

Abstract
Using forward and reverse genetics and global gene expression analyses, we explored the crosstalk between the IκB kinase β (IKKβ) and the transforming growth factor β (TGFβ) signaling pathways. We show that in vitro ablation of Ikkβ in fibroblasts led to progressive ROS accumulation and TGFβ activation, and ultimately accelerated cell migration, fibroblast-myofibroblast transformation and senescence. Mechanistically, the basal IKKβ activity was required for anti-oxidant gene expression and redox homeostasis. Lacking this activity, IKKβ-null cells showed ROS accumulation and activation of stress-sensitive transcription factor AP-1/c-Jun. AP-1/c-Jun activation led to up-regulation of the Tgfβ2 promoter, which in turn further potentiated intracellular ROS through the induction of NADPH oxidase (NOX). These data suggest that by blocking the autocrine amplification of a ROS-TGFβ loop IKKβ plays a crucial role in the prevention of fibroblast-myofibroblast transformation and senescence.
Electronic supplementary material
The online version of this article (doi:10.1007/s13238-015-0241-6) contains supplementary material, which is available to authorized users.
Keywords: IkB kinase β (IKKβ), nuclear factor κB (NF-κB), transforming growth factors β (TGFβ), reactive oxygen species (ROS), myofibroblast, senescence
INTRODUCTION
The IκB kinase β (IKKβ) is a key catalytic subunit of the IKK complex, involved in inflammatory responses. It is robustly activated by cytokines, bacterial and viral products and metabolic stresses. IKKβ activation leads to phosphorylation of inhibitor of κB (IκB), and subsequently, translocation of the nuclear factor κB (NF-κB) to nucleus. The nuclear NF-κB binds to the κB elements in gene promoters and enhancers to either activate or repress gene expression (Perkins, 2007). By regulating genes coding for cytokines, chemokines, enzymes and molecules with microbicidal activity, the IKK-NF-κB cascade offers important protection against stress and danger signals (Vallabhapurapu and Karin, 2009). Persistent and unrestrained activation of the cascade, on the other hand, leads to chronic inflammation that may be the underlying cause of detrimental and life-threatening diseases, such as rheumatoid arthritis, atherosclerosis and cancer (Luo et al., 2005; Kim et al., 2006; Chariot, 2009). For this reason, inhibition of IKK signaling is widely considered as a promising strategy for treating many illnesses; the challenge however is to fully recognize, and develop means to offset, the potential harmful consequences of pathway inactivation (Baldwin, Jr., 2001; Li et al., 2002; Bacher and Schmitz, 2004; Courtois and Gilmore, 2006; Karin, 2006).
IKKβ maintains low static activity in the absence of external stimuli. This is associated with slow IκB degradation and equilibrium NF-κB activity (O’Dea et al., 2007). The basal activity is important for redox homeostasis, thus IKKβ inactivation renders cells or tissues vulnerable to oxidative damage (Gerondakis et al., 2006). For example, when Ikkβ is knocked out in hepatocytes, the livers of the knockout mice have normal development, but exhibit elevated levels of reactive oxygen species (ROS). In addition, IKKβ-defective livers are susceptible to injuries by carcinogens, concanavalin A and bacterial infection (Lavon et al., 2000; Maeda et al., 2005). When IKKβ is knocked out in fibroblasts, the null cells have elevated ROS levels and are sensitive to damage by stress and injury (Maeda et al., 2005; Chen et al., 2006; Giorgio et al., 2007; May and Madge, 2007; Sen and Roy, 2010). These observations suggest that IKKβ may be involved in a plethora of physiological processes through the regulation of redox homeostasis (Karin, 2008; Pasparakis, 2009).
In the present work, we investigated the role of IKKβ through global gene expression analyses and identified a crosstalk interaction between IKKβ and TGFβ signaling. We showed that loss of IKKβ in fibroblasts led to TGFβ activation, which in turn modulated cell motility, myofibroblast transformation and senescence. These results suggest that IKKβ can act as a repressor of the TGFβ pathway.
RESULTS
IKKβ represses TGFβ signaling
To explore the roles of IKK and NF-κB signaling in fibroblasts, we examined global gene expression in wild type and cells lacking IKKα, IKKβ or the p65 subunit of NF-κB. Comparison of differentially expressed genes between wild type and knockout cells, we found that genes up-regulated in the wild type cells were enriched for the terpenoid backbone biosynthesis pathway, whereas genes down-regulated in the wild type cells were enriched for the focal adhesion and vascular smooth muscle contraction pathways (Table 1).
Table 1.
Biological pathways affected by the IKK-NF-κB cascade*
| WT vs. Ikkα −/− | WT vs. Ikkβ −/− | WT vs. p65 −/− | |
|---|---|---|---|
| Up-regulated genes enriched pathways | |||
| Terpenoid backbone biosynthesis | 0.003663 | 1.53 × 10−5 | 0.014653 |
| Down-regulated genes enriched pathways | |||
| Focal adhesion | 1.78 × 10−8 | 0.039390 | 3.14 × 10−10 |
| Vascular smooth muscle contraction | 9.00 × 10−7 | 0.000589 | 1.66 × 10−5 |
* Each entry is the False Discovery Rate (FDR) adjusted P-values for the pathway in the corresponding row in the comparison in the corresponding column. The P-values were calculated by R package CLEAN using the KEGG pathway database
We further examined differential gene expression between IKKβ-competent (Ikkβ−/−/Ad-IKKβ) and -deficient (Ikkβ−/−/Ad-β-Gal and Ikkβ−/−) cells using the same strategy. Genes up-regulated in the IKKβ-competent cells were, as expected, enriched for pathways involved in immunity and inflammation, such as antigen processing and presentation, rheumatoid arthritis, and B cell receptor signaling pathway and allograft rejection, but intriguingly, genes down-regulated in the IKKβ-competent cells were enriched for focal adhesion, ECM-receptor interaction and, and the TGFβ signaling pathways (Table 2).
Table 2.
The IKKβ-regulated biological pathways*
| Ad-IKKβ vs. uninfected | Ad-IKKβ vs. Ad-β-Gal | |
|---|---|---|
| Up-regulated genes enriched pathways | ||
| Antigen processing and presentation | 3.74 × 10−6 | 4.02 × 10−6 |
| Leishmaniasis | 3.69 × 10−5 | 3.29 × 10−12 |
| Phagosome | 4.00 × 10−5 | 0.002085 |
| Rheumatoid arthritis | 1.51 × 10−9 | |
| B cell receptor signaling pathway | 0.000402 | 0.000641 |
| Graft-versus-host disease | 0.002208 | 0.002457 |
| Allograft rejection | 0.003035 | 0.004726 |
| Type I diabetes mellitus | 0.009985 | 0.022062 |
| Autoimmune thyroid disease | 0.013477 | 0.025376 |
| Down-regulated genes enriched pathways | ||
| Focal adhesion | 0.000511 | 6.21 × 10−6 |
| TGFβ signaling pathway | 0.009496 | 0.028866 |
| ECM-receptor interaction | 4.73 × 10−8 | 4.70 × 10−7 |
| Protein digestion and absorption | 1.18 × 10−7 | 0.001664 |
| Amoebiasis | 5.40 × 10−6 | 0.000540 |
* Each entry is the False Discovery Rate (FDR) adjusted P-values for the pathway in the corresponding row in the comparison in the corresponding column. The P-values were calculated by R package CLEAN using the KEGG pathway database
We validated the array data focusing on IKKβ-repressed genes of the TGFβ pathway. Compared to the wild type, the Ikkβ−/− cells had elevated Tgfβ2 and Tgfβ3 mRNA transcripts (Fig. 1A), corresponding to higher gene promoter activities (Fig. 1B). They also exhibited increased SMAD transcriptional activity (Fig. 1C) and phosphorylation (Fig. 1D), as well as increased expression of a number of SMAD target genes, such as Smad6, Ctgf and Acta2 (Figs. 1E and S1). In addition, we observed the expression of myofibroblast marker α smooth muscle actin (α-SMA), the product of Acta2, in IKKβ-null but not wild type cells (Fig. 1D). Adenoviral-mediated expression of IKKβ, but not of GFP used as control, in the null cells repressed Tgfβ expression and promoter activity, decreased SMAD activity and target gene expression, similar to the effects of Ad-SMAD7 and reached the levels same as that in the wild type cells (Fig. 1A–C and 1E). These results indicate that loss of IKKβ leads to the activation of TGFβ expression and signaling.
Figure 1.
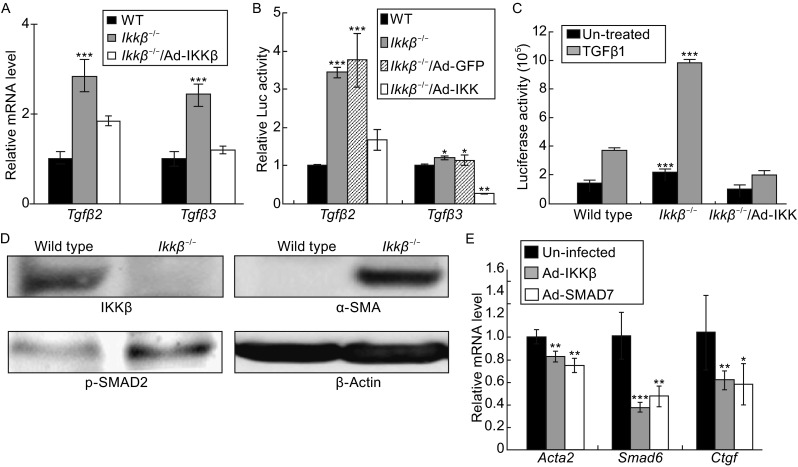
Loss of IKKβ upregulates TGFβ expression and activity. The IKKβ-competent, i.e. wild type and Ikkβ −/−/Ad-IKKβ, and IKKβ-deficient, i.e. Ikkβ −/− and Ikkβ −/− /Ad-GFP, fibroblasts were examined for Tgfβ2 and 3 (A) mRNA expression and (B) promoter activity, and for (C) basal (un-treated) and TGFβ1-induced SMAD activity (SBE-luc) and (D) SMAD phosphorylation, and IKKβ, α-SMA and β-actin expression. (E) The Ikkβ −/− cells, either uninfected or infected with Ad-IKKβ and Ad-SMAD7, were examined for the expression of SMAD-target genes, i.e. Acta2, Smad6 and Ctgf. Results represent the mean values ± SD from at least three independent experiments. *P < 0.05, **P < 0.01 and ***P < 0.001 were considered significantly different from the wild type or control samples
TGFβ upregulation leads to migration and myofibroblast transformation of IKKβ-null cells
TGFβ plays a pivotal role in cell proliferation, differentiation, wound healing and extracellular matrix production, and it induces growth arrest and myofibroblast transformation in fibroblasts (Datto et al., 1999; Phan, 2002). Chen, et al. have reported that the IKKβ-deficient cells grow slower, but migrate faster (Chen et al., 2006). We confirmed these observations (Fig. 2A and 2B), and furthermore, we showed that the migration rate of the null cells was significantly reduced by expression of IKKβ and inhibitory SMAD7, and by treatment with SB505124, a TGFβ receptor inhibitor (Fig. 2B and 2C).
Figure 2.
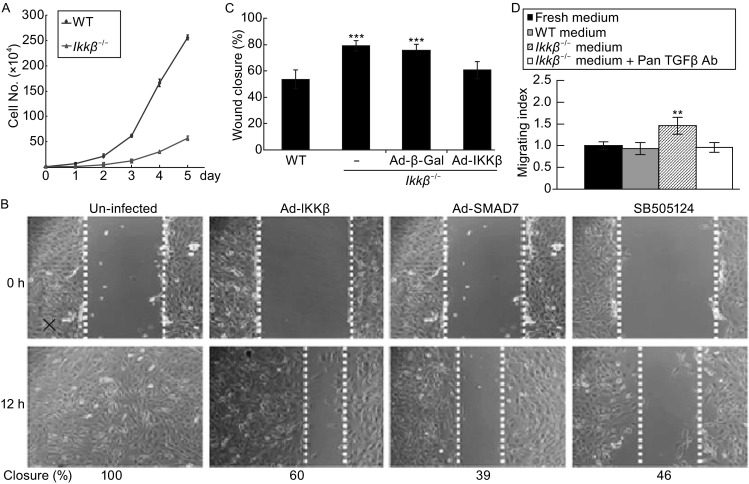
IKKβ loss induces TGFβ expression and cell migration. (A) The wild type and Ikkβ −/− fibroblasts were examined for growth rate. The in vitro wound healing assay was performed on wild type and Ikkβ −/− fibroblasts either uninfected or infected with Ad-GFP or Ad-IKKβ, or pre-treated with SB505124 for 24 h. (B) The cells were photographed at 0 and 12 h after the scratch wound, and (C) the speed of healing was calculated. (D) The in vitro wound healing assay was performed on wild type cells in normal growth medium, condition-medium collected from wild type cells or condition-medium collected from Ikkβ −/− cells with or without anti-pan-TGFβ. The healing rate was calculated at 12 h after injury. Results represent the mean values ± SD from at least three independent experiments. **P < 0.01 and ***P < 0.001 were considered significantly different from the wild type or control samples
To assess if promoted migration was due to TGFβ secretion, we collected conditioned medium from wild type and Ikkβ−/− cultures and examined its effects on migration of the wild type cells. The wild type-conditioned medium had no effect, but the Ikkβ−/−-conditioned medium accelerated migration by 50% (Fig. 2D). Additionally, the migration stimulatory activity was abolished by TGFβ neutralizing antibodies, supporting the notion that TGFβ secreted by the IKKβ-null cells contributed to the stimulation of fibroblast migration.
Progressive ROS accumulation and TGFβ activation following IKKβ ablation
Infection of IkkβF/F embryonic fibroblasts with Ad-Cre could ablate the Ikkβ gene in vitro. Using this approach, we generated the IkkβF/F/Ad-Cre cells, in which IKKβ expression, NF-κB activity, and NF-κB target gene expression were abolished or significantly reduced (Figs. 3A and S2A–S2E). Infection of the IkkβF/F/Ad-Cre cells with Ad-IKKβ, but not Ad-GFP, restored NF-κB activity and target gene expression (Fig. S2D and S2E).
Figure 3.
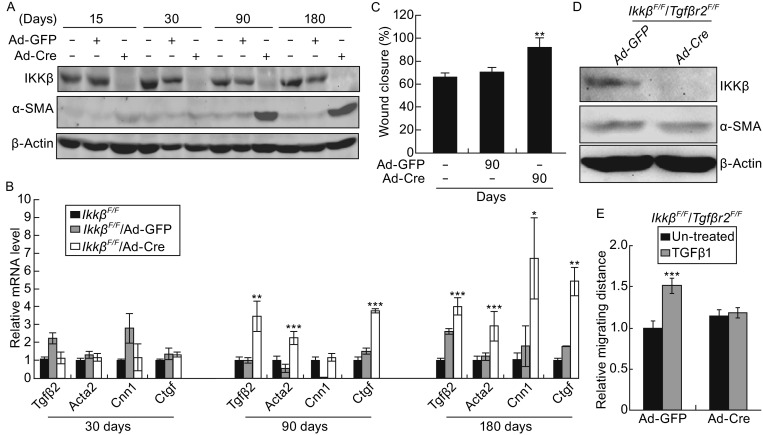
IKKβ ablation leads to progressive activation of TGFβ and cell migration. The Ikkβ F/F fibroblasts were infected with Ad-GFP or Ad-Cre and the cells were maintained in culture for various days as indicated. The cells were examined for (A) IKKβ, α-SMA and β-actin protein, (B) mRNA for Tgfβ2 and SMAD-target genes, and (C) rate of wound healing. The Ikkβ F/F/Tgfbr2 F/F fibroblasts were infected with Ad-GFP or Ad-Cre. At 3 months after infection, the cells were examined for (D) IKKβ, α-SMA and β-actin protein and (E) rate of in vitro wound healing in the presence or absence of TGFβ1. Results represent the mean values ± SD from at least three independent experiments. *P < 0.05, **P < 0.01 and ***P < 0.001 were considered significantly different from the wild type or control samples
The IkkβF/F/Ad-Cre cells lacked IKKβ, but surprisingly, they did not have detectable α-SMA expression immediately following Ad-Cre infection (Fig. 3A). These cells instead displayed a gradual increase in the expression of α-SMA, TGFβ2 and SMAD-target genes (Fig. 3A and 3B), and they exhibited faster migration only after 90 days of Ad-Cre infection (Fig. 3C). Simultaneous ablation of IKKβ and TGFβ receptor 2 reduced α-SMA upregulation and TGFβ1-induced migration (Figs. 3D, 3E and S3). The data derived from the in vitro gene ablation system suggest that IKKβ loss leads to a gradual activation of TGFβ signaling and progressive myofibroblast conversion.
Loss of IKKβ leads to activation of the ROS-TGFβ-NOX cascade
Consistent with the notion that IKKβ represses reactive oxygen species (ROS) (Tanaka et al., 1999; Maeda et al., 2005), we showed that the H2O2 level, measured by 2′,7′-Dichlorodihydrofluorescein diacetate (CM-H2DCFDA) labeling, was high in Ikkβ−/− but low in wild type cells (Fig. S4A). In addition, the expression of the oxidative stress-inducible biomarker gene Heme oxygenase 1 (Ho-1) was more abundant in Ikkβ−/− than wild type cells (Fig. 4A). The IKKβ- and p65-deficient cells have similar gene expression signatures and faster migration phenotype (Table 1 and Fig. S5), and like the IKKβ-null cells, the p65−/− cells also had increased Ho-1 expression (Fig. 4B). Furthermore, we detected in the p65−/− cells decreased expression of superoxide dismutase 2 (Sod2), encoding for a crucial redox scavenger. Correspondingly, compared to the IKKβ-competent, i.e. wild type and Ikkβ−/−/Ad-IKKβ, cells, the IKKβ-deficient Ikkβ−/−cells had decreased level of RNA pol II recruitment to the Sod2 promoter and reduced p65 bound at the gene enhancer (Fig. 4C).
Figure 4.
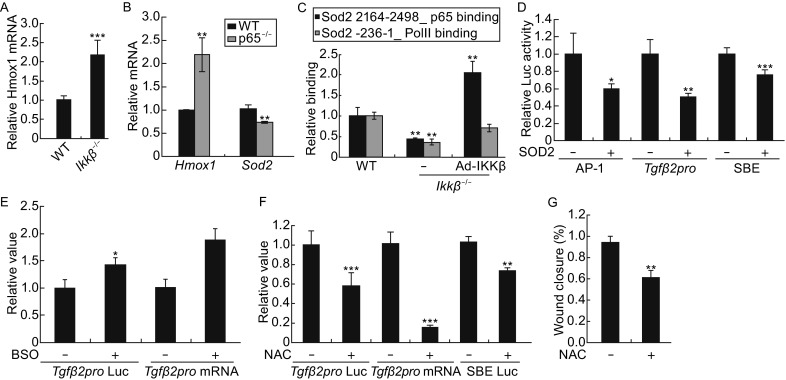
ROS accumulation leads to TGFβ activation in IKKβ-null cells. (A) Wild type and Ikkβ −/− cells and (B) wild type and p65 −/− cells were examined for mRNA of oxidative stress marker Hmox-1 and/or redox scavenger gene Sod2. (C) The wild type and Ikkβ −/− cells with or without Ad-IKKβ infection were examined for p65 binding of the Sod2 enhancer and RNA Pol II occupancy of the Sod2 promoter. (D) The Ikkβ −/− cells were transfected with luciferase reporters for AP-1, SMAD and Tgfβ2 promoter, together with either an empty vector or SOD2 expression plasmids. The luciferase activities in SOD2 transfected cells were compared to those in empty vector transfected cells, designated as 1. (E) The wild type cells were either un-transfected or transfected with Tgfβ2 promoter reporter, and treated with pro-oxidant BSO. The Tgfβ2 mRNA expression and promoter activity were examined. The Ikkβ −/− cells were either un-transfected or treanfected with the luciferase reporter plasmids for Tgfβ2 promoter or SMAD (SEB-luc). The cells were treated with anti-oxidant NAC, and were examined for (F) Tgfβ2 mRNA expression and luciferase activity, and (G) migration by the in vitro wound healing assays. Results represent the mean values ± SD from at least three independent experiments. *P < 0.05, **P < 0.01 and ***P < 0.001 were considered significantly different from the wild type or control samples
To evaluate if SOD2 reduction contributed to TGFβ activation, we expressed SOD2 in IKKβ-null cells and observed a significant decrease of Tgfβ2 promoter and SMAD activity (Fig. 4D). We further showed that SOD2 expression caused down-regulation of ROS-sensitive AP-1 activity, raising the possibility that the TGFβ signaling was actually modulated by cellular redox status (Bataller et al., 2003; Fleckenstein et al., 2007; Roy et al., 2011). To test the possibility, we treated the wild type cells with pro-oxidant L-Buthionine sulphoximine (BSO), and the Ikkβ−/− cells with anti-oxidant N-acetyl cysteine (NAC). As predicted by the hypothesis, BSO increased Tgfβ2 gene expression and promoter activity and NAC significantly attenuated them, and reduced SMAD activity and migration of the IKKβ-deficient cells (Fig. 4E–G). These data suggest that SOD2 reduction and ROS accumulation contribute to TGFβ activation in the IKKβ-null cells.
TGFβ, on the other hand, has been shown to activate NADPH oxidases (NOX), which could further augment ROS (Hecker et al., 2009; Bondi et al., 2010). By monitoring intracellular glutathione (GSH), the most abundant redox scavenger, we observed that treating cells with TGFβ1 caused GSH depletion, whereas treating cells with TGFβ inhibitors restored GSH in IKKβ-null and TGFβ1 treated wild type cells (Anderson, 1998) (Fig. 5A). Furthermore, the IKKβ-null cells had high levels of NOX1 and NOX4 expression and NOX inhibitors abolished TGFβ-induced GSH depletion in these cells (Armstrong et al., 2002; Bedard and Krause, 2007) (Fig. 5B and 5C).
Figure 5.
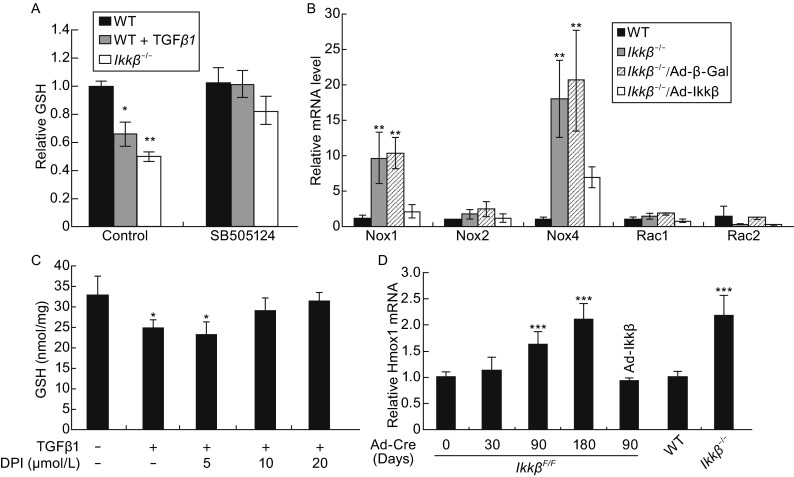
Activation of ROS-TGFβ-NOX amplification loop in the IKKβ-null cells. The intracellular GSH in wild type, TGFβ1 treated wild type and Ikkβ −/− cells, (A) in the presence or absence of the TGFβ receptor inhibitor SB505124, or (C) in the presence or absence of the NOX inhibitor DPI. (B) The mRNA of genes coding for components of the NADH complexes in IKKβ-competent, i.e. wild type and Ikkβ −/−/Ad-IKKβ, and IKKβ-deficient, i.e. Ikkβ −/− and Ikkβ −/−/Ad-β-Gal, cells. (D) The mRNA for oxidative stress marker Hmox-1 in wild type, Ikkβ −/− and in Ikkβ F/F fibroblasts infected with Ad-Cre for 0 to 180 days with or without Ad-IKKβ. Results represent the mean values ± SD from at least three independent experiments. *P < 0.05, **P < 0.01 and ***P < 0.001 were considered significantly different from the wild type or control samples
Taken together, the above data suggest a scenario that reduction of redox scavengers in IKKβ-null cells could lead to ROS accumulation; the oxidative stresses in turn might activate the TGFβ-NOX cascade to further augment ROS. IKKβ ablation therefore leads to the activation of an autocrine cycle of ROS amplification. Consistent with the conclusion, we found that Ad-Cre infection of the IkkβF/F cells led to a gradual ROS increase. While 36% IkkβF/F/Ad-Cre cells displayed high H2O2 level at 30 days of Ad-Cre infection, the number increased to almost 50% at 90 days after infection (Fig. S4B and S4C). Similarly, the HO-1 expression increased gradually after Ad-Cre infection of IkkβF/F cells, and by 180 days, it reached the levels similar to that in Ikkβ−/− cells and twice that in wild type or Ad-IKKβ-infected IkkβF/F/Ad-Cre cells (Fig. 5D).
AP-1 is involved in ROS-induced TGFβ expression
To identify the molecular link between ROS and TGFβ, we scanned the Tgfβ2 promoter for transcription factor binding sites and found two potential AP-1-cJun binding sites (Fig. S6A). AP-1 is a stress responsive transcription factor; we tested its activation with a luciferase reporter bearing an AP-1 binding site and found that luciferase expression was induced by IKKβ ablation, but repressed by IKKβ expression and NAC treatment (Fig. 6A). In addition, AP-1 binding to the Tgfβ2 promoter, as measured by chromatin immunoprecipitation, was increased, associated with the transcriptionally active H3K4me3 modification on the Tgfβ2 promoter, in IKKβ-deficient cells (Fig. S6B and S6C). Both AP-1 binding and H3K4me3 were potentiated by IKKβ ablation and BSO treatment, but reduced by IKKβ over-expression and NAC treatment (Fig. 6B and 6C).
Figure 6.
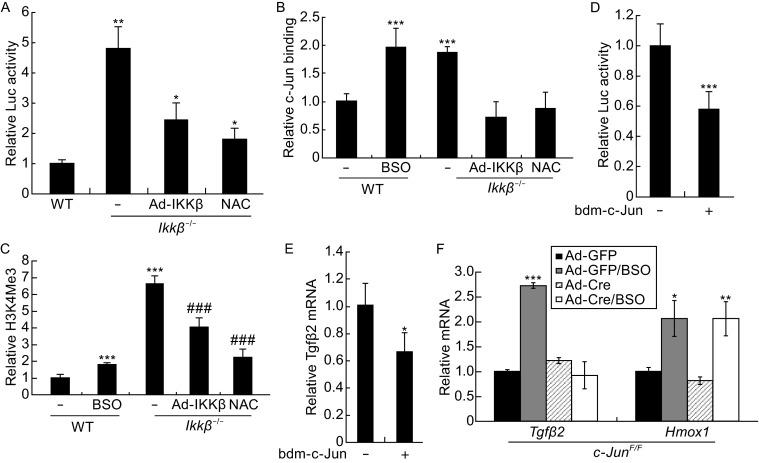
c-Jun regulates TGFβ expression in IKKβ-null cells. The wild type cells with or without BSO treatment, and the Ikkβ −/− cells with or without Ad-IKKβ infection or NAC treatment were examined for (A) luciferase activity following AP-1-luc plasmid transfection, and (B and C) ChIP assays for (B) c-Jun binding of the Tgfβ2 enhancer and (C) H3K4me3 modification of the Tgfβ2 promoters. The Ikkβ −/− cells were transfected with a dominant negative c-Jun (bdm-c-Jun) expression plasmids, and (D) together with Tgfb2-luc and examined for the luciferase activities, and (E) examined for the Tgfb2 mRNA. (F) The mRNA for Tgfb2 and Hmox-1 was examined in c-Jun F/F cells infected with Ad-Cre or Ad-GFP and treated with BSO. Results represent the mean values ± SD from at least three independent experiments. *P < 0.05, **P < 0.01 and ***P < 0.001 were considered significantly different from the un-treated wild type samples; ###P < 0.001 was significantly different from the un-treated Ikkβ −/− samples
To validate the role of AP-1-c-Jun, we expressed a dominant negative mutant c-Jun (bdm-c-Jun) in the Ikkβ−/− cells and found that its expression repressed Tgfβ2 promoter activity and gene expression (Fig. 6D and 6E). We further used c-Jun-competent (c-JunF/F/Ad-GFP) and -deficient (c-JunF/F/Ad-Cre) cells and showed that while c-Jun ablation did not affect HO-1 induction, it abolished Tgfβ2 induction under the oxidative stress conditions created by BSO treatment (Fig. 6F). Collectively, our data suggest that the ROS may act upstream to activate AP-1/c-Jun, which in turn can induce Tgfβ promoter and gene expression in the Ikkβ-null cells.
Loss of IKKβ leads to senescence
Chronic oxidative stress can induce, stabilize and amplify senescence, leading ultimately to the detrimental effects of aging (Passos et al., 2010; Nelson et al., 2012). To assess if IKKβ ablation could lead to senescence, we examined the expression of senescence-associated β-Galactosidase (SA-β-Gal) (Dimri et al., 1995). SA-β-Gal activity was low in IkkβF/F cells, but gradually increased following Ad-Cre infection; by 180 days after infection the activity reached approximately 50% of the level in Ikkβ−/−cells (Fig. 7A). In Ad-Cre infected IkkβF/F cells, there was also a progressive increase of the cell cycle regulator cyclin-dependent kinase inhibitor 1A (p21) (Cdkn1a), the extracellular matrix component Fibronectin (Fn1), and γH2AX, a histone modification associated with DNA double strand damage (Dumont et al., 2000; Debacq-Chainiaux et al., 2008; Weyemi et al., 2011) (Fig. 7B and 7C). Furthermore, there was a slight but gradual increase of telomere shortening, suggesting that IKKβ loss may lead to irreversible DNA damage and a senescent phenotype (Balaban et al., 2005; Giorgio et al., 2007) (Fig. 7D). Hence, by repressing the ROS-AP-1-TGFβ axis IKKβ may prevent senescence in fibroblasts (Fig. 7E).
Figure 7.
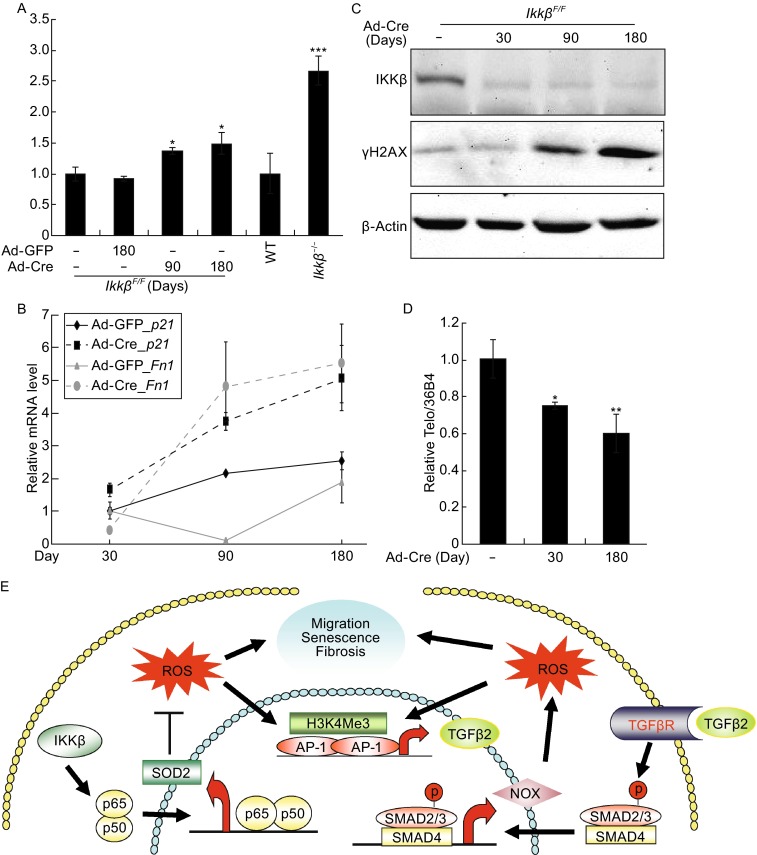
Phenotype of the IKKβ-deficient cells. The wild type, Ikkβ −/−, Ikkβ F/F/Ad-GFP and Ikkβ F/F/Ad-Cre cells were examined for (A) SA-β-gal activity, (B) expression of senescent markers, p21 and Fn, (C) expression of IKKβ, β-actin and γH2AX, a marker for DNA damage, and (D) the telomere length. Results represent the mean values ± SD from at least three independent experiments. * P < 0.05, ** P < 0.01 and *** P < 0.001 were considered significantly different from the wild type or control samples. (E) A proposed model depicting the role of IKKβ in the regulation of the ROS-TGFβ autocrine amplification loop. Specifically, IKKβ acts through p65 to regulate expression of anti-oxidant genes, such as SOD2. Loss of IKKβ decreases SOD2 expression and dampens the scavenger capacity, resulting in ROS accumulation and AP-1/c-Jun activation. The AP-1/c-Jun regulates TGFβ expression thereby activating the TGFβ-NOX axis to further potentiate ROS accumulation. The amplification of the ROS-TGFβ-NOX axis eventually leads to increased cell migration, myofibroblast transformation and senescence
DISCUSSION
The global gene expression signatures provide an initial clue that loss of IKKβ or key components of the NF-κB pathways may lead to activation of TGFβ signaling in fibroblasts. Following this lead, we have identified a molecular link between the IKKβ and TGFβ pathways. We show that the IKKβ-NF-κB cascade sustains the expression of anti-oxidant genes and that inactivation of this cascade impedes the scavenge capacity and results in ROS accumulation. Elevated ROS in turn triggers the feed-forward activation of the ROS-AP-1-TGFβ-NOX loop that leads ultimately to increased motility, fibroblast-myofibroblast transformation, and senescence (Fig. 7E).
The antagonistic relationship between IKK and TGFβ signaling has been reported in other experimental settings. For example, in osteoclasts and head and neck cancers, TGFβ is found acting through the TGFβ activated kinase 1 to activate IKK and NF-κB, whereas NF-κB up-regulates SMAD7 to inhibit TGFβ signaling (Gingery et al., 2008; Freudlsperger et al., 2013). The IKK-NF-κB pathway is also found to modulate transcription factors/cofactors and attenuate SMAD activity (Dennler et al., 2000; Nagarajan et al., 2000; Bitzer et al., 2000; Verrecchia et al., 2001). Here we describe a unique mechanism where the crosstalk of IKK and TGFβ is mediated by ROS. Specifically, the elevated ROS in IKKβ-null cells induce c-Jun binding and activation of the Tgfβ promoter.
There are at least two sources for the ROS in IKKβ-null cells. First, IKKβ ablation results in insufficient ROS removal due to down-regulation of antioxidant genes, in agreement with previous reports (Chen et al., 2003; Sakon et al., 2003; Peng et al., 2007; Peng et al., 2010). Second, IKKβ ablation causes increased ROS production as the result of TGFβ induced NOX4 expression and NADH activity. Interestingly, the TGFβ-NOX axis itself is also activated by ROS, and thus, this axis and ROS may form an autocrine loop to amplify each other. Such feed-forward signal amplification is likely to be responsible for the progressive ROS accumulation and TGFβ activation in fibroblasts following IKKβ ablation. When the TGFβ signals reach a threshold level, it is able to induce cell migration and myofibroblast transformation; when the chronic ROS reach a threshold level, they may contribute to premature senescence, as it also happens in cells deficient in GSH (Chen et al., 2009).
As the IKK-NF-κB cascade is a major player of the inflammatory response, its inhibition is a promising strategy for treating a vast number of diseases associated with inflammation (McIntyre et al., 2003; Ruocco et al., 2005; Polzer et al., 2008). In particular, this cascade is considered a molecular link between inflammation and cancer; therefore, targeting the cascade has become an attractive rationale in cancer therapy (Vallabhapurapu and Karin, 2009; DiDonato et al., 2012). The caveat is that such treatment may have adverse effects due to disruption of the cascade’s pleiotropic physiological functions (DiDonato et al., 2012). Our data in fibroblasts echo this concern and suggest that complete, irreversible and long-term inhibition of IKKβ may lead to chronic oxidative stress, and increase the risks for fibrogenesis and senescence.
MATERIALS AND METHODS
Viruses, plasmids, reagents and antibodies
The adenoviral expression vectors for IKKβ, SMAD7, β-GAL, GFP and GFP-Cre were from Drs. Yi Zheng at the Cincinnati Children’s Hospital, Yinling Hu at the National Cancer Institute, and Chia-yang Liu at Indiana University. The reporter plasmids, NF-κB-luc, SBE-Luc, AP-1-Luc, and the Tgfβ1, Tgfβ2 and Tgfβ3 promoter-luc were obtained from Drs. Edward B. Leof at Mayo Clinic and Alvaro Puga at the University of Cincinnati (Tojima et al., 2000). Expression vector for SOD2 was from Dr. Shanglin Shi at the University of Kentucky and Bdm-c-Jun was described before (Geh et al., 2011). TGFβ1 was from PeproTech, NAC, BSO and DPI were from Sigma-Aldrich, and SB505124 was from EMD Millipore. The following antibodies were used in the study: anti- IKKα, -IKKβ, -IkBα, and -p-SMAD2 (Ser-465, 467) from Cell Signaling, anti-pan TGFβ from R&D Systems, anti-α-SMA from Abcam, anti-β-actin from Sigma-Aldrich, anti-γH2AX from Novus Biologicals, anti-PolII, -H3, -H3K27Me3, H3K9Me2, H3K9Ac and H3K4Me3 from EMD Millipore, and anti-p65, -c-Jun, and IgG from Santa Cruz Biotechnologies.
Mouse fibroblasts, cell culture, transfection, infection and luciferase assays
The wild type, fibroblasts deficient in IKKβ, IKKα and p65 were gifts from Drs. Karin and Zandi, and were maintained under culture conditions as described (Chen et al., 2006). The IkkβF/F, IkkβF/F/Tgfbr2F/F and c-JunF/F fibroblasts were prepared using E13.5 embryos following standard 3T3 protocol (Aaronson and Todaro, 1968). The cells were cultured in DMEM supplemented with 10% FBS, 50 U/mL penicillin, 50 mg/mL streptomycin for less than 10 passages before used for experiments or adenoviral infection. Some of adenoviral infected cells were allowed to grow for 6 months with approximately 50 passages. Adenoviruses were used at 100–500 PFU to infect 70% confluent cells as described before (Peng et al., 2010). Cells were transfected using the lipofectamin plus method and Firefly and Renilla luciferase activities were measured 24 to 48 h after transfection following the manufacture’s protocols (Thermo Fisher Scientific).
Western blotting, ROS measurements, SA-β-Gal activity and in vitro wound healing assays
The SA-β-Gal activities were measured at the PH 6.0 using Beta-Glo Assay system (Promega), Western blotting, measurement of ROS and GSH, and the in vitro wound healing assays were done as previously described (Zhang et al., 2003; Peng et al., 2010). The conditional medium used for in vitro wound healing assays was derived from fresh medium overlaid on wild type or Ikkβ−/− cells for 24 h.
RNA isolation, reverse transcription and gene expression profiling
RNA was extracted, labeled and hybridized to Affymetrix Mouse Genome 430 2.0 Arrays using standard protocol (Medvedovic et al., 2009). Data was processed by performing background correction, quantile normalization, and calculation of expression set summaries using the Robust Multichip Average (RMA) protocol (Irizarry et al., 2003) as implemented in the Bioconductor affy package. Differentially expressed genes between two groups were identified by two-group comparison using intensity-based empirical Bayes method (IBMT) (Sartor et al., 2006). Pathway enrichment analysis was performed using the LRpath methodology (Sartor et al., 2009) implemented in the CLEAN package (Freudenberg et al., 2009).
Quantitative RT-PCR (qRT-PCR), chromatin-immunopreciptation (ChIP) and telomere measurement
qRT-PCR was performed using a DNA Engine Opticon2 Real-Time PCR Detection System (MJ Research) and SYBR Green qPCR MasterMix (Applied Biosystems) and primers for the genes of interest as listed in Table S1. All experiments were performed at least in triplicates. The relative differences in qRT-PCR among samples were determined by the ΔCT value as described previously (Schnekenburger et al., 2007). Hence, the ΔCT value for each sample was calculated by subtracting cycle threshold (CT) value (obtained from the means of replicates) of the input DNA (or Gapdh signal) from that of each sample in order to normalize ChIP assay (or to normalize gene expression) results. The ΔΔCT value was calculated by subtracting control ΔCT values from the corresponding experimental ΔCT values. The resulting values were converted to fold changes over control by raising 2 to the power of −ΔCT values.
ChIP was performed following the protocol described previously (Schnekenburger et al., 2007). Briefly, cells were fixed for 10 min with 1% formaldehyde, followed by addition of 0.125 mol/L glycine for 5 min to stop cross-linking. Cells were washed with ice-cold PBS and harvested in cell lysis buffer (5 mmol/L PIPES [pH 8.0], 85 mmol/L KCl, 0.5% NP-40, and protease inhibitor cocktail [Roche]) for 10 min on ice. The nuclei were pelleted, resuspended in nucleus lysis buffer (50 mmol/L Tris-HCl [pH 8.1], 10 mmol/L EDTA, 1% SDS, and protease inhibitor cocktail), and incubated on ice for 10 min. Chromatin was sheared to a size range of 0.3 to 0.8 kb by sonication. After centrifugation to remove cell debris, chromatin was precleared for 1 h at 4°C with a 50% gel slurry of protein A-agarose beads saturated with salmon sperm DNA (Upstate), and then diluted three times in IP dilution buffer (16.7 mmol/L Tris-Cl [pH 8.1], 167 mmol/L NaCl, 1.2 mmol/L EDTA, 1.1% Triton X-100, 0.01% sodium dodecyl sulfate) with 10% of the supernatants used as input. The diluted chromatin was incubated with antibodies specific for the proteins of interest for 2 h at 4°C, followed by addition of a 50% gel slurry of protein A-agarose and incubation overnight (Upstate). The agarose beads were pelleted and washed twice with 1× dialysis buffer (50 mmol/L Tris-HCl [pH 8.0], 2 mmol/L EDTA, 0.2% Sarkosyl) and four times with IP wash buffer (100 mmol/L Tris-HCl [pH 9.0], 500 mmol/L LiCl, 1% NP-40, 1% deoxycholic acid). Precipitated chromatin complexes were removed from the beads by incubation with elution buffer (50 mmol/L NaHCO3, 1% SDS) with mild vortexing. This step was repeated, and the eluates were combined. Cross-linking was reversed by adding NaCl to a final concentration of 0.3 mol/L and incubating overnight at 65°C in the presence of RNase A. Samples were then digested with proteinase K at 45°C for 1.5 h. DNA was purified by chromatography on QIAquick columns (QIAGEN) and eluted in double-distilled water for further qPCR analysis.
The telomere length was measured using Q-PCR as described (Callicott and Womack, 2006). Briefly, the genomic DNA were extracted using a QIAmp DNA micro Kit (Qiagen, Valencia, CA, USA) and quantified.
PCR reactions were performed on the ABI Prism 7700 Sequence Detection System (Applied Biosystems), using telomeric primers for the reference control gene (mouse 36B4 single copy gene). The telomere signal was normalized to the signal from the single-copy gene to generate a relative telomere to single copy gene (T/S) ratio indicative of relative telomere length. Equal amounts of DNA (300 pg) were used for each reaction with several repeats and average telomere length was calculated.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ACKNOWLEDGEMENTS
This work is supported in part by NIH grants EY15227 (YX) and Albert J. Ryan Fellowship (LC); YX, LN and MM are members of Center of Environmental Genetics (P30-ES06096). We thank Drs. Michael Karin for Ikka −/− and Ikkβ −/− fibroblasts and Ikkβ F/F mice, Ebrahim Zandi for p65 −/− fibroblasts, Yinling Hu, Yi Zheng, Chia-yang Liu for providing adenoviral vectors, Dr. Alvaro Puga for SMAD7 adenovirus and Tgfβ1, Tgfβ2 and Tgfβ3 promoter-luc, Dr. Edward B. Leof for SBE-luc plasmid, Dr. Xianglin Shi for SOD2 expression vector and Drs. Michael Karin and Harold Moses for the Ikkβ F/F and Tgfbr2 F/F mice.
ABBREVIATIONS
α-SMA, α smooth muscle actin; AP-1, activating protein-1; BSO, L-Buthionine sulphoximine; GSH, glutathione; HO-1, heme oxygenase; IKKβ, IκB kinase β; NAC, N-acetyl cysteine, NF-κB, nuclear factor Κb; NOX, nicotinamide adenine dinucleotide phosphate oxidase; ROS, reactive oxygen species; SOD2, superoxide dismutase 2; TGFβ, transforming growth factor β.
COMPLIANCE WITH ETHICS GUIDELINES
Liang Chen, Zhimin Peng, Qinghang Meng, Maureen Mongan, Jingcai Wang, Maureen Sartor, Jing Chen, Liang Niu, Mario Medvedovic, Winston Kao and Ying Xia declare that they have no conflict of interest. All institutional and national guidelines for the care and use of laboratory animals were followed.
REFERENCES
- Aaronson SA, Todaro GJ. Development of 3T3-like lines from Balb-c mouse embryo cultures: transformation susceptibility to SV40. J Cell Physiol. 1968;72:141–148. doi: 10.1002/jcp.1040720208. [DOI] [PubMed] [Google Scholar]
- Anderson ME. Glutathione: an overview of biosynthesis and modulation. Chem Biol Interact. 1998;111–112:1–14. doi: 10.1016/S0009-2797(97)00146-4. [DOI] [PubMed] [Google Scholar]
- Armstrong JS, Steinauer KK, Hornung B, Irish JM, Lecane P, Birrell GW, Peehl DM, Knox SJ. Role of glutathione depletion and reactive oxygen species generation in apoptotic signaling in a human B lymphoma cell line. Cell Death Differ. 2002;9:252–263. doi: 10.1038/sj.cdd.4400959. [DOI] [PubMed] [Google Scholar]
- Bacher S, Schmitz ML. The NF-kappaB pathway as a potential target for autoimmune disease therapy. Curr Pharm Des. 2004;10:2827–2837. doi: 10.2174/1381612043383584. [DOI] [PubMed] [Google Scholar]
- Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Baldwin AS., Jr Series introduction: the transcription factor NF-kappaB and human disease. J Clin Investig. 2001;107:3–6. doi: 10.1172/JCI11891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bataller R, et al. NADPH oxidase signal transduces angiotensin II in hepatic stellate cells and is critical in hepatic fibrosis. J Clin Investig. 2003;112:1383–1394. doi: 10.1172/JCI18212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- Bitzer M, von Gersdorff G, Liang D, Dominguez-Rosales A, Beg AA, Rojkind M, Bottinger EP. A mechanism of suppression of TGF-beta/SMAD signaling by NF-kappa B/RelA. Genes Dev. 2000;14:187–197. [PMC free article] [PubMed] [Google Scholar]
- Bondi CD, Manickam N, Lee DY, Block K, Gorin Y, Abboud HE, Barnes JL. NAD(P)H oxidase mediates TGF-beta1-induced activation of kidney myofibroblasts. J Am Soc Nephrol. 2010;21:93–102. doi: 10.1681/ASN.2009020146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callicott RJ, Womack JE. Real-time PCR assay for measurement of mouse telomeres. Comp Med. 2006;56:17–22. [PubMed] [Google Scholar]
- Chariot A. The NF-kappaB-independent functions of IKK subunits in immunity and cancer. Trends Cell Biol. 2009;19:404–413. doi: 10.1016/j.tcb.2009.05.006. [DOI] [PubMed] [Google Scholar]
- Chen F, Castranova V, Li Z, Karin M, Shi X. Inhibitor of nuclear factor kappaB kinase deficiency enhances oxidative stress and prolongs c-Jun NH2-terminal kinase activation induced by arsenic. Cancer Res. 2003;63:7689–7693. [PubMed] [Google Scholar]
- Chen F, Lu Y, Castranova V, Li Z, Karin M. Loss of Ikkbeta promotes migration and proliferation of mouse embryo fibroblast cells. J Biol Chem. 2006;281:37142–37149. doi: 10.1074/jbc.M603631200. [DOI] [PubMed] [Google Scholar]
- Chen Y, Johansson E, Fan Y, Shertzer HG, Vasiliou V, Nebert DW, Dalton TP. Early onset senescence occurs when fibroblasts lack the glutamate–cysteine ligase modifier subunit. Free Radic Biol Med. 2009;47:410–418. doi: 10.1016/j.freeradbiomed.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtois G, Gilmore TD. Mutations in the NF-kappaB signaling pathway: implications for human disease. Oncogene. 2006;25:6831–6843. doi: 10.1038/sj.onc.1209939. [DOI] [PubMed] [Google Scholar]
- Datto MB, Frederick JP, Pan L, Borton AJ, Zhuang Y, Wang XF. Targeted disruption of Smad3 reveals an essential role in transforming growth factor beta-mediated signal transduction. Mol Cell Biol. 1999;19:2495–2504. doi: 10.1128/MCB.19.4.2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debacq-Chainiaux F, Pascal T, Boilan E, Bastin C, Bauwens E, Toussaint O. Screening of senescence-associated genes with specific DNA array reveals the role of IGFBP-3 in premature senescence of human diploid fibroblasts. Free Radic Biol Med. 2008;44:1817–1832. doi: 10.1016/j.freeradbiomed.2008.02.001. [DOI] [PubMed] [Google Scholar]
- Dennler S, Prunier C, Ferrand N, Gauthier JM, Atfi A. c-Jun inhibits transforming growth factor beta-mediated transcription by repressing Smad3 transcriptional activity. J Biol Chem. 2000;275:28858–28865. doi: 10.1074/jbc.M910358199. [DOI] [PubMed] [Google Scholar]
- DiDonato JA, Mercurio F, Karin M. NF-kappaB and the link between inflammation and cancer. Immunol Rev. 2012;246:379–400. doi: 10.1111/j.1600-065X.2012.01099.x. [DOI] [PubMed] [Google Scholar]
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumont P, Burton M, Chen QM, Gonos ES, Frippiat C, Mazarati JB, Eliaers F, Remacle J, Toussaint O. Induction of replicative senescence biomarkers by sublethal oxidative stresses in normal human fibroblast. Free Radic Biol Med. 2000;28:361–373. doi: 10.1016/S0891-5849(99)00249-X. [DOI] [PubMed] [Google Scholar]
- Fleckenstein K, Zgonjanin L, Chen L, Rabbani Z, Jackson IL, Thrasher B, Kirkpatrick J, Foster WM, Vujaskovic Z. Temporal onset of hypoxia and oxidative stress after pulmonary irradiation. Int J Radiat Oncol Biol Phys. 2007;68:196–204. doi: 10.1016/j.ijrobp.2006.12.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freudenberg JM, Joshi VK, Hu Z, Medvedovic M. CLEAN: clustering enrichment analysis. BMC Bioinform. 2009;10:234. doi: 10.1186/1471-2105-10-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freudlsperger C, Bian Y, Contag WS, Burnett J, Coupar J, Yang X, Chen Z, Van WC. TGF-beta and NF-kappaB signal pathway cross-talk is mediated through TAK1 and SMAD7 in a subset of head and neck cancers. Oncogene. 2013;32:1549–1559. doi: 10.1038/onc.2012.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geh E, Meng Q, Mongan M, Wang J, Takatori A, Zheng Y, Puga A, Lang RA, Xia Y. Mitogen-activated protein kinase kinase kinase 1 (MAP3K1) integrates developmental signals for eyelid closure. Proc Natl Acad Sci USA. 2011;108:17349–17354. doi: 10.1073/pnas.1102297108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerondakis S, Grumont R, Gugasyan R, Wong L, Isomura I, Ho W, Banerjee A. Unravelling the complexities of the NF-kappaB signalling pathway using mouse knockout and transgenic models. Oncogene. 2006;25:6781–6799. doi: 10.1038/sj.onc.1209944. [DOI] [PubMed] [Google Scholar]
- Gingery A, Bradley EW, Pederson L, Ruan M, Horwood NJ, Oursler MJ. TGF-beta coordinately activates TAK1/MEK/AKT/NFkB and SMAD pathways to promote osteoclast survival. Exp Cell Res. 2008;314:2725–2738. doi: 10.1016/j.yexcr.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgio M, Trinei M, Migliaccio E, Pelicci PG. Hydrogen peroxide: a metabolic by-product or a common mediator of ageing signals? Nat Rev Mol Cell Biol. 2007;8:722–728. doi: 10.1038/nrm2240. [DOI] [PubMed] [Google Scholar]
- Hecker L, Vittal R, Jones T, Jagirdar R, Luckhardt TR, Horowitz JC, Pennathur S, Martinez FJ, Thannickal VJ. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat Med. 2009;15:1077–1081. doi: 10.1038/nm.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441:431–436. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- Karin M. The IkappaB kinase: a bridge between inflammation and cancer. Cell Res. 2008;18:334–342. doi: 10.1038/cr.2008.30. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Hawke N, Baldwin AS. NF-kappaB and IKK as therapeutic targets in cancer 3. Cell Death Differ. 2006;13:738–747. doi: 10.1038/sj.cdd.4401877. [DOI] [PubMed] [Google Scholar]
- Lavon I, et al. High susceptibility to bacterial infection, but no liver dysfunction, in mice compromised for hepatocyte NF-kappaB activation. Nat Med. 2000;6:573–577. doi: 10.1038/75057. [DOI] [PubMed] [Google Scholar]
- Li X, Massa PE, Hanidu A, Peet GW, Aro P, Savitt A, Mische S, Li J, Marcu KB. IKKalpha, IKKbeta, and NEMO/IKKgamma are each required for the NF-kappa B-mediated inflammatory response program. J Biol Chem. 2002;277:45129–45140. doi: 10.1074/jbc.M205165200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo JL, Kamata H, Karin M. IKK/NF-kappaB signaling: balancing life and death–a new approach to cancer therapy. J Clin Investig. 2005;115:2625–2632. doi: 10.1172/JCI26322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis 1. Cell. 2005;121:977–990. doi: 10.1016/j.cell.2005.04.014. [DOI] [PubMed] [Google Scholar]
- May MJ, Madge LA. Caspase inhibition sensitizes inhibitor of NF-kappaB kinase beta-deficient fibroblasts to caspase-independent cell death via the generation of reactive oxygen species. J Biol Chem. 2007;282:16105–16116. doi: 10.1074/jbc.M611115200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntyre KW, Shuster DJ, Gillooly KM, Dambach DM, Pattoli MA, Lu P, Zhou XD, Qiu Y, Zusi FC, Burke JR. A highly selective inhibitor of I kappa B kinase, BMS-345541, blocks both joint inflammation and destruction in collagen-induced arthritis in mice. Arthritis Rheum. 2003;48:2652–2659. doi: 10.1002/art.11131. [DOI] [PubMed] [Google Scholar]
- Medvedovic M, et al. Influence of fatty acid diets on gene expression in rat mammary epithelial cells. Physiol Genom. 2009;38(1):80–88. doi: 10.1152/physiolgenomics.00007.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajan RP, Chen F, Li W, Vig E, Harrington MA, Nakshatri H, Chen Y. Repression of transforming-growth-factor-beta-mediated transcription by nuclear factor kappaB. Biochem J. 2000;348(Pt 3):591–596. doi: 10.1042/bj3480591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson G, Wordsworth J, Wang C, Jurk D, Lawless C, Martin-Ruiz C, von Zglinicki T. A senescent cell bystander effect: senescence-induced senescence. Aging Cell. 2012;11:345–349. doi: 10.1111/j.1474-9726.2012.00795.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Dea EL, Barken D, Peralta RQ, Tran KT, Werner SL, Kearns JD, Levchenko A, Hoffmann A. A homeostatic model of IkappaB metabolism to control constitutive NF-kappaB activity. Mol Syst Biol. 2007;3:111. doi: 10.1038/msb4100148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasparakis M. Regulation of tissue homeostasis by NF-kappaB signalling: implications for inflammatory diseases. Nat Rev Immunol. 2009;9:778–788. doi: 10.1038/nri2655. [DOI] [PubMed] [Google Scholar]
- Passos JF, et al. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol Syst Biol. 2010;6:347. doi: 10.1038/msb.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Z, Peng L, Fan Y, Zandi E, Shertzer HG, Xia Y. A critical role for IkappaB kinase beta in metallothionein-1 expression and protection against arsenic toxicity. J Biol Chem. 2007;282:21487–21496. doi: 10.1074/jbc.M702510200. [DOI] [PubMed] [Google Scholar]
- Peng Z, Geh E, Chen L, Meng Q, Fan Y, Sartor M, Shertzer HG, Liu ZG, Puga A, Xia Y (2010) IkB kinase b regulates redox homeostasis by controlling the constitutive levels of glutathione. Mol Pharmacol 77(5):784–792 [DOI] [PMC free article] [PubMed]
- Perkins ND. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol. 2007;8:49–62. doi: 10.1038/nrm2083. [DOI] [PubMed] [Google Scholar]
- Phan SH. The myofibroblast in pulmonary fibrosis. Chest. 2002;122:286S–289S. doi: 10.1378/chest.122.6_suppl.286S. [DOI] [PubMed] [Google Scholar]
- Polzer K, Baeten D, Soleiman A, Distler J, Gerlag DM, Tak PP, Schett G, Zwerina J. Tumour necrosis factor blockade increases lymphangiogenesis in murine and human arthritic joints. Ann Rheum Dis. 2008;67:1610–1616. doi: 10.1136/ard.2007.083394. [DOI] [PubMed] [Google Scholar]
- Roy S, Dontamalla SK, Mondru AK, Sannigrahi S, Veerareddy PR. Downregulation of apoptosis and modulation of TGF-beta1 by sodium selenate prevents streptozotocin-induced diabetic rat renal impairment. Biol Trace Elem Res. 2011;139:55–71. doi: 10.1007/s12011-010-8635-z. [DOI] [PubMed] [Google Scholar]
- Ruocco MG, Maeda S, Park JM, Lawrence T, Hsu LC, Cao Y, Schett G, Wagner EF, Karin M. IkB kinase IKKb, but not IKKa, is a critical mediator of osteoclast survival and is required for inflammation-induced bone loss. J Exp Med. 2005;201:1677–1687. doi: 10.1084/jem.20042081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakon S, et al. NF-kappaB inhibits TNF-induced accumulation of ROS that mediate prolonged MAPK activation and necrotic cell death. EMBO J. 2003;22:3898–3909. doi: 10.1093/emboj/cdg379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartor MA, Tomlinson CR, Wesselkamper SC, Sivaganesan S, Leikauf GD, Medvedovic M. Intensity-based hierarchical Bayes method improves testing for differentially expressed genes in microarray experiments. BMC Bioinform. 2006;7:538. doi: 10.1186/1471-2105-7-538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartor MA, Leikauf GD, Medvedovic M. LRpath: a logistic regression approach for identifying enriched biological groups in gene expression data. Bioinformatics. 2009;25:211–217. doi: 10.1093/bioinformatics/btn592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnekenburger M, Peng L, Puga A. HDAC1 bound to the Cyp1a1 promoter blocks histone acetylation associated with Ah receptor-mediated trans-activation. Biochim Biophys Acta. 2007;1769:569–578. doi: 10.1016/j.bbaexp.2007.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen CK, Roy S. Oxygenation state as a driver of myofibroblast differentiation and wound contraction: hypoxia impairs wound closure. J Investig Dermatol. 2010;130:2701–2703. doi: 10.1038/jid.2010.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M, Fuentes ME, Yamaguchi K, Durnin MH, Dalrymple SA, Hardy KL, Goeddel DV. Embryonic lethality, liver degeneration, and impaired NF-kappa B activation in IKK-beta-deficient mice. Immunity. 1999;10:421–429. doi: 10.1016/S1074-7613(00)80042-4. [DOI] [PubMed] [Google Scholar]
- Tojima Y, et al. NAK is an IkappaB kinase-activating kinase. Nature. 2000;404:778–782. doi: 10.1038/35008109. [DOI] [PubMed] [Google Scholar]
- Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- Verrecchia F, Vindevoghel L, Lechleider RJ, Uitto J, Roberts AB, Mauviel A. Smad3/AP-1 interactions control transcriptional responses to TGF-beta in a promoter-specific manner. Oncogene. 2001;20:3332–3340. doi: 10.1038/sj.onc.1204448. [DOI] [PubMed] [Google Scholar]
- Weyemi U. ROS-generating NADPH oxidase NOX4 is a critical mediator in oncogenic H-Ras-induced DNA damage and subsequent senescence. Oncogene. 2011;31(9):1117–1129. doi: 10.1038/onc.2011.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Wang W, Hayashi Y, Jester JV, Birk DE, Gao M, Liu CY, Kao WW, Karin M, Xia Y. A role for MEK kinase 1 in TGF-beta/activin-induced epithelium movement and embryonic eyelid closure. EMBO J. 2003;22:4443–4454. doi: 10.1093/emboj/cdg440. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.