revealed a novel p52 half-site binding sequence that specifically maps to the C250T TERT promoter.24 The association of p52 to this half-site was subsequently validated using electrophoretic mobility shift assays and ChIP experiments in GBM cell lines, which predominantly carry TERT promoter mutations. Non-canonical NF-κB signaling via TWEAK/Fn14 receptor activation induced TERT expression and telomerase activity selectively in GBM cells harboring C250T but not C228T TERT mutations and CRISPR/Cas9-targeted reversal of C250T mutation abolished p52-mediated TERT reactivation.24 Furthermore, constitutive activation of NF-κB-inducing kinase (NIK), which generates the active p52 subunit and is concordant with increased telomerase expression in C250T-mutant GBM tumors, promoted in vitro as well as in vivo GBM tumor cell growth.24
While both the C250T and C228T mutant TERT promoters generate a new ETS binding site and several native ETS consensus motifs are located on the WT TERT promoter region, our data suggests that p52 binds cooperatively with ETS1/2 preferentially at the C250T TERT promoter to mediate TERT reactivation. This is due to the critical juxtaposition of an adjacent p52 half-site to the ETS binding motif which is created by the C250T promoter mutation. This combination and favorable proximity of binding sites were essential for p52 and ETS dimerization on the C250T TERT promoter, thereby leading to efficient activation of ETS-dependent TERT transcription.24 In contrast, such co-occurrence of both binding motifs was absent in the wild-type and C228T TERT promoter sequences, thereby highlighting the distinct regulation of TERT transcription at C250T and C228T TERT mutations as well as wild-type TERT promoter.
Although our findings appear to contradict the observations by Bell et al. and Stern et al., the apparent controversy in the candidate transcription factor(s) or ETS factor responsible for mutant TERT promoter activation may be attributed to differences in cancer cell lines (and their tissue-of-origin) used in each study. Bell and colleagues selected two C228T-mutant GBM cell lines to perform their siRNA screen while the investigation by Stern et al. was conducted primarily in C228T-mutant HCC cell lines. Moreover, Stern and co-authors only checked for the recruitment of two ETS factors - GABPA and ELF1 to mutant TERT allele as a follow-up to Bell et al.’s findings but did not address the role of other ETS family members in their analysis. In contrast, our study focused on the specific regulation of C250T TERT promoter by p52 through cooperation with ETS1/2 at the C250T mutation site in GBM cell lines. Hence, culture adaptation of the various established cell lines as well as the aberrant expression of certain ETS or NF-κB transcription factors in specific cancer cell lines and tissue types may be contributing factors in the conflicting data presented. Till date, there are at least three transcription factors which are important for TERT reactivation at the mutant TERT promoter – GABP, ETS1 and p52. It would be pertinent for future investigators to examine the regulation of these factors and/or signaling pathways in primary tumors in order to resolve the current controversy.
Non-canonical NF-κB signaling is well documented to be activated in gliomas, plausibly through overexpression of Fn14 receptor, and its de-regulation has been implicated to serve as an oncogenic driver of GBM progression.26,27 Thus, in GBM tumors which display a high prevalence of TERT promoter mutations,13 non-canonical NF-κB pathway may be one of the key mechanisms that drive telomerase reactivation. Given the gradual but persistent nature of non-canonical NF-κB signaling, in contrast to canonical NF-κB activation, it is likely that tumor cells utilize the former mechanism to keep the TERT promoter constitutively active for sustained telomerase expression. While the above studies have sought to address the molecular mechanisms underscoring telomerase reactivation in cancers displaying recurrent TERT promoter mutations, major questions remain as to why these somatic mutations occur in a selective spectrum of human solid tumors despite their high prevalence in cancers. For instance, TERT promoter mutations are rarely detected in tumors originating from highly proliferative tissues such as colorectal cancer and leukemia but are frequently found in tumors derived from tissues with low self-renewal rates such as GBM and urothelial cancer.13 The predominance of TERT promoter mutations in these tumor types suggests that a selective growth advantage is conferred by overexpression of the TERT gene during cancer development.
In a recent investigation, Chiba and co-authors provided insight to the significance of TERT promoter mutations in cancer. Using the CRISPR/Cas9 genome editing tool to introduce the mutant TERT promoter in human embryonic stem (ES) cells, the group demonstrated that mutant cells did not undergo transcriptional repression of TERT following somatic cell differentiation. In contrast to differentiated cells carrying wild-type TERT promoter, cells with TERT promoter mutations retained telomerase activity and longer telomeres despite undergoing several cycles of replication post-differentiation.28 A similar phenotype was maintained when these cells were transplanted in immune-deficient mice to induce teratoma tumor formation.28 These observations demonstrate the significant role of TERT promoter mutations in the immortalization of cancer cells, through the circumvention of telomere shortening, which leads to replicative senescence. The causal role of TERT promoter mutations in the maintenance of telomerase activity in cancer cells is further corroborated by studies from various groups including ours. Reversal of either C228T or C250T mutations to wild-type TERT promoter via genome editing tools resulted in the substantial reduction of endogenous telomerase activity, telomere length and proliferation of cancer cells.24,29,30 Hence, these studies collectively provide evidence for the instrumental role of TERT promoter mutations in sustaining telomerase expression that drives cancer cell immortalization and progression.
The various studies illustrated in this review have raised important implications for TERT promoter mutations as potential biomarkers of cancer prognosis. From the therapeutic perspective, elucidating the molecular mechanisms underscoring telomerase reactivation during TERT promoter mutations will allow future clinicians to target tumor cell survival selectively, without impairing normal stem cell functions since these mutations are restricted to human cancer cells. Although several recent studies have shed light on some of the major mechanisms controlling transcriptional activation at mutant TERT promoters, many key questions remain to be addressed. The human TERT promoter is an essential regulatory element which modulates telomerase expression and is known to contain several G-rich repeats that can potentially form G-quadruplexes.31 Both the C228T and C250T mutations are located within such a repeat sequence.19 It is currently unknown how these mutations affect formation of the predicted G-quadruplex and whether this secondary DNA structure plays a role in the recruitment of GABP or other relevant ETS factors during differential activating signals to mediate TERT transcription. From the current literature, it is also unclear whether the mechanisms of TERT reactivation are governed by histone modifying proteins or chromatin regulators that modulate the epigenetic switch, which in turn facilitate recruitment of ETS factors to the mutant allele. In view of the recent work by several independent groups, we present a plausible model (Fig. 1) for the mechanisms of telomerase reactivation at mutant TERT promoters in human cancers with the hope that future efforts can unveil the missing links to these pertinent questions.
Figure 1.
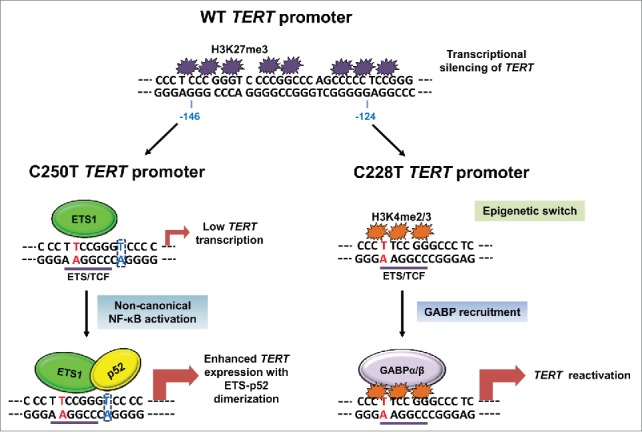
Current model for the transcriptional activation of TERT expression at mutant TERT promoters in cancers. The wild-type (WT) TERT promoter is enriched with repressive histone marks such as H3K27me3, which is associated with transcriptional silencing of TERT gene.23 During TERT promoter mutations (mutated residues are depicted in red) such as C228T mutation, an epigenetic switch occurs resulting in the association of active H3K4me2/3 marks and GABP recruitment on the mutant allele.23 Stabilization of GABP, an ETS family transcription factor, on mutant TERT promoter leads to TERT reactivation.19,23 In the context of C250T TERT promoter mutation, a proximal ETS binding motif is created next to a p52 half-site which facilitates cooperative binding of ETS1 and p52.24 The critical residues (in blue) required for dimerization of ETS and p52 are denoted in a dashed rectangle and are absent in both WT and C228T TERT promoters.24 Thus, during de-regulated non-canonical NF-κB signaling in C250T-mutant cancers, stabilization of ETS-p52 dimer on mutant TERT promoter results in elevated TERT expression.24
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work is funded by IMCB, A*STAR. Y.L. is supported by an A*STAR fellowship and the IMCB Early Career Researcher grant.
References
- [1].Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, Coviello GM, Wright WE, Weinrich SL, Shay JW. Specific association of human telomerase activity with immortal cells and cancer. Science 1994; 266:2011-5; PMID:7605428; http://dx.doi.org/ 10.1126/science.7605428 [DOI] [PubMed] [Google Scholar]
- [2].Li Y, Tergaonkar V. Noncanonical Functions of Telomerase: Implications in Telomerase-Targeted Cancer Therapies. Cancer Res 2014; 74:1639-44; PMID:24599132; http://dx.doi.org/ 10.1158/0008-5472.CAN-13-3568 [DOI] [PubMed] [Google Scholar]
- [3].Greider CW, Blackburn EH. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell 1985; 43:405-13; PMID:3907856; http://dx.doi.org/ 10.1016/0092-8674(85)90170-9 [DOI] [PubMed] [Google Scholar]
- [4].Low KC, Tergaonkar V. Telomerase: central regulator of all of the hallmarks of cancer. Trends Biochem Sci 2013; 38:426-34; PMID:23932019; http://dx.doi.org/ 10.1016/j.tibs.2013.07.001 [DOI] [PubMed] [Google Scholar]
- [5].Akincilar SC, Unal B, Tergaonkar V. Reactivation of telomerase in cancer. Cell Mol Life Sci 2016; 73:1659-70; PMID:26846696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ghosh A, Saginc G, Leow SC, Khattar E, Shin EM, Yan TD, Wong M, Zhang Z, Li G, Sung WK, et al.. Telomerase directly regulates NF-κB-dependent transcription. Nat Cell Biol 2012; 14:1270-81; PMID:23159929; http://dx.doi.org/ 10.1038/ncb2621 [DOI] [PubMed] [Google Scholar]
- [7].Greider CW. Molecular biology wnt regulates TERT–putting the horse before the cart. Science 2012; 336:1519-20; PMID:22723405; http://dx.doi.org/ 10.1126/science.1223785 [DOI] [PubMed] [Google Scholar]
- [8].Hoffmeyer K, Raggioli A, Rudloff S, Anton R, Hierholzer A, Del Valle I, Hein K, Vogt R, Kemler R. Wnt/β-catenin signaling regulates telomerase in stem cells and cancer cells. Science 2012; 336:1549-54; PMID:22723415; http://dx.doi.org/ 10.1126/science.1218370 [DOI] [PubMed] [Google Scholar]
- [9].Koh CM, Khattar E, Leow SC, Liu CY, Muller J, Ang WX, Li Y, Franzoso G, Li S, Guccione E, et al.. Telomerase regulates MYC-driven oncogenesis independent of its reverse transcriptase activity. J Clin Invest 2015; 125:2109-22; PMID:25893605; http://dx.doi.org/ 10.1172/JCI79134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zhu J, Zhao Y, Wang S. Chromatin and epigenetic regulation of the telomerase reverse transcriptase gene. Protein Cell 2013; 1:22-32; http://dx.doi.org/ 10.1007/s13238-010-0014-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Horn S, Figl A, Rachakonda PS, Fischer C, Sucker A, Gast A, Kadel S, Moll I, Nagore E, Hemminki K, et al.. TERT Promoter Mutations in Familial and Sporadic Melanoma. Science 2013; 339:959-61; PMID:23348503; http://dx.doi.org/ 10.1126/science.1230062 [DOI] [PubMed] [Google Scholar]
- [12].Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA. Highly Recurrent TERT Promoter Mutations in Human Melanoma. Science 2013; 339:957-9; PMID:23348506; http://dx.doi.org/ 10.1126/science.1229259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Killela PJ, Reitman ZJ, Jiao Y, Bettegowda C, Agrawal N, Diaz LA, Friedman AH, Friedman H, Gallia GL, Giovanella BC, et al.. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci 2013; 110:6021-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Rachakonda PS, Hosen I, de Verdier PJ, Fallah M, Heidenreich B, Ryk C, Wiklund NP, Steineck G, Schadendorf D, Hemminki K, et al.. TERT promoter mutations in bladder cancer affect patient survival and disease recurrence through modification by a common polymorphism. Proc Natl Acad Sci 2013; 110:17426-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Vinagre J, Almeida A, Pópulo H, Batista R, Lyra J, Pinto V, Coelho R, Celestino R, Prazeres H, Lima L, et al.. Frequency of TERT promoter mutations in human cancers. Nat Commun 2013; 4:2185; PMID:23887589 [DOI] [PubMed] [Google Scholar]
- [16].Borah S, Xi L, Zaug AJ, Powell NM, Dancik GM, Cohen SB, Costello JC, Theodorescu D, Cech TR. Cancer. TERT promoter mutations and telomerase reactivation in urothelial cancer. Science 2015; 347:1006-10; PMID:25722414; http://dx.doi.org/ 10.1126/science.1260200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Heidenreich B, Rachakonda PS, Hemminki K, Kumar R. TERT promoter mutations in cancer development. Curr Opin Genetics Dev 2014; 24:30-7; PMID:24657534; http://dx.doi.org/ 10.1016/j.gde.2013.11.005 [DOI] [PubMed] [Google Scholar]
- [18].Hollenhorst PC, Ferris MW, Hull MA, Chae H, Kim S, Graves BJ. Oncogenic ETS proteins mimic activated RAS/MAPK signaling in prostate cells. Genes Dev 2011:25:2147-57; PMID:22012618; http://dx.doi.org/ 10.1101/gad.17546311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bell RJA, Rube HT, Kreig A, Mancini A, Fouse SD, Nagarajan RP, Choi S, Hong C, He D, Pekmezci M, et al.. The transcription factor GABP selectively binds and activates the mutant TERT promoter in cancer. Science 2015; 348:1036-9; PMID:25977370; http://dx.doi.org/ 10.1126/science.aab0015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].LaMarco K, Thompson CC, Byers BP, Walton EM, McKnight SL. Identification of Ets- and notch-related subunits in GA binding protein. Science 1991; 253:789-92; PMID:1876836; http://dx.doi.org/ 10.1126/science.1876836 [DOI] [PubMed] [Google Scholar]
- [21].Thompson CC, Brown TA, McKnight SL. Convergence of Ets- and notch-related structural motifs in a heteromeric DNA binding complex. Science 1991; 253:762-8; PMID:1876833; http://dx.doi.org/ 10.1126/science.1876833 [DOI] [PubMed] [Google Scholar]
- [22].Wei GH, Badis G, Berger MF, Kivioja T, Palin K, Enge M, Bonke M, Jolma A, Varjosalo M, Gehrke AR, et al.. Genome‐wide analysis of ETS‐family DNA‐binding in vitro and in vivo. EMBO J 2010; 29:2147-60; PMID:20517297; http://dx.doi.org/ 10.1038/emboj.2010.106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Stern JL, Theodorescu D, Vogelstein B, Papadopoulos N, Cech TR. Mutation of the TERT promoter, switch to active chromatin, and monoallelic TERT expression in multiple cancers. Genes Dev 2015; 29:2219-24; PMID:26515115; http://dx.doi.org/ 10.1101/gad.269498.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Li Y, Zhou QL, Sun W, Chandrasekharan P, Cheng HS, Ying Z, Lakshmanan M, Raju A, Tenen DG, Cheng SY, et al.. Non-canonical NF-κB signalling and ETS1/2 cooperatively drive C250T mutant TERT promoter activation. Nat Cell Biol 2015; 17:1327-38; PMID:26389665; http://dx.doi.org/ 10.1038/ncb3240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Tergaonkar V. NF-κB drives TERT promoter reactivation in cancer. Cell Cycle 2015; 15:156-7; PMID:24905827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Cherry EM, Lee DW, Jung JU, Sitcheran R. Tumor necrosis factor-like weak inducer of apoptosis (TWEAK) promotes glioma cell invasion through induction of NF-kappaB-inducing kinase (NIK) and noncanonical NF-kappaB signaling. Mol Cancer 2015; 14:9; PMID:25622756; http://dx.doi.org/ 10.1186/s12943-014-0273-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Tran NL, McDonough WS, Donohue PJ, Winkles JA, Berens TJ, Ross KR, Hoelzinger DB, Beaudry C, Coons SW, Berens ME. The human Fn14 receptor gene is up-regulated in migrating glioma cells in vitro and overexpressed in advanced glial tumors. Am J Pathol 2003; 162:1313-21; PMID:12651623; http://dx.doi.org/ 10.1016/S0002-9440(10)63927-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Chiba K, Johnson JZ, Vogan JM, Wagner T, Boyle JM, Hockemeyer D. Cancer-associated TERT promoter mutations abrogate telomerase silencing. Elife 2015; 4:e07918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Li C, Wu S, Wang H, Bi X, Yang Z, Du Y, He L, Cai Z, Wang J, Fan Z. The C228T mutation of TERT promoter frequently occurs in bladder cancer stem cells and contributes to tumorigenesis of bladder cancer. Oncotarget 2015; 6:19542-51; PMID:26143634; http://dx.doi.org/ 10.18632/oncotarget.4295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Xi L, Schmidt JC, Zaug AJ, Ascarrunz DR, Cech TR. A novel two-step genome editing strategy with CRISPR-Cas9 provides new insights into telomerase action and TERT gene expression. Genome Biol 2015; 16:231; PMID:26553065; http://dx.doi.org/ 10.1186/s13059-015-0791-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Daniel M, Peek GW, Tollefsbol TO. Regulation of the human catalytic subunit of telomerase (hTERT). Gene 2012; 498:135-46; PMID:22381618; http://dx.doi.org/ 10.1016/j.gene.2012.01.095 [DOI] [PMC free article] [PubMed] [Google Scholar]