

Abstract
We describe a large Lebanese family with two affected members, a young female proband and her male cousin, who had multisystem involvement including profound global developmental delay, severe hypotonia and weakness, respiratory insufficiency, blindness, and lactic acidemia—findings consistent with an underlying mitochondrial disorder. Whole-exome sequencing was performed on DNA from the proband and both parents. The proband and her cousin carried compound heterozygous mutations in the PMPCA gene that encodes for α-mitochondrial processing peptidase (α-MPP), a protein likely involved in the processing of mitochondrial proteins. The variants were located close to and postulated to affect the substrate binding glycine-rich loop of the α-MPP protein. Functional assays including immunofluorescence and western blot analysis on patient's fibroblasts revealed that these variants reduced α-MPP levels and impaired frataxin production and processing. We further determined that those defects could be rescued through the expression of exogenous wild-type PMPCA cDNA. Our findings link defective α-MPP protein to a severe mitochondrial disease.
Keywords: diffuse cerebellar atrophy, generalized hypotonia due to defect at the neuromuscular junction, hydrocephalus, mitochondrial encephalopathy
INTRODUCTION
The majority of mitochondrial proteins are encoded as precursor proteins from nuclear DNA, synthesized in the cytosol, and then imported into the mitochondria. These precursor proteins contain amino-terminal extensions called targeting peptides (TPs) that function as import signals and are recognized by import receptors present on the mitochondrial membrane. Once imported into the mitochondria, the TP is cleaved off and the protein can fold and fully mature, allowing for optimal protein function. The majority of these essential cleavage reactions are catalyzed by MPP (mitochondrial processing peptidase), a matrix-localized, soluble heterodimer of α-MPP and β-MPP subunits, encoded by PMPCA and PMPCB, respectively (Kucera et al. 2013). In budding yeast, when either MPP subunit is eliminated, the cells continue to import proteins into the mitochondria, but they fail to cleave TPs, the precursor proteins accumulate, and the cells fail to grow (Geli and Glick 1990). The limited studies available on PMPCA and PMPCB have suggested that the catalytic activity of MPP is confined to the β-MPP subunit, whereas the α-MPP subunit seems to be involved in substrate recognition (Luciano et al. 1997; Gakh et al. 2002). Existing data have suggested that a flexible, glycine-rich loop within the α-MPP subunit is largely responsible for substrate affinity and MPP functionality (Taylor et al. 2001). This loop is a key structural element of MPP that moves the precursor protein substrate toward the active site through a multistep process (Kucera et al. 2013).
As appropriate cleavage by processing peptidases is necessary for protein stability, correct localization, and proper assembly of protein complexes, some have hypothesized a link between MPP dysfunction and human disease (Teixeira and Glaser 2013). One such example is the association between MPP and Friedreich's ataxia (FRDA), an autosomal-recessive neurodegenerative disease in which the mitochondrial protein frataxin is deficient (Palau 2001; Wilson 2003; Lim et al. 2007). It has been shown that frataxin is processed by MPP and mutations in frataxin alter this interaction, resulting in frataxin deficiency and impaired function (Koutnikova et al. 1998; Cavadini et al. 2000). Further evidence for this hypothesis is suggested by the finding that without proper processing by MPP, precursor proteins, generally more unstable than their processed mature form, are readily degraded (Mukhopadhyay et al. 2007). Here we describe two first cousins with a complex multisystem mitochondrial disease carrying compound heterozygous missense variants in PMPCA associated with a marked reduction in the levels of processed frataxin. We also demonstrated rescue in the proband's fibroblasts by overexpressing wild-type (WT) PMPCA using a lentiviral vector.
RESULTS
Clinical Presentation
We describe a large family with two members presenting with a severe mitochondrial disease. The pedigree is shown in Figure 1A. The proband is a 6-yr-old female of Lebanese descent who was hospitalized at the age of 6 mo with developmental delay and failure to thrive. Her neurological examination revealed bilateral ptosis, ophthamoplegia, mild generalized hypotonia, and weakness. Laboratory tests revealed lactic acidemia and elevated transaminases. A brain magnetic resonance imaging (MRI) revealed cerebellar and mild diffuse cerebral atrophy (Fig. 1B,C). Magnetic resonance spectroscopy revealed a lactate peak (data not shown). Over the subsequent years, her condition deteriorated and by 6 yr she was completely blind and had severe optic atrophy. She was minimally responsive and has been immobile because of profound generalized weakness. She has chronic hypoventilation and a tracheostomy was placed for ventilator support. She has persistent lactic acidemia, whereas her transaminases gradually normalized. MRIs over the years have shown progression of generalized brain atrophy, with diffuse parenchymal volume loss and areas of gliosis and severe ventriculomegaly involving both of the lateral ventricles as well as the third and fourth ventricles (Fig. 1D,E).
Figure 1.

Clinical and histopathological findings in a family with PMPCA mutations. (A) Pedigree of the extended family carrying PMPCA mutations, Arrow indicates the proband. (B–G) Magnetic resonance imaging (MRI) head findings in the proband (B–E) and affected cousin (F,G). MRI at 6 mo of age revealed cerebellar atrophy with enlarged interfolial spaces (B,C), whereas at 3 yr of age there was marked cerebral and cerebellar atrophy with enlarged ventricles (D,E). The cousin's head MRI at 8 mo of age revealed cerebral and cerebellar atrophy (F,G). (H–N) Histopathological findings from the muscle biopsy in the proband (H), the affected cousin (I–K), and electron microscopic findings in the proband (L–N). Fiber size variation was noted on H&E (hematoxylin and eosin) staining (H), whereas trichrome staining revealed coarse granular staining suggesting enlarged mitochondria (marked by arrows) (I), and was also noted on NADH (nicotinamide adenine dinucleotide) staining (J). Toludine blue staining showed large prominent lipid droplets (arrows) (K). Electron microscopy revealed scattered structurally abnormal mitochondria with electron dense aggregates within cristae (arrows) (L–N).
Her muscle biopsy findings included mild variation in fiber size (Fig. 1H) and an unusual staining pattern with cytochrome oxidase (COX) characterized by clumping of the reaction product in both subsarcolemmal and intermyofibrillar distribution suggestive of a mitochondrial myopathy. Similar findings were present on staining by succinate dehydrogenase, and electron microscopy revealed scattered structurally abnormal mitochondria. The mitochondria were enlarged and elongated with electron dense aggregates within cristae (Fig. 1L–N). Mitochondrial respiratory electron transport chain (ETC) enzyme analysis was performed on skin fibroblasts, and no enzymatic defect was detected in any component of the respiratory complex. Furthermore, activities of pyruvate carboxylase, phosphoenolpyruvate carboxykinase, and citrate synthase in cultured skin fibroblasts were normal. The whole mitochondrial genome sequence of the patient was negative for point mutations or deletions. Additional DNA sequencing was performed for several mitochondrial nuclear genes followed by comprehensive mitochondrial nuclear gene panel analysis of 101 genes that revealed no pathogenic mutations. Her chromosomal microarray analysis did not reveal any significant copy-number variations.
Patient 2 is the first cousin of the proband. Their fathers are brothers and their mothers are first cousins (Fig. 1A). His presentation at 6 mo of age was similar to that of the proband with developmental delay, weakness, and ataxia. He had persistent lactic academia, as well. Brain MRIs showed marked cerebellar and mild cerebral atrophy (Fig. 1F,G). He developed severe hypertrophic left ventricular cardiomyopathy and liver failure and died at 14 mo from respiratory failure in the setting of having multiple medical complications. A limited autopsy including liver and skeletal muscles was performed. Skeletal muscle findings revealed the presence of multiple fibers with subsarcolemmal clusters of abnormal mitochondria (Fig. 1I,J) and lipid droplets indicative of mitochondrial dysfunction (Fig. 1K). The reported electron microscopic (EM) abnormalities included giant mitochondria (megaconial) and mitochondria with concentric cristae. ETC analysis on the muscle showed normal activity of complexes I, II, III, and IV. Liver histopathology revealed mild hepatic fibrosis, marked cholestasis, and microvesicular steatosis. The electron microscopy revealed glycogen depletion in hepatocytes. Mitochondria appeared swollen with increased matrix and margination of cristae, with matrical inclusions of neutral fat and vacuolar structures. The ETC activity in the liver specimen was suggestive of defects in complexes II and III.
Exome Sequencing
Exome sequencing and analysis was performed on the proband and both parents as described in the Methods section and the coverage is described in Table 1. Two variants in the PMPCA gene were identified in the proband that segregated appropriately in the parents as described in Table 2. PMPCA was a prime candidate given that it has been ascribed a role in the mitochondria based on a large integrative genomic database of putative mitochondrial genes (Pagliarini et al. 2008). The proband and the cousin were confirmed to be compound heterozygous for the two missense PMPCA mutations (Chr 9: 139313082; NM_015160, c.1066G>A; p.G356S and Chr 9: 139313299; NM_01560, c.1129G>A; p.A377T) by Sanger sequencing with appropriate parental segregation of the mutant alleles. (Fig. 2A). Several unaffected siblings (III:1, III:3, III:4, III:5, and III:6) were WT or heterozygous for a single variant. The identified mutations were not present in any of the publicly available databases including the 1000 Genomes, Exome Variant Server, and Exome Aggregation Consortium (ExAC) databases. Several in silico missense variant prediction methods, including PolyPhen-2, MutationTaster, and SIFT, suggested that the missense variants we identified were likely to be deleterious and the altered amino acids were extremely well conserved across evolution (Fig. 2B). The locations of the mutations suggest that both could affect the functionality of the α-MPP glycine-rich loop. The significance of these residues is further corroborated by the fact that p.A377 is conserved across vertebrates and invertebrates, whereas p.G356 is conserved across vertebrates, invertebrates, and monera.
Table 1.
Whole-exome sequencing coverage
| Sample | Total aligned reads | Average read coveragea | % Coding genesa with >10-fold coverage |
|---|---|---|---|
| Proband | 31,863,723 | 41.9 | 84.1 |
| Father | 42,287,619 | 54.6 | 87.1 |
| Mother | 33,224,612 | 42.9 | 84.4 |
aBased upon the consensus coding DNA sequence (CCDS) database.
Table 2.
PMPCA (NM_015160) variants
| Chr:Position GRCh37 (hg19) | HGVS cDNA | HGVS protein | Predicted effect | dbSNP ID | Genotype | Parent | MAF (%) ExAC database |
|---|---|---|---|---|---|---|---|
| 9:139313082 | c.1066G>A | p.G356S | Missense | Absent | Heterozygous | Mother | 0 |
| 9:139313299 | c.1129G>A | p.A377T | Missense | Absent | Heterozygous | Father | 0 |
HGVS, Human Genome Variation Society; dbSNP, Database for Short Genetic Variations; MAF, minor allele frequency; ExAC, Exome Aggregation Consortium.
Figure 2.
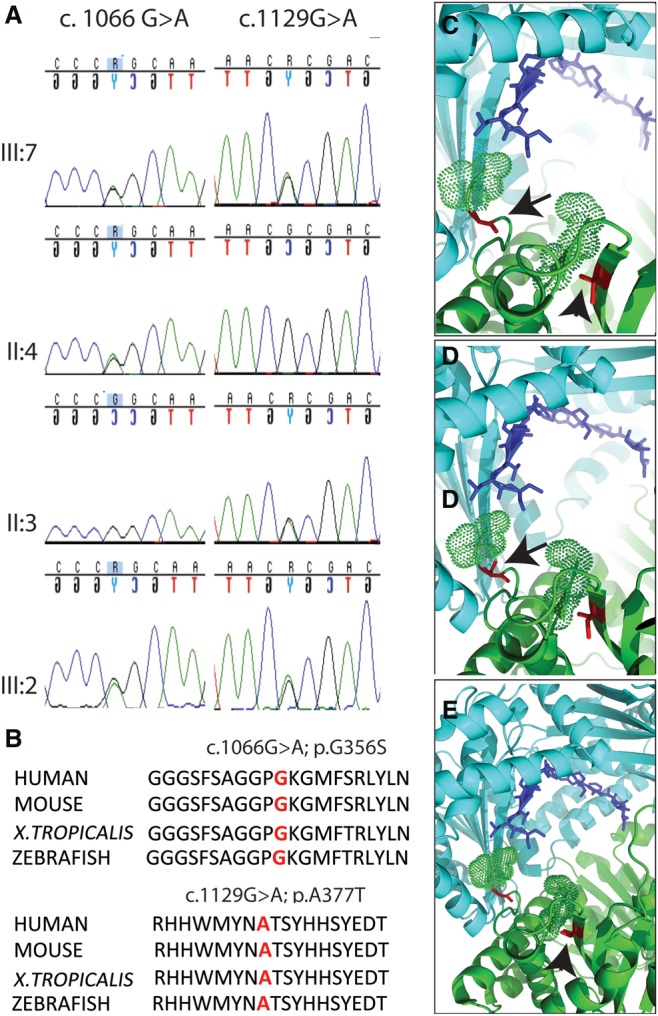
Molecular findings in the family with PMPCA mutations. (A) Distribution of the two heterozygous mutations c.1066G>A (p.G356S) and c. 1129G>A (p.A377T) in the proband, her affected cousin, and both their sets of parents. (B) Conservation of the amino acid residues across the vertebrates. (C–E) The crystal structure of yeast PMPCA with its substrate (PDB: 1HR8) is presented to show the location of the mutations. (C) WT-PMPCA is a heterodimer that consists of two subunits, α and β. The β-subunit is colored in cyan, whereas the α-subunit is colored in green. The blue stick represents a substrate. Part of the catalytic loop (residues 345–355) is shown in dots, with a few residues missing in the middle because of high flexibility. (D,E) The two mutation sites colored in red, depicted by arrow p.G356S (D) and arrowhead A377T (E), are located in and 20 residues downstream from the catalytic loop. The increased side chain volume caused by the mutations likely restricts the dynamic of the catalytic loop, which affects the catalytic activity of PMPCA.
Computer modeling revealed that the mutations were located in p.G356S or 20 residues downstream (p.A377T) of the glycine-rich catalytic loop (residues 345–358), which would be predicted to restrict the dynamic movements of this catalytic loop and thereby affect the catalytic activity or substrate binding of PMPCA (Fig. 2C–E). This loop is highly conserved among vertebrates and invertebrates, and its depletion has been shown to reduce the enzyme's affinity for substrate peptides and its activity (Nagao et al. 2000; Kucera et al. 2013). The glycine-rich loop is positioned at the opening to the space between the subunits, such that it is exposed to the external substrate and the zinc-binding pocket. The loop needs to be highly flexible as the process of substrate translocation causes it to undergo major conformational changes (Kucera et al. 2013). The intraloop p.G356S mutation may directly alter the conformation and affinity of the loop by replacing a small glycine with a bulkier and more nucleophilic serine residue. This could interfere with the flexibility of the loop, as well as with the ability of the loop to recognize and bind to substrates, change conformation following early TP cleavage in multistep processing, and maintain heterodimer stability. The downstream p.A377T mutation replaces the alanine residue with a threonine bearing an alcohol group. Moreover, the side chain of threonine points toward to the catalytic loop. The enhanced polarity and bulkier size of p. A377T mutation could additionally affect the flexibility of the catalytic loop and the stability of the heterodimer.
Functional Effects of the Mutations
To evaluate the functional effects of PMPCA mutations, fibroblasts from the proband and two age-matched controls were obtained. Immunofluorescence experiments were performed using anti-PMPCA and anti-mitochondrial antibodies that revealed a reduction in PMPCA staining along with several abnormally enlarged mitochondria (Fig. 3A–F).
Figure 3.

Effects of PMPCA mutations on fibroblasts cultured from the proband and rescue experiments. (A–F) Immunofluorescence showing swollen mitochondria (arrows) and abnormal PMPCA staining compared with control. (G) Western blot analysis revealed a reduction in mature frataxin (18 kDa) and an increase in unprocessed frataxin (23 kDa). (H) WT PMPCA cDNA overexpression in the proband's fibroblasts resulted in normalization of frataxin processing with high amounts of processed frataxin and reduction in unprocessed form. Empty vector rescue was used as control showing absent processed frataxin and a high amount of unprocessed frataxin. Further, PMPCA levels are much higher in the rescued cells than in the one rescued by the empty vector.
One of the proteins extensively processed inside mitochondria is frataxin, encoded by FXN, and known to interact with MPP (Koutnikova et al. 1998; Branda et al. 1999; Gordon et al. 1999; Cavadini et al. 2000). Frataxin deficiency causes FRDA, an autosomal-recessive neurodegenerative disease (Palau 2001; Wilson 2003; Lim et al. 2007). An imbalance in the ratio of immature to mature frataxin and general depletion of the processed forms characterizes FRDA (Palau 2001; Wilson 2003; Lim et al. 2007). Studies have shown that FXN mutations alter its interaction with MPP, resulting in impaired frataxin function (Koutnikova et al. 1998). We performed experiments on cultured fibroblasts from the proband and two age-matched controls for frataxin processing and found abnormal processing in the proband. The fibroblasts from the proband contained higher levels of the immature form of frataxin (23 kDa) and lower levels of the processed or mature form (18 kDa) (Fig. 3G).
To directly show that the observed reduction in mature frataxin is due to the PMPCA mutations, we performed rescue experiments using lentiviral transduction of fibroblasts from Patient 1. The PMPCA-rescued cells from the proband revealed a reduction in the unprocessed form of frataxin (23 kDa) and an increase in the levels of the processed (mature) form (18 kDa) in comparison to the control rescue (Fig. 3H). The levels of PMPCA increased appropriately in the PMPCA-rescued cells as compared with the control (Fig. 3H). This directly demonstrates that the mutant alleles in PMPCA are loss of function and can be rescued by transduction of the WT gene in primary patient cells. Moreover, this also shows that the processing defect observed in the patient cells is due to loss of function of PMPCA.
DISCUSSION
This study describes a severe mitochondrial disease due to PMPCA mutations in a large family, which we show is associated with altered levels of mature frataxin. Whereas a recent study described cerebellar ataxia as a presentation of PMPCA mutations (Jobling et al. 2015), the phenotype in our family is more severe and typical of a mitochondrial disease. The increased severity of disease in our family may be due to direct perturbation of the glycine-rich catalytic loop, which was not seen in the patients described by Jobling et al. (2015). The p.A377T variant, present in homozygous state in their patients, is downstream from the catalytic loop and may have milder effects. The proband had severe muscle weakness, extensive brain atrophy, visual impairment, and respiratory defects, and the affected cousin in addition had cardiomyopathy and liver complications and died at an early age from resultant complications. The observed phenotype may be related to impaired PMPCA function due to a reduction in its level and the resultant abnormal processing of frataxin and other mitochondrial proteins. Importantly, the observed processing defects were rescued by ectopic expression of the wild-type form of PMPCA. The mutations observed in both affected family members appear to affect the critical glycine-rich catalytic loop within the α-subunit of the MPP enzyme, which is responsible for moving the precursor protein substrates toward the active site on the β-subunit for processing to mature forms (Taylor et al. 2001; Kucera et al. 2013).
We have described abnormal frataxin processing in this report, but there are more than 1000 nuclear-encoded proteins that undergo mitochondrial import and amino-terminal TP processing in the mitochondria. We expect that the maturation of several additional mitochondrial proteins may be affected by an α-MPP functional deficit and therefore contribute to the severe phenotype observed in the family. We have tested several additional mitochondrial proteins for abnormal processing using the patient's fibroblasts. One of them is PINK1, potentially affected by PMPCA levels (Greene et al. 2012), but we did not find a change in PINK1 amounts in patient's fibroblasts (data not shown). Similarly, members of the electron transport chain family (COX4I1, ATP5A1, and ATP5B) were tested without any evidence of a defect in their processing (data not shown). These findings indicate that either the above proteins are processed independently of MPP or that the mutations in PMPCA may not affect their recognition by the MPP for processing. A search for additional mitochondrial proteins that may be affected by PMPCA mutations is currently underway.
In summary, by combining exome-sequencing and variant-segregation analysis across a family with a unique syndrome along with comparative genomic, structural, and functional rescue experiments, we have been able to demonstrate causality for a mitochondrial disease resulting from PMPCA disruption (MacArthur et al. 2014). Additionally, by testing a multitude of candidate mitochondrial import proteins, we will be able to establish the mechanism by which this disruption leads to specific phenotypes, as observed in these patients with a multisystemic disorder.
METHODS
Genetic Studies
The proband (Patient 1), her parents, the affected cousin (Patient 2) and his parents, and other family members were enrolled in an institutional review board (IRB)-approved study at Boston Children's Hospital (BCH) to perform whole-exome sequencing. Blood samples were collected and processed for DNA extraction by the Research Connection Biobank Core at BCH. DNA from the proband and both parents was sent for whole-exome sequencing (WES) to Axeq Technologies. Samples were prepared as an Illumina sequencing library and enriched for exonic sequences using the Illumina Exome Enrichment protocol. The captured libraries were sequenced using Illumina HiSeq 2000 Sequencers. The reads were mapped to the human genome assembly UCSC hg19 using Burrows–Wheeler Alignment (BWA version 0.5.8). Single-nucleotide polymorphisms (SNPs) and small insertions/deletions were called with SAMtools (version 0.1.7). The resulting VCF (variant call format) files were filtered to include nonsynonymous, splice site, and indel variants with an allele frequency <0.001 in the National Heart, Lung, and Blood Institute (NHLBI) Exome Variant Server (EVS) database (http://evs.gs.washington.edu/EVS/) or <0.01 in 1000 Genomes Project, phase 3 (http://www.1000genomes.org). The Variant Explorer pipeline developed by the authors (K.S.-A. and K.M.) was used for annotating all variants according to the minor-allele-frequency data described above. Polymerase chain reaction (PCR) was performed to amplify the PMPCA mutations using the primers PMPCA_F: CTTCATCCCCTTTGCAGTGT and PMPCA_R: TACAACGTGGCTGTCTCCG, and the amplified DNA returned was sent for Sanger sequencing. The results were analyzed using the Sequencer 5.0 software and aligned with the WT gene sequence available from NCBI Blast.
Protein Expression Study
Western blot was performed to study the level of PMPCA protein on the fibroblasts available from the patient. Fibroblasts from two control human cell lines (ATCC-2127 and ATCC-2104) were used as controls. Anti-PMPCA antibody (NBP1-89126, 1:100 dilution, Novus Biologicals) was used to probe the specific protein band at 58 kDa. The results of the study were visualized using enhanced chemiluminescence using the program Quantity One 4.2.1 (Bio-Rad) and were compared with appropriate age- and tissue-matched controls normalized for the loading control glyceraldehyde-3-phosphate dehydrogenase (GAPDH) or β-tubulin.
To identify the probable interactions of PMPCA with other proteins that may be processed in the mitochondria, we also performed quantitative protein analysis for CO4I1, frataxin, PINK1, and a mitochondrial antibody cocktail (ab3298, Abcam).
Immunofluorescence on Skin Fibroblasts
Cultured patient fibroblasts were transferred onto chambered slides and grown to 50%–60% confluence. The fibroblasts were fixed with 4% paraformaldehyde for 20 min and 10% Triton-X was used to permeabilize the cells before staining. The fixed slides were stored and used up to a week. Rabbit anti-PMPCA from Novus Biologicals (SAB1303187; 100 µL) made in rabbit was used as a primary antibody at a ratio of 1:100 overnight at 4°C. Anti-mitochondrial antibody (mouse) from Abcam (ab 3298) was used for costaining at 1:200 overnight at 4°C.
Abcam frataxin antibody (mouse) (17A11) (ab113691) was also used at 1:200 with Novus PMPCA for colocalization studies.
Viral Transfection of Skin Fibroblasts for Rescue
Human PMPCA cDNA clone (HsCD00334734) was ordered from the Plasmid ID database and the gene was cloned into HMD lentiviral transfection vector with a green fluorescent protein (GFP) tag (to assess for those cells that have successfully been lentivirally transduced) (Ludwig et al. 2014) by restriction digestion using the following primers: PMPCA_ cloning_F:TAAGCAGGATCCATGGCGGCTGTGGTGCTGGCG and PMPCA_cloning_R: TGCTTACTCGAGCTACCGGAAGAGCCGTGCGCA.
0059-1 (patient) and 2127 (control) fibroblasts were each then transfected with HMD-PMPCA (rescue) viral transfection vector and an empty HMD vector (control) without the PMPCA insert as another control. Cells that were successfully transfected were separated from the other cells using fluorescence-activated cell sorting (FACS). The transfected cells (0059_HMD-PMPCA, 0059-HMD only, 2127_HMD-PMPCA and 2127_HMD only) were then allowed to grow and washed with media two to three times before splitting and kept frozen at −140°C for future use. Western blot was repeated with the transfected fibroblasts using PMPCA and frataxin antibodies to study the effects of the rescue.
ADDITIONAL INFORMATION
Ethics Statement
The Institutional Review Board of Boston Children's Hospital approved this research protocol (10-02-0053). Written informed consent was received from proband's parents, parents of the cousin, and other family members.
Data Deposition and Access
Our patient consent does not allow patient sequence data to be uploaded to a data repository. The NM_015160: c.1066G>A; p.G356S and c.1129G>A; p.A377T missense variants have been deposited to ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/) under accession numbers SCV000262557 and SCV000262558 and MSeqDR (https://mseqdr.org/) under accession numbers MSCV_0000001 and MSCV_0000002.
Acknowledgments
We thank the family for their support and enrollment.
Author Contributions
M.J. drafted and revised the manuscript for content, including medical writing for content, analysis or interpretation of data, and acquisition of data. I.A. contributed to study concept and design, drafting and revising the manuscript for content, including medical writing for content, analysis, or interpretation of data. J.S. contributed to the acquisition of data, analysis and interpretation of data, and revision of the manuscript for content. T.A.B., M.T., and S.K. contributed to acquisition of data. K.S.-A. contributed to analysis or interpretation of data and statistical analysis. L.C. contributed to drafting and revising the manuscript for content. F.C.G. contributed vital reagents and acquisition of data. K.M. contributed to analysis and interpretation of data and statistical analysis. H.G.L. and R.F. contributed to analysis and interpretation of data and revised the manuscript for content. V.G.S. drafted and revised the manuscript for content and contributed vital reagents and acquired, analyzed, and interpreted data. P.B.A. contributed to the study concept and design, obtained funding, drafted and revised the manuscript for content, analysis, and interpretation of data, and supervised and coordinated study.
Funding
This work was made possible by grants from the following institutions: P.B.A. was supported by the William F. Milton fund from Harvard Medical School, Boston, 1R01AR068429-01 from the National Institute of Arthritis and Musculoskeletal and Skeletal Diseases (NIAMS) of the National Institute of Health (NIH), and U19HD077671 from National Institute of Child Health and Human Development (NICHD)/National Human Genome Research Institute (NHGRI)/NIH. The Gene Discovery Core of The Manton Center for Orphan Disease Research, Boston Children's Hospital also supported the work. Sanger sequencing was performed by the Molecular Genetics Core Facility of the IDDRC at Boston Children's Hospital, which was supported by National Institutes of Health grant P30 HD18655.
Competing Interest Statement
The authors have declared no competing interest.
Referees
Neil Sondheimer
Anonymous
REFERENCES
- Branda SS, Cavadini P, Adamec J, Kalousek F, Taroni F, Isaya G. 1999. Yeast and human frataxin are processed to mature form in two sequential steps by the mitochondrial processing peptidase. J Biol Chem 274: 22763–22769. [DOI] [PubMed] [Google Scholar]
- Cavadini P, Adamec J, Taroni F, Gakh O, Isaya G. 2000. Two-step processing of human frataxin by mitochondrial processing peptidase. Precursor and intermediate forms are cleaved at different rates. J Biol Chem 275: 41469–41475. [DOI] [PubMed] [Google Scholar]
- Gakh O, Cavadini P, Isaya G. 2002. Mitochondrial processing peptidases. Biochim Biophys Acta 1592: 63–77. [DOI] [PubMed] [Google Scholar]
- Geli V, Glick B. 1990. Mitochondrial protein import. J Bioenerg Biomembr 22: 725–751. [DOI] [PubMed] [Google Scholar]
- Gordon DM, Shi Q, Dancis A, Pain D. 1999. Maturation of frataxin within mammalian and yeast mitochondria: one-step processing by matrix processing peptidase. Hum Mol Genet 8: 2255–2262. [DOI] [PubMed] [Google Scholar]
- Greene AW, Grenier K, Aguileta MA, Muise S, Farazifard R, Haque ME, McBride HM, Park DS, Fon EA. 2012. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep 13: 378–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jobling RK, Assoum M, Gakh O, Blaser S, Raiman JA, Mignot C, Roze E, Durr A, Brice A, Levy N, et al. 2015. PMPCA mutations cause abnormal mitochondrial protein processing in patients with non-progressive cerebellar ataxia. Brain 138: 1505–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutnikova H, Campuzano V, Koenig M. 1998. Maturation of wild-type and mutated frataxin by the mitochondrial processing peptidase. Hum Mol Genet 7: 1485–1489. [DOI] [PubMed] [Google Scholar]
- Kucera T, Otyepka M, Matuskova A, Samad A, Kutejova E, Janata J. 2013. A computational study of the glycine-rich loop of mitochondrial processing peptidase. PLoS One 8: e74518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim F, Palomo GM, Mauritz C, Gimenez-Cassina A, Illana B, Wandosell F, Diaz-Nido J. 2007. Functional recovery in a Friedreich's ataxia mouse model by frataxin gene transfer using an HSV-1 amplicon vector. Mol Ther 15: 1072–1078. [DOI] [PubMed] [Google Scholar]
- Luciano P, Geoffroy S, Brandt A, Hernandez JF, Geli V. 1997. Functional cooperation of the mitochondrial processing peptidase subunits. J Mol Biol 272: 213–225. [DOI] [PubMed] [Google Scholar]
- Ludwig LS, Gazda HT, Eng JC, Eichhorn SW, Thiru P, Ghazvinian R, George TI, Gotlib JR, Beggs AH, Sieff CA, et al. 2014. Altered translation of GATA1 in Diamond–Blackfan anemia. Nat Med 20: 748–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur DG, Manolio TA, Dimmock DP, Rehm HL, Shendure J, Abecasis GR, Adams DR, Altman RB, Antonarakis SE, Ashley EA, et al. 2014. Guidelines for investigating causality of sequence variants in human disease. Nature 508: 469–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay A, Yang CS, Wei B, Weiner H. 2007. Precursor protein is readily degraded in mitochondrial matrix space if the leader is not processed by mitochondrial processing peptidase. J Biol Chem 282: 37266–37275. [DOI] [PubMed] [Google Scholar]
- Nagao Y, Kitada S, Kojima K, Toh H, Kuhara S, Ogishima T, Ito A. 2000. Glycine-rich region of mitochondrial processing peptidase α-subunit is essential for binding and cleavage of the precursor proteins. J Biol Chem 275: 34552–34556. [DOI] [PubMed] [Google Scholar]
- Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong SE, Walford GA, Sugiana C, Boneh A, Chen WK, et al. 2008. A mitochondrial protein compendium elucidates complex I disease biology. Cell 134: 112–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palau F. 2001. Friedreich's ataxia and frataxin: molecular genetics, evolution and pathogenesis (Review). Int J Mol Med 7: 581–589. [DOI] [PubMed] [Google Scholar]
- Taylor AB, Smith BS, Kitada S, Kojima K, Miyaura H, Otwinowski Z, Ito A, Deisenhofer J. 2001. Crystal structures of mitochondrial processing peptidase reveal the mode for specific cleavage of import signal sequences. Structure 9: 615–625. [DOI] [PubMed] [Google Scholar]
- Teixeira PF, Glaser E. 2013. Processing peptidases in mitochondria and chloroplasts. Biochim Biophys Acta 1833: 360–370. [DOI] [PubMed] [Google Scholar]
- Wilson RB. 2003. Frataxin and frataxin deficiency in Friedreich's ataxia. J Neurol Sci 207: 103–105. [DOI] [PubMed] [Google Scholar]