

Abstract
A number of epidemiological studies have implicated calcium (Ca2+) signaling as a major factor in obesity that contributes to aberrant systems metabolism. Somewhat paradoxically, obesity correlates with decreased circulating Ca2+ levels, leading to increased release of intracellular Ca2+ stores from the endoplasmic reticulum. These findings suggest that insulin resistance associated with the obese state is linked to activation of canonical Ca2+ signaling pathways. Mechanistically, increased intracellular Ca2+ binds calmodulin (CaM) to activate a set of Ca2+/CaM-dependent protein kinases. In this research resource, we explore the metabolic functions and implications of Ca2+/CaM-dependent protein kinase kinase 2 (CaMKK2) as a metabolic effector of Ca2+/CaM action. We reveal the importance of CaMKK2 for gating insulin release from pancreatic β-cells while concomitantly influencing the sensitivity of insulin-responsive tissues. To provide a better understanding of the metabolic impact of CaMKK2 loss, we performed targeted metabolomic analyses of key metabolic byproducts of glucose, fatty acid, and amino acid metabolism in mice null for CaMKK2. We quantified amino acids and acyl carnitines in 3 insulin-sensitive tissues (liver, skeletal muscle, plasma) isolated from CaMKK2−/− mice and their wild-type littermates under conditions of dietary stress (low-fat diet, normal chow, high-fat diet, and fasting), thereby unveiling unique metabolic functions of CaMKK2. Our findings highlight CaMKK2 as a molecular rheostat for insulin action and emphasize the importance of Ca2+/CaM/CaMKK2 in regulation of whole-body metabolism. These findings reveal that CaMKK2 may be an attractive therapeutic target for combatting comorbidities associated with perturbed insulin signaling.
Obesity and diabetes have emerged as 2 of the most significant public health problems in modern times. Obesity, defined as a low-grade inflammatory disease, predisposes individuals for hepatic steatosis, insulin resistance, and comorbidities of metabolic syndrome (1). In fact, obesity substantially increases the risk for type 2 diabetes, which is characterized by insulin insensitivity in peripheral tissues and the inability of pancreatic islets to produce and secrete insulin in response to elevated blood glucose (2). Production and secretion of insulin from pancreatic β-cells coupled with its action on peripheral tissues is balanced by a variety of counter-regulatory signals and the mechanism of action of many of these signals remain poorly understood.
Calcium (Ca2+) represents one such signal that has been linked to obesity and insulin resistance (3, 4). Numerous epidemiological studies implicate Ca2+ signaling as a major determinant in obesity that contributes to aberrant systems metabolism (4–7). The actions of Ca2+ are mediated through interaction with its ubiquitous receptor, calmodulin (CaM) and the Ca2+/CaM complex mediates amplification of Ca2+ signals through activation of a tightly regulated set of Ca2+/CaM-dependent protein kinases. Central to this kinase “cascade” is Ca2+/CaM-dependent protein kinase kinase 2 (CaMKK2), which becomes activated and directs the downstream actions of Ca2+/CaM-dependent protein kinase 1 (CaMKI), CaMKIV, and AMP-activated protein kinase (AMPK) upon binding Ca2+/CaM (8). We have previously reported that ablation of CaMKK2 protects against high-fat diet (HFD)-induced obesity partly through regulation of hypothalamic AMPKα signaling to control production of the orexigenic hormone, neuropeptide Y (9). In the liver, disruption of CaMKK2 also leads to loss of gluconeogenic gene expression and subsequent hypoglycemia (10). Moreover, CaMKK2-null mice maintain normal circulating insulin levels and remain glucose responsive under HFD conditions (10). These findings implicate CaMKK2 as an important determinant for maintenance of glucose homeostasis.
There has been an increasing demand to identify definitive links between metabolic anomalies and complex disorders such as obesity, diabetes, and even cancer. Considering the critical roles of Ca2+/CaM signaling cascades and CaMKK2 in these diseases, a more global and encompassing examination of the role of CaMKK2 in metabolism was needed. Addressing this need, we report here the first comprehensive, systems biology assessment of CaMKK2 function in vivo. Our findings reveal previously unappreciated roles for CaMKK2 in pancreatic β-cell function wherein CaMKK2 functions as a rheostat for insulin secretion. In peripheral insulin-sensitive tissues, expression of CaMKK2 contributes to lowering insulin sensitivity, suggesting its involvement in the metabolic progression to diseases such as obesity and type 2 diabetes. To empirically evaluate the impact of CaMKK2 on intermediate metabolites of amino acid (AA) and fatty acid oxidation in key metabolic tissues (liver, skeletal muscle, and plasma), we performed targeted quantitative metabolomics. Using this technology, we define the effect of CaMKK2 ablation on circulating plasma hormone and metabolite levels in response to a variety of metabolic stresses (feeding vs fasting and low-fat diet [LFD] vs HFD). Collectively, our findings reveal that although loss of CaMKK2 fails to alter basic islet structure, mice lacking CaMKK2 display markedly increased insulin sensitivity, contributing to their protection against insulin resistance and HFD-induced obesity. This research resource underscores the importance of Ca2+ signaling in the regulation of systems metabolism and highlights CaMKK2 as a potential therapeutic target for combatting diseases arising from perturbed insulin action.
Materials and Methods
Animal experiments
All animal experiments were performed according to protocols approved by the Animal Care Research Committee at either Baylor College of Medicine or Duke University School of Medicine. Generation of CaMKK2−/− mice has been described previously (9). CaMKK2−/− mice were maintained on a pure C57BL/6J genetic background. Generation of Tg(CaMKK2-EGFP)DF129Gsat mice has been previously described (11). CaMKK2-enhanced green fluorescent protein (EGFP) mice were used exclusively for analysis of CaMKK2 pancreatic expression. For all studies only male, age-matched (10- to 16-wk-old) mice were used to avoid metabolic complications of unsynchronized estrous in female mice. Mice were maintained in a temperature controlled (23°C) facility with a 12-hour light, 12-hour dark cycle. CaMKK2+/+ and CaMKK2−/− mice were fed either 2920X Teklad Global rodent chow, HFD or control LFD ad libitum or subjected to 24–48 hours of fasting, but with free access to water as indicated (see Table 1). After fasting or ad libitum feeding, mice were weighed and blood glucose was measured using a handheld glucometer (One Touch Ultra; Lifescan).
Table 1.
Composition of Diets Used in the Study
| Diet | Description | Nutrients | g% | kcal% |
|---|---|---|---|---|
| 5001 | NC | Protein | 25 | 29.83 |
| Carbohydrate | 47.5 | 56.74 | ||
| Fat | 11.4 | 13.43 | ||
| Total | 100 | |||
| kcal/gm | 4.09 | |||
| D12328 | LFD | Protein | 16.8 | 16.4 |
| Carbohydrate | 74.3 | 73.1 | ||
| Fat | 4.8 | 10.5 | ||
| Total | 100 | |||
| kcal/gm | 4.07 | |||
| D12330 | HFD | Protein | 23 | 16.4 |
| Carbohydrate | 35.5 | 25.5 | ||
| Fat | 35.8 | 58 | ||
| Total | 100 | |||
| kcal/gm | 5.56 | |||
| TD0.06414 | HFD unmatched | Protein | 23.5 | 18.3 |
| Carbohydrate | 27.3 | 21.4 | ||
| Fat | 34.3 | 60.3 | ||
| Total | 100 | |||
| kcal/gm | 5.1 |
Hormone profiling
Plasma glucose, insulin, leptin, glycerol, triglycerides, and adiponectin levels were measured by ELISA (Millapore and Randox). Plasma free fatty acids (FFAs) were measured with a FFA C Test kit (Wako). Plasma cholesterol and Low-Density Lipoprotein and High-Density Lipoprotein cholesterol were determined with a Cholesterol Quantitation kit (Thermo Electron - Infinity).
Body composition
Magnetic resonance imaging analysis of body composition was performed using an EchoMRI Whole Body Composition Analyzer (Echo Medical Systems) as previously described (12).
Homeostatic model assessment for insulin resistance (HOMA-IR), glucose tolerance test (GTT), and insulin tolerance test (ITT)
Calculations for HOMA-IR were made based on the following equation for insulin resistance as previously described: insulin (μU/mL)/22.5 × glucose (mg/dL)/18 (13). For glucose tolerance, mice were fasted for 17 hours and then injected ip with glucose at 2 g/kg of body weight. For insulin sensitivity, mice were fasted for 3 hours and injected ip with regular insulin (100 U/mL), resulting in a final concentration of 1.0 U/kg of body weight. Blood was collected before insulin injection and at 15, 30, 60, and 120 minutes after injection. Glucose levels were monitored before and after injection using a handheld glucometer (One Touch Ultra; Lifescan).
Tissue histology and immunohistochemistry (IHC)
Pancreas, liver, and skeletal muscle tissues were isolated from CaMKK2+/+ and CaMKK2−/− mice followed by fixation in 10% neutral buffered formalin and paraffin embedded. Five-micrometer serial sections were cut on a rotary microtome, mounted on glass slides, and allowed to air-dry overnight. Sections were then deparaffinized by subjecting the slides to an organic solvent (Hemo-de) for 10 minutes followed by rehydration of the sections through graded ethanol steps starting at 100% ethanol, then 95% ethanol, 70% ethanol, 50% ethanol before finishing at 20% ethanol. Slides were then transferred to sterile H2O. Hematoxylin and eosin (H&E)-stained pancreas, liver, and skeletal muscle sections were visualized and photographed on a Zeiss Axiovert microscope at representative magnifications as indicated in the figure legends.
IHC to detect the CaMKK2-EGFP protein was performed as follows: sections were blocked in 3% goat serum for 30 minutes at room temperature (RT) followed by the application of the primary antibody (anti-rabbit EGFP; diluted 1:100) at 4°C for 12 hours. Sections were washed 3× 5 minutes in PBS at RT. Secondary antibody (Biotinylated anti-rabbit IgG; diluted 1:1000) was then applied for 1 hour at RT. Sections were washed 3× 5 minutes in PBS at RT. Pre-diluted streptavidin was then applied to the sections for 30 minutes at RT and washed 3× 5 minutes in PBS. Visualization of the signal was performed using the 3,3′-diaminobenzidine method according to manufacturer's instructions. As a final step, H&E staining was performed before dehydrating the sections and mounting. Control sections were treated identically except for the omission of the primary antibody. IHC-stained tissue sections were visualized and photographed on a Zeiss Axiovert microscope at a magnification of 20× unless noted otherwise.
IHC to detect endogenous CaMKK2 protein was performed as follows: 7-μm sections were cut on a rotary microtome and then blocked for 1 hour at RT with 3% normal donkey serum vol/vol (Jackson ImmunoResearch), Antibodies used were obtained as follows: anti-insulin (AM029 5M) and anti-glucagon (AR039–5R); both pre-titered (Biogenex); and anti-CaMKK2 diluted 1:100 (Santa Cruz Biotechnology, Inc). Sections were then incubated with long chain biotin spacer secondary antibodies (Jackson ImmunoResearch) as follows: anti-mouse 1:1000 (insulin), anti-rabbit 1:1000 (glucagon), and anti-goat (CaMKK2) for 1 hour at RT. After washing, sections were incubated with either streptavidin-conjugated fluorescein 1:1000 (insulin and glucagon) or streptavidin-conjugated CY5 1:1000 (CaMKK2) (Jackson ImmunoResearch). Sections were washed in PBS, mounted, and visualized on a Zeiss Axiovert microscope (Carl Zeiss).
RIA for insulin content
Islet insulin content was quantified by RIA methods previously described (14) using a commercially available Rat Insulin RIA kit (RI-13K; EMD Millipore).
Insulin secretion assays
CaMKK2+/+ and CaMKK2−/− islets were isolated according to previously published methods with the exception that Liberase-R1 (Roche) was substituted for collagenase-P (15). Purified islets were cultured overnight in RPMI 1640 containing 10% Fetal Bovine Serum, 11mM glucose, 0.5% penicillin, 0.5% streptomycin, and 10mM HEPES (pH 7.4). Stimulatory responses of islets were assayed in Krebs-Ringer bicarbonate buffer supplemented with 15mM HEPES (pH 7.4) and RIA grade BSA. Before the start of any secretion assay, islets were equilibrated for 1 hour in Krebs-Ringer bicarbonate buffer containing 2.8mM glucose to establish a baseline. At the end of the assay, islets were sonicated in acidified ethanol for quantification of total insulin content. Secretion data are expressed as percentage of the total available insulin secreted per minute.
Plasma isolation
Whole blood was collected and transferred into a pre-chilled microcentrifuge tube containing 10 μL of 0.5M EDTA (pH 8.0) for isolation of EDTA plasma, which was transferred to a clean microcentrifuge tube and stored at −80°C until used.
Tissue preparation
Frozen pieces of mouse liver and skeletal muscle were pulverized with a liquid nitrogen-chilled 3-lb sledgehammer. The resulting pulverized tissue (∼130 mg) was placed into a pre-chilled 14-mL tube, followed by the addition of 1.17 mL of pre-chilled HPLC-grade water. All samples were then homogenized on ice using a Polytron homogenizer. An aliquot of each homogenized tissue was used to perform a bicinchoninic acid assay protein concentration assay to normalize the resulting metabolites to total protein. The remainder of the homogenate was used to perform metabolomic profiling as described below.
Metabolomic profiling
Methods used for metabolomics analysis have been described in detail elsewhere (16–18). Briefly, AAs and acyl carnitines (ACs) were measured using stable isotope dilution techniques. AA and AC species were measured using flow injection tandem mass spectrometry (MS) and sample preparation methods described previously (19). Data were acquired using a Micromass Quattro micro TM system equipped with a model 2777 autosampler, a model 1525 μ HPLC solvent delivery system, and a data system controlled by MassLynx 4.0 operating system (Waters). For each tissue, 16 AAs were measured in liver and skeletal muscle. Additionally, 16 AA and 45 AC metabolites were measured in EDTA plasma. The number of AC species analyzed for each tissue varied as some species of AC are not detectable in all tissues. A total of 55 ACs were measured in skeletal muscle. A complete list of the specific metabolites analyzed is listed in Supplemental Tables 1 and 2. All MS analyses employed stable-isotope dilution. Addition of clusters of internal standards specific to the AA and AC analyte “modules” facilitates identification of each analyte peak and provides the reference for quantifying their levels. The stable-isotope internal standards used were obtained from Isotec, Cambridge Isotope Laboratories, and CDN Isotopes, and a complete list of standards has been published previously (19). In addition to mass, analytes were identified on the basis of the particular MS/MS transitions monitored for each class of metabolites.
Statistical analyses
Tissue-specific MS-based metabolomic analyses of liver, skeletal muscle and plasma AA and AC from CaMKK2+/+ and CaMKK2−/− male mice (n ≥ 5 each genotype) were analyzed using Partek Genomics Suite software. To compare only the effects of genotype (HFD vs LFD; fed vs fasted), an ANOVA analysis with contrasts of each set of metabolites was performed. We used the Benjamini and Hochberg method for estimation of false discovery rate (20). The cut-offs for differentially expressed metabolites was false discovery rate = 0.1. Zero values observed in the raw data were replaced with uniformly distributed random values between zero and minimum values for the specific metabolite. Metabolites with more than 10% missing data have been excluded from the analysis. Raw data values were log-transformed before analysis to establish distributions closer to the Gaussian. The total number of metabolites analyzed for each tissue was the following: plasma, 60; liver, 67; and skeletal muscle, 77. The results are presented as heat maps and Venn diagrams. Heat maps were generated using dChip (http://www.hsph.harvard.edu/cli/complab/dchip) (21). Standardized z-scores of the log2-transformed values were used for heat maps of raw data with blue representing low z-score values and red representing high z-score values, respectively.
Graphical representations of raw data are also presented as the average of the sum (Σ) of all of a subclass of metabolite for each group of mice of a given genotype (CaMKK2+/+ and CaMKK2−/−) ± SEM as indicated. Standard statistical comparison of different groups with Gaussian distribution was carried out using a 2-tailed unpaired Student's t test. Differences of P ≤ .05 were considered statistically significant. For all mouse studies, the experimental number used is indicated in each figure legend.
Database availability
The entire raw datasets obtained from the MS-based metabolomics analyses performed in this study will be made publically available through the Nuclear Receptor Signaling Atlas data repository at http://www.nursa.org/. An analyzed version of these datasets is also included as Supplemental Dataset 1. For more details, see also Supplemental Methods.
Results
Loss of CaMKK2 protects against HFD-induced obesity, alters circulating hormone levels, including insulin, and confers protection against insulin resistance
Immediately after weaning (3 wk of age), independent cohorts of CaMKK2+/+ and CaMKK2−/− mice were placed on normal chow (NC) (5001) (Table 1) or HFD (D12330) (Table 1) for 20 weeks, after which body weight, fat mass, and lean mass measurements (Figure 1A, i–iii) as well as hormone levels (Figure 1B, i–iii) were determined. Additional metadata were also collected and are presented in Supplemental Figure 1. We found an attenuated increase in the body weight of CaMKK2−/− mice on HFD, which is associated with, and is likely due to, the decrease in fat mass and concomitant increase in lean mass in the CaMKK2−/− mice fed HFD (Figure 1A, i–iii), consistent with previously reported findings (9, 10). Moreover, we observed that HFD feeding in CaMKK2+/+ mice led to severe insulin resistance as indicated by HOMA-IR, increased leptin and reduced adiponectin (Figure 1B, i–iii), yet CaMKK2−/− mice were refractory to such changes (Figure 1B, i–iii).
Figure 1.

Loss of CaMKK2 protects against HFD-induced obesity, alters circulating hormone levels, including insulin, and confers protection against insulin resistance. A, Graphical representation of body weight (i), fat mass (ii), and lean mass (iii) measurements from CaMKK2+/+ and CaMKK2−/− male mice fed either NC or HFD. B, HOMA-IR score (i), leptin (ii), and adiponectin (iii) levels from the CaMKK2+/+ and CaMKK2−/− cohorts fed either NC or HFD. C, i and iv, Serum glucose levels in CaMKK2+/+ and CaMKK2−/− mice fed NC (i) and HFD (iv) after an ITT. C, ii, iii, v, and vi, Serum glucose levels (ii and v) and serum insulin levels (iii and vi) in CaMKK2+/+ and CaMKK2−/− mice fed NC (ii and iii) and HFD (v and vi) after a glucose tolerance test (GTT). Data are represented as mean ± SEM. *, P ≤ .05; **, P ≤ .01; ***, P ≤ .001.
The protection against HFD-induced obesity and normal HOMA-IR score observed upon ablation of CaMKK2 prompted us to test for differences in insulin sensitivity or glucose clearance between CaMKK2+/+ and CaMKK2−/− mice in response to chronic overnutrition. We performed ITT on approximately 20-week-old CaMKK2+/+ and CaMKK2−/− mice fed NC or HFD and observed no difference in insulin sensitivity between the 2 genotypes fed NC (Figure 1Ci). Both CaMKK2+/+ and CaMKK2−/− mice demonstrated nearly identical glucose tolerance and displayed no overt differences in circulating insulin levels on NC (Figure 1C, ii and iii). On HFD, we observed a trend toward increased insulin resistance in CaMKK2+/+ mice compared with CaMKK2−/− mice, especially at the 30- and 60-minute timepoints (Figure 1Civ). In fact, we found significantly lower levels of serum insulin in CaMKK2−/− mice on HFD compared with CaMKK2+/+ at all timepoints tested even though there were comparable serum glucose levels in CaMKK2+/+ and CaMKK2−/− mice (Figure 1C, v and vi). These data are consistent with previous reports demonstrating that loss of CaMKK2 provides protection against insulin resistance and HFD-induced obesity (9, 10).
CaMKK2-deficient islet cells display enhanced insulin secretion
Given the observation that CaMKK2−/− mice display markedly improved insulin sensitivity, we more closely examined the function of CaMKK2 in insulin-producing islets. Wild-type pancreatic islets express the CaMKK2 transcript (Supplemental Figure 2A), whereas expression of EGFP in Tg(CaMKK2-EGFP)DF129Gsat pancreatic sections confirms CaMKK2 protein expression within islets (Figure 2A, i and ii). Morphologically, CaMKK2-null islets appeared normal and comparable with those from CaMKK2+/+ mice (Figure 2A, iii and iv) and loss of CaMKK2 in β-cells failed to impact their polarity within pancreatic rosette clusters (Supplemental Figure 2B, i and ii).
Figure 2.

CaMKK2-deficient islets display enhanced insulin content and secretion. A, i, Immunohistochemical staining for EGFP expression in pancreatic sections from Tg(CaMKK2-EGFP)DF129Gsat (CaMKK2:EGFP) mice (11) wherein EGFP expression is driven by the endogenous CaMKK2 regulatory promoter. EGFP expression, indicative of CaMKK2 protein expression, is observed within pancreatic islets. A, ii, Immunostaining for EGFP within normal, non-transgenic CaMKK2+/+ pancreatic sections shows no signal and serves as a negative control. A, iii and iv, H&E-stained sections showing architecture of pancreatic islets from CaMKK2+/+ (iii) and CaMKK2−/− (iv) mice. Scale bars, 20 μm. B, Immunohistochemical staining for glucagon, insulin, and CaMKK2 in pancreatic α- and β-cells from CaMKK2+/+ and CaMKK2−/− mice. Scale bars, 20 μm. C, Quantification of insulin content via RIA in isolated pancreatic islets from CaMKK2+/+ and CaMKK2−/− mice. D, Static insulin secretion assay using pancreatic islets isolated from CaMKK2+/+ and CaMKK2−/− mice. E, Perifusion insulin secretion assay using pancreatic islets isolated from CaMKK2+/+ and CaMKK2−/− mice. A graphical representation of the area under the curve (AUC) for data in E is shown in the accompanying inset. Data are represented as mean ± SEM. ***, P ≤ .001.
Furthermore, we found that although CaMKK2 only marginally co-localized with glucagon-producing α-cells, CaMKK2 showed strong colocalization with insulin-producing β-cells (Figure 2B). Pancreatic α- and β-cells still express glucagon and insulin, respectively, even in the absence of CaMKK2, suggesting that CaMKK2 expression is not required for production of these pancreatic hormones (Figure 2B). We failed to observe any significant differences in insulin-1 (Ins1) and Ins2 transcript levels isolated from islets of CaMKK2+/+ and CaMKK2−/− mice as assessed by quantitative Polymerase Chain Reaction analysis (Supplemental Figure 2C). Assessment of insulin and glucagon content in islets isolated from CaMKK2+/+ and CaMKK2−/− mice fed either NC, LFD, or HFD revealed only marginal differences upon CaMKK2 ablation (Supplemental Figure 2, D–E). Consistent with our IHC results showing increased fluorescence of insulin immunostaining in CaMKK2-deficient islets (Figure 2B), RIA analysis demonstrated a quantitative increase in insulin content per islet in CaMKK2−/− tissue compared with those from CaMKK2+/+ mice (Figure 2C).
Because the ability of CaMKK2-null islets to produce insulin is not impaired, and in fact appears to be enhanced, we next assessed functionality of islet cells from CaMKK2+/+ and CaMKK2−/− mice by performing glucose-stimulated insulin secretion assays. In a static setting, insulin secretion was significantly higher in CaMKK2−/− mice compared with CaMKK2+/+ mice at higher glucose concentrations (16.8mM), and more than doubled in the presence of high extracellular concentrations of K+ (Figure 2D). We also observed a marked biphasic response in insulin secretion in CaMKK2-deficient islets, the first in response to glucose administration and the second in response to K+-induced depolarization (Figure 2E). Quantification of the area under the curve from Figure 2E reveals increased insulin secretion in islets isolated from CaMKK2−/− mice (Figure 2E, inset). Next, we inhibited CaMKK2 activity in human INS-1 cells using a selective CaMKK2 inhibitor, 7-Oxo-7H-benzimidazo[2,1-a]benz[de]isoquinoline-3-carboxylic acid acetate (STO-609) in a static insulin secretion assay and observed increased insulin secretion in the presence of the inhibitor (Supplemental Figure 2F), recapitulating our findings in islets isolated from CaMKK2−/− mice. Importantly, inhibition of CaMKK2 activity in vitro had a minimal effect on INS-1 cell growth (Supplemental Figure 2G), but eliminated the potentiating effect of tolbutamide on insulin secretion in INS-1 cells (Supplemental Figure 2F). In all, both static and perifusion assays indicate that either loss or inhibition of CaMKK2 not only leads to increased insulin protein production, but also potentiates insulin secretion.
Assessment of metabolic changes in CaMKK2−/− mice subjected to dietary or feeding/fasting stress
To more carefully examine the impact of CaMKK2 function on peripheral insulin signaling, we fed cohorts of CaMKK2+/+ and CaMKK2−/− mice the Surwit diet (Table 1), which consists of calorically paired LFD or HFD, and compared them with an independent cohort of mice fed NC. We analyzed a number of circulating hormones, FFAs, ketones, and glycerol in CaMKK2+/+ and CaMKK2−/− mice fed these 3 diets and represented the findings in heat map format (Figure 3A). Consistent with our hormonal profiling of HFD-fed CaMKK2+/+ and CaMKK2−/− mice in Figure 1, circulating leptin, insulin, and adiponectin levels were all reduced in CaMKK2-null mice (Figure 3, A and B, i–iii). Conversely, ablation of CaMKK2 showed no effect on FFA or glycerol levels but revealed a statistically significant increase in ketones on HFD as previously reported (Supplemental Figure 2D) (10).
Figure 3.

Summary of metabolic changes upon ablation of CaMKK2. A, Heat map representation of levels of selected circulating hormones (leptin, insulin, and adiponectin) from CaMKK2+/+ and CaMKK2−/− male mice fed NC, LFD, or HFD. B, Graphical representation of raw data from A representing the average of leptin (i), insulin (ii), and adiponectin (iii) from CaMKK2+/+ and CaMKK2−/− male mice fed NC, LFD, or HFD. Data are graphed as mean ± SEM. C, Venn diagram comparison of total metabolites (AAs and ACs) analyzed via MS-based metabolomics from plasma, liver, and muscle from CaMKK2+/+ and CaMKK2−/− mice fed NC, LFD, or HFD. The number of individual metabolites whose values were statistically different between CaMKK2+/+ and CaMKK2−/− mice fed a specific diet is indicated within each colored circle representing the diet tested, NC (red), LFD (green), and HFD (blue), with overlapping metabolites indicated in the intersecting regions of the Venn diagram. D, Tabular representation of the number of individual metabolites whose values were statistically different between CaMKK2+/+ and CaMKK2−/− mice fed each diet (NC, LFD, and HFD) and the direction of change (increase or decrease) relative to wild-type mice. E, Summary of MS-based metabolomics analysis of CaMKK2+/+ and CaMKK2−/− mice fed different diets (NC, LFD, and HFD). The number of individual metabolites (AA, AC) whose values were statistically different between CaMKK2+/+ and CaMKK2−/− mice fed each specific diet (NC, LFD, and HFD) are provided for the 3 tissues analyzed (plasma, liver, and muscle). Data are represented as mean ± SEM. *, P ≤ .05; **, P ≤ .01; ***, P ≤ .001.
Using quantitative MS-based metabolomics, we next determined the AA and AC profiles of 3 insulin-responsive tissues (plasma, liver, and skeletal muscle) in CaMKK2+/+ and CaMKK2−/− mice. To determine whether loss of CaMKK2 influenced levels of these metabolites (15 AAs, 45 ACs), we performed MS analyses on these tissues from 4–12 mice of either genotype fed each one of the 3 diets previously described (NC, LFD, and HFD) (Table 1) or as a function of feeding states (ad libitum fed, 24 h fasted, and 48 h fasted). We found significant changes in the concentration of various metabolites in these tissues in the absence of CaMKK2 (summarized in Figure 3, C–E, and Supplemental Figure 3).
Taking into account metabolite levels from all 3 tissues, mice on NC yielded the fewest number of metabolites whose corresponding levels were statistically changed between CaMKK2+/+ and CaMKK2−/− mice (Figure 3, C–E). Conversely, CaMKK2−/− mice fed LFD yielded the greatest number of statistically significant changes in metabolite levels (Figure 3, C–E). Only 5 metabolites reflected significant changes in CaMKK2−/− mice regardless of diet and were all increased specifically in CaMKK2-deficient skeletal muscle compared with CaMKK2+/+ mice (Figure 3A and Supplemental Dataset 1). Figure 3D summarizes the number of AAs and ACs that either increased (↑, highlighted in red) or decreased (↓, highlighted in blue) in the CaMKK2−/− relative to CaMKK2+/+ mice with respect to each diet. Figure 3E summarizes the number of AAs and ACs species significantly altered between the 2 genotypes within each tissue (plasma, liver, and skeletal muscle). The accompanying pie chart above each column represents the diets tested in this study and provides a graphical summary of the data in Supplemental Dataset 1.
Interestingly, we found most statistically significant changes resulting from CaMKK2 ablation were increases in AA and AC levels, regardless of the dietary condition (Figure 3D). On the NC diet, 10 out of 15 AAs showed higher levels in the CaMKK2-null mice, and these increases were observed solely in skeletal muscle (Figure 3, D and E). In the liver, none of the metabolite species analyzed reflected significant changes in CaMKK2−/− mice fed NC (Figure 3E). Only the plasma and skeletal muscle reflect varying metabolite levels between the 2 genotypes on the NC diet. However, differences in hepatic metabolite levels were observed in mice fed either LFD or HFD (Figure 3E). For all diets tested, skeletal muscle displayed the most significant number of metabolites that were altered due to the absence of CaMKK2 (Figure 3E).
In the feeding to fasting transition, we observed the highest number of metabolite changes (57) in the CaMKK2−/− mice that were fasted for 48 hours, which was twice the number of changes observed for mice fed ad libitum or fasted for 24 hours (Supplemental Figure 3, A and C). All 8 metabolites that were statistically altered in CaMKK2−/− mice in all 3 feeding states were observed in the liver, and all were decreased relative to CaMKK2+/+ metabolite levels (Supplemental Figure 3B and Supplemental Dataset 1). For mice fasted for 24 hours, all 13 metabolites that were statistically different between CaMKK2+/+ and CaMKK2−/− mice were uniquely observed in the liver (Supplemental Figure 3B and Supplemental Dataset 1). AAs were lower in CaMKK2−/− mice regardless of diet or tissue (Supplemental Figure 3C). Similar to the effect of diet (Figure 3E), AC levels tended to be elevated in CaMKK2−/− mice in all feeding states and in all 3 tissues analyzed (Supplemental Figure 3B). In the ad libitum fed or 24 hour-fasted state, we observed a greater proportion of metabolites to be significantly altered in CaMKK2-null mice in the liver (Supplemental Figure 3C). In contrast, approximately half of the metabolites that were significantly changed in CaMKK2−/− mice were observed in the plasma after a 24 hour-fasting period (Supplemental Figure 3C).
Hepatic metabolism
Because liver represents a major target of insulin action, we characterized the effects of CaMKK2 ablation on hepatic AA metabolism and fatty acid oxidation in mice subjected to chronic metabolic stress (LFD or HFD). A summary of these data are presented in Figure 3, A–C, which highlight the number of metabolites that are significantly different between CaMKK2+/+ and CaMKK2−/− mice in response to each specific diet. Our analysis identified significant differences between CaMKK2+/+ and CaMKK2−/− livers as a function of each diet. Specifically, there were no alterations in the levels of hepatic metabolites assessed in this study between CaMKK2+/+ and CaMKK2−/− mice fed NC, whereas 19 were significantly altered when fed LFD and 9 metabolites showed significant changes in mice fed HFD (Figure 3E).
The direction of metabolite changes for hepatic AA and AC levels between CaMKK2+/+ and CaMKK2−/− mice is represented as a heat map (Figure 4A). H&E sections of livers isolated from CaMKK2+/+ and CaMKK2−/− mice fed LFD display a fatty liver morphology in the absence of CaMKK2, whereas CaMKK2−/− mice fed HFD are protected from hepatic steatosis (Figure 4B). The sums of the concentration of AA or subgroups of AC were averaged for CaMKK2+/+ and CaMKK2−/− mice fed each of the 3 diets (NC, LFD, and HFD). This graphical representation revealed that the cumulative medium chain AC were significantly higher in CaMKK2-null mouse liver compared with those in CaMKK2+/+ mice fed HFD (Figure 4, C and D). These data are consistent with a previous report from our laboratory demonstrating increased hepatic lipid metabolism in CaMKK2−/− mice and perhaps explain the reduction of liver steatosis observed in CaMKK2−/− mice fed HFD (Figure 4B).
Figure 4.

Metabolomics analysis upon CaMKK2 ablation shows alterations in global hepatic metabolism. A, Heat map representation of z-score transformed raw data from liver-specific MS-based metabolomics analysis of AAs and ACs from CaMKK2+/+ and CaMKK2−/− mice fed NC, LFD, or HFD. B, H&E-stained liver sections from CaMKK2+/+ and CaMKK2−/− mice fed LFD and HFD. Scale bars, 100 μm. C, Enlarged heat map representation of selected z-score transformed raw data from metabolomics analysis of medium chain ACs from livers of CaMKK2+/+ and CaMKK2−/− mice fed HFD. D, Graphical representation of data from C representing the average of the sum (Σ) of medium chain AC levels from CaMKK2+/+ and CaMKK2−/− mice fed a HFD. Data are graphed as mean ± SEM. **, P ≤ .01.
When fed a LFD, both the cumulative AA and short chain AC were significantly higher in CaMKK2−/− liver (Supplemental Figure 4, A and B). Just as comparison of levels of individual metabolites in livers of CaMKK2+/+ and CaMKK2−/− mice fed NC failed to reflect any significant changes (Figure 3C), no differences were observed when comparing the cumulative sums of metabolite groups.
In general, we observed a trend toward increased sums of subgroups of AC in CaMKK2−/− liver compared with CaMKK2+/+ liver as the mice transitioned from the fed to the fasted state (Supplemental Figure 3, B and C). This effect was apparent especially for the medium chain AC and long chain AC species (Supplemental Figure 4, C and D). Consistent with previously published data from our laboratory, these data suggest increased fatty acid oxidation in the liver of CaMKK2−/− mice (10). CaMKK2−/− mice also demonstrated significantly lower cumulative hepatic AA compared with CaMKK2+/+ mice, regardless of feeding state (Supplemental Figures 3, B and C, and 4, E–H).
Skeletal muscle metabolism
Using the same method of analysis as performed for liver, we identified 17 metabolites that significantly differ between CaMKK2+/+ and CaMKK2−/− skeletal muscle when fed NC (Figure 3E). This number more than doubled when mice were fed LFD and rose to 20 altered metabolites in skeletal muscle when mice were fed HFD (Figure 3E).
For skeletal muscle, the direction of total metabolite changes for AA and AC between CaMKK2+/+ and CaMKK2−/− mice is represented as a heat map (Figure 5A). H&E sections of skeletal muscle isolated from CaMKK2+/+ and CaMKK2−/− mice fed either LFD or HFD display comparable tissue morphology (Figure 5B). The sums of the concentration of AA or subgroups of AC in skeletal muscle were averaged for CaMKK2+/+ and CaMKK2−/− mice fed each of the 3 diets (NC, LFD, and HFD). These data revealed that medium chain and very long chain AC levels were significantly higher in CaMKK2-null livers compared with those in CaMKK2+/+ mice fed HFD (Figure 5A and Supplemental Figure 5A). We also observed a general trend towards increased cumulative AA and AC in CaMKK2-deficient skeletal muscle for all diets assessed in this study (Figure 3, D and E, and Supplemental Figure 5A). For mice fed LFD, all AC subgroups (short, medium, long, and very long AC) showed levels that were significantly higher in the CaMKK2−/− vs CaMKK2+/+ skeletal muscle (Supplemental Figure 5A). Only the sum of AA was significantly increased for the CaMKK2-null skeletal muscle in mice fed NC diet (Supplemental Figure 5A).
Figure 5.

Metabolomic analysis upon CaMKK2 ablation shows alterations in global skeletal muscle metabolism. A, Heat map representation of z-score transformed raw data from skeletal muscle-specific MS-based metabolomics analysis of AAs and ACs from CaMKK2+/+ and CaMKK2−/− mice fed NC, LFD, or HFD. B, H&E-stained skeletal muscle sections from CaMKK2+/+ and CaMKK2−/− mice fed LFD and HFD. Scale bars, 100 μm. C, Enlarged heat map representation of selected z-score transformed raw data from metabolomics analysis of very long chain ACs from skeletal muscle of CaMKK2+/+ and CaMKK2−/− mice fed HFD. C, Graphical representation of data from C representing the average of the sum (Σ) of very long chain AC levels from CaMKK2+/+ and CaMKK2−/− mice fed HFD. Data are graphed as mean ± SEM. **, P ≤ .01.
Similarly, the cumulative level of AC species within subgroups (short, medium, long, and very long AC) increased in CaMKK2−/− skeletal muscle compared with CaMKK2+/+ as the mice transitioned from the fed to fasted states (Supplemental Figure 5A). This increase was significant for the short chain AC species after both 24 hours (Supplemental Figure 5B) and 48 hours of fasting (Supplemental Figure 5, C–E). Additionally, cumulative AA levels were significantly decreased in CaMKK2−/− mice after a 48 hour fast (Supplemental Figure 5B).
Plasma metabolites
Surprisingly, we observed very few metabolites that were significantly changed in plasma between CaMKK2+/+ and CaMKK2−/− mice fed any of the 3 diets tested (NC, LFD, and HFD). Specifically, we identified only 5 metabolites whose levels were altered for CaMKK2-null mice fed NC, 3 fed LFD and 2 fed HFD (Figure 3E). We again represented the direction of these changes in the levels of AA and AC in the plasma as a heat map and graphed the sum of concentrations of AA or groups of AC species for each genotype under each of the 3 diets (Figure 6, A–C). None of the sums for any of the metabolite groups were significantly increased or decreased in the plasma isolated from CaMKK2−/− in response to any of the diets tested in this study, although medium chain and long chain AC species displayed an increasing trend (Figure 6, B and C).
Figure 6.

Metabolomic analysis upon CaMKK2 ablation shows alterations in global plasma metabolism. A, Heat map representation of z-score transformed raw data from plasma specific MS-based metabolomics analysis of AAs and ACs from CaMKK2+/+ and CaMKK2−/− mice fed NC, LFD, or HFD. B, Enlarged heat map representation of selected z-score transformed raw data from metabolomics analysis of long chain ACs from plasma of CaMKK2+/+ and CaMKK2−/− mice fed HFD. C, Graphical representation of data from C representing the average of the sum (Σ) of long chain AC levels from CaMKK2+/+ and CaMKK2−/− mice fed HFD. Data are graphed as mean ± SEM.
Relative to those of CaMKK2+/+ mice, plasma samples from CaMKK2−/− mice fasted for 48 hours showed increased cumulative medium and long chain AC species (Supplemental Figure 6, A and C–E). The increase in serum AC perhaps provides additional evidence for the increase in hepatic lipid oxidation in CaMKK2-deficient mice, consistent with previously reported findings (10). Meanwhile, a decreasing trend of plasma AA was observed in CaMKK2−/− mice compared with that of CaMKK2+/+ mice in the transition from the fed to the fasted state (Supplemental Figure 6, B and C).
Discussion
Disruption of Ca2+ signaling has been linked to obesity and insulin resistance (3–6). Central to the downstream actions of Ca2+ signaling is CaMKK2, which phosphorylates and activates other kinases such as CaMKI, CaMKIV, and AMPK in response to an increase in intracellular Ca2+. We have previously reported that mice with germ line disruption of CaMKK2 are protected from obesity (9), maintain normal circulating insulin levels and remain glucose responsive when fed a HFD (10). Aside from its regulatory role in energy homeostasis, CaMKK2 is also important for adipocyte differentiation (22), macrophage functions (23), as well as lipid and carbohydrate metabolism in the liver (10). However, the metabolic implications of CaMKK2 function in the pancreas and the influence of CaMKK2 in insulin-responsive tissues remained largely unexplored before this study. Although loss of CaMKK2 failed to alter basic islet structure or polarity, insulin levels in CaMKK2−/− islets were significantly higher than those in CaMKK2+/+ islets, suggesting that CaMKK2 and Ca2+ signaling contribute to the regulation of insulin production in pancreatic β-cells. Although static insulin secretion was unaffected by loss of CaMKK2 in response to basal glucose, elevated glucose levels or high extracellular K+ markedly increased insulin secretion from islets devoid of CaMKK2. Similarly, pharmacological inhibition of CaMKK2 in INS-1 cells was sufficient to increase insulin secretion. Aside from a potential role in regulating insulin production, CaMKK2 may also be a component of the Ca2+-mediated response to glucose signaling in pancreatic islets, and its absence promotes glucose-mediated insulin secretion. Moreover, a biphasic, punctuated response in insulin secretion from CaMKK2-deficient islets was observed when we measured dynamic insulin release. These observations suggest that perhaps inhibition or loss of CaMKK2 results in either a greater number of secretory granules and/or more granules that are poised for immediate insulin release at the plasma membrane.
In all, the function of CaMKK2 in pancreatic β-cells appears to be 2-fold, regulating not only insulin production, but also glucose-mediated insulin secretion. Under chronic metabolic stress (eg, HFD), CaMKK2 provides an instructive signal in the pancreas to overproduce and/or secrete insulin, eventually leading to insulin resistance and glucose intolerance. In mice lacking CaMKK2, insulin production and secretion are already increased compared with wild-type mice, albeit at relatively high physiological levels that permit normal metabolic function when maintained on a standard diet. Chronic metabolic stress fails to further induce insulin production or secretion in CaMKK2-deficient mice due to the absence of this “molecular rheostat” function (Figure 7), which could be an underlying reason why CaMKK2−/− mice are protected from HFD-induced glucose intolerance and obesity. The exact mechanism responsible for CaMKK2 action to control both insulin production and secretion by pancreatic β-cells remains to be fully explored, but the findings presented here highlight the importance of such future studies.
Figure 7.
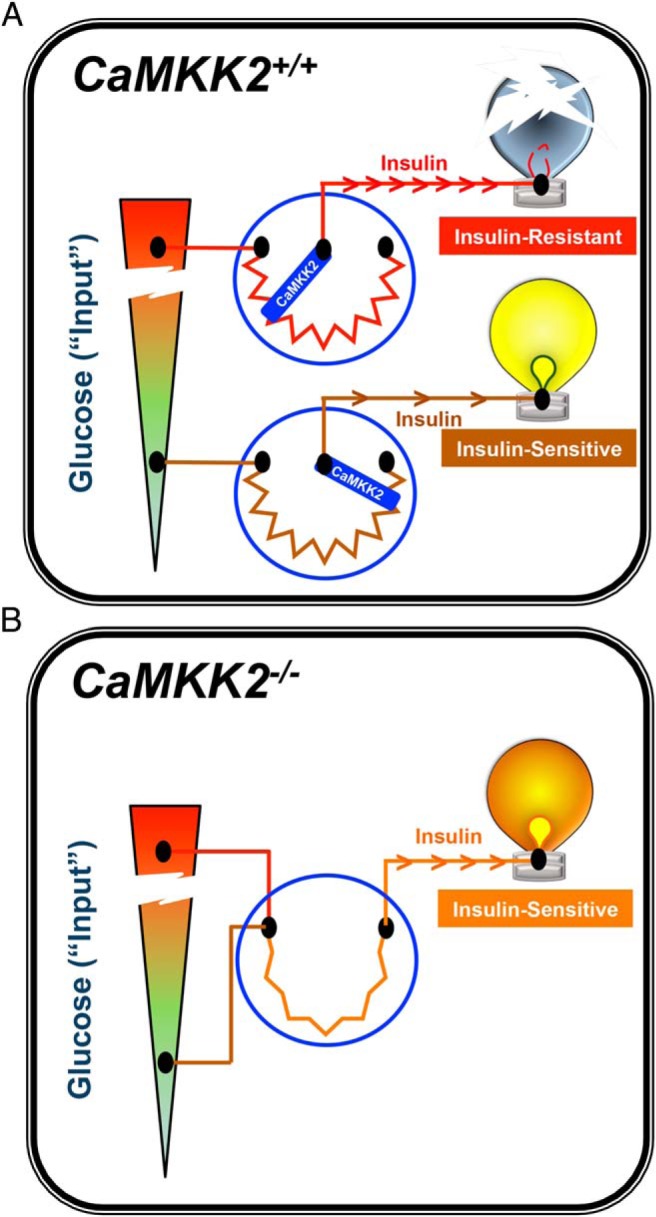
Model summarizing the role of CaMKK2 as a molecular rheostat for insulin action. A, CaMKK2+/+ mice respond to overnutrition by overcompensating production and secretion of insulin due to elevated glucose levels. CaMKK2 is an integral component of the molecular mechanism in pancreatic β-cells whereby increasing demand for insulin leads to insulin resistance, glucose intolerance, and inevitably gives rise to type 2 diabetes (symbolized by the burnt out light bulb). B, In contrast, CaMKK2−/− mice generate higher, yet steady state levels of insulin (still within the physiological range) even upon exposure to overnutrition. Ablation of CaMKK2 renders insulin-producing pancreatic β-cells more acutely responsive to glucose while simultaneously conferring improved insulin sensitivity to peripheral metabolic tissues. Our findings support the notion that inhibitors of CaMKK2 may serve as therapeutic agents for combatting diet-induced insulin resistance.
The present study is the first to use metabolomics to evaluate the inherent metabolic differences between wild-type and CaMKK2-deficient liver, skeletal muscle, and plasma. Our quantitative analyses present an overarching view of how loss of CaMKK2 alters the metabolic landscape of these 3 insulin-sensitive tissues and provides a comprehensive resource for tissue- and nutrition-specific responses. The differences observed in the tissue-specific metabolic profiles of wild-type and CaMKK2-null mice subjected to various dietary stresses likely reflects both cell-autonomous responses to CaMKK2 loss as well as responses to circulating factors such as, but not limited to, insulin. Loss of CaMKK2 showed the greatest impact on skeletal muscle metabolism regardless of diet, whereas CaMKK2-null livers were essentially metabolically similar to wild-type livers only in mice fed a normal diet. Similarly, there were no major effects of CaMKK2 loss on plasma metabolites in mice fed any one of the 3 diets we tested. These and other pertinent observations became evident upon comparison of tissue- and nutrition-specific metabolite levels in the presence or absence of CaMKK2. As a research resource, we now have comprehensive metabolic profiles for liver, skeletal muscle, and plasma in mice under controlled states of metabolic stress (diet or fasting). Further integration of these metabolomics data with other available OMICs data will provide a better understanding of CaMKK2 function not only in metabolism but also in pathological conditions wherein chronic exposure to metabolic insults create predispositions for more severe disease outcomes.
We showed that blocking CaMKK2 activity using its selective inhibitor, STO-609, resulted in increased insulin secretion. These data encourage further investigation of whether pharmacological inhibition of CaMKK2 is sufficient to protect mice from diet-induced glucose intolerance and insulin resistance. A previous report from our group demonstrated that intracerebroventricular administration of STO-609 to wild-type mice attenuated food intake and promoted weight loss after a brief fasting period similar to CaMKK2-deficient mice (9). Also, in vivo injection of STO-609 to wild-type mice harboring hepatic tumors significantly regressed tumor burden (24). Altogether, these data demonstrate the potential for a pharmacologic intervention targeted against CaMKK2 to mediate symptoms and pathologies downstream of Ca2+-induced perturbations in systems metabolism. In addition to the practical nature of this Research Resource, this study offers numerous long-term possibilities for use of tissue-specific metabolomics profiling for early diagnosis of metabolic diseases and encourages development and assessment of selective pharmacologic inhibitors against CaMKK2 as putative therapeutic agents for treatment of type 2 diabetes, obesity, and even cancer.
Acknowledgments
We thank the joint participation by Adrienne Helis Malvin Medical Research Foundation through its direct engagement in the continuous active conduct of medical research in conjunction with Baylor College of Medicine and the CaMKK2 Inhibitors for Therapeutic Treatment of Hepatic Cancer program.
This work was supported by the National Institutes of Health Grant GM033976 (to A.R.M.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- AA
- amino acid
- AC
- acyl carnitine
- AMPK
- AMP-activated protein kinase
- Ca2+
- calcium
- CaM
- calmodulin
- CaMKI
- Ca2+/CaM-dependent protein kinase 1
- CaMKK2
- Ca2+/CaM-dependent protein kinase kinase 2
- EGFP
- enhanced green fluorescent protein
- FFA
- free fatty acid
- H&E
- hematoxylin and eosin
- HFD
- high-fat diet
- HOMA-IR
- homeostatic model assessment for insulin resistance
- IHC
- immunohistochemistry
- Ins1
- insulin-1
- ITT
- insulin tolerance test
- LFD
- low-fat diet
- MS
- mass spectrometry
- NC
- normal chow
- RT
- room temperature
- STO-609
- 7-Oxo-7H-benzimidazo[2,1-a]benz[de] isoquinoline-3-carboxylic acid acetate.
References
- 1. Faloia E, Michetti G, De Robertis M, Luconi MP, Furlani G, Boscaro M. Inflammation as a link between obesity and metabolic syndrome. J Nutr Metab. 2012;2012:476380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. van Greevenbroek MM, Schalkwijk CG, Stehouwer CD. Obesity-associated low-grade inflammation in type 2 diabetes mellitus: causes and consequences. Neth J Med. 2013;71:174–187. [PubMed] [Google Scholar]
- 3. Schrager S. Dietary calcium intake and obesity. J Am Board Fam Pract. 2005;18:205–210. [DOI] [PubMed] [Google Scholar]
- 4. dos Santos LC, de Padua Cintra I, Fisberg M, Martini LA. Calcium intake and its relationship with adiposity and insulin resistance in post-pubertal adolescents. J Hum Nutr Diet. 2008;21:109–116. [DOI] [PubMed] [Google Scholar]
- 5. da Silva Ferreira T, Torres MR, Sanjuliani AF. Dietary calcium intake is associated with adiposity, metabolic profile, inflammatory state and blood pressure, but not with erythrocyte intracellular calcium and endothelial function in healthy pre-menopausal women. Br J Nutr. 2013;110:1079–1088. [DOI] [PubMed] [Google Scholar]
- 6. Parikh SJ, Yanovski JA. Calcium intake and adiposity. Am J Clin Nutr. 2003;77:281–287. [DOI] [PubMed] [Google Scholar]
- 7. Torres MR, Ferreira Tda S, Carvalho DC, Sanjuliani AF. Dietary calcium intake and its relationship with adiposity and metabolic profile in hypertensive patients. Nutrition. 2011;27:666–671. [DOI] [PubMed] [Google Scholar]
- 8. Racioppi L, Means AR. Calcium/calmodulin-dependent protein kinase kinase 2: roles in signaling and pathophysiology. J Biol Chem. 2012;287:31658–31665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Anderson KA, Ribar TJ, Lin F, et al. Hypothalamic CaMKK2 contributes to the regulation of energy balance. Cell Metab. 2008;7:377–388. [DOI] [PubMed] [Google Scholar]
- 10. Anderson KA, Lin F, Ribar TJ, et al. Deletion of CaMKK2 from the liver lowers blood glucose and improves whole-body glucose tolerance in the mouse. Mol Endocrinol. 2012;26:281–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gong S, Zheng C, Doughty ML, et al. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature. 2003;425:917–925. [DOI] [PubMed] [Google Scholar]
- 12. York B, Yu C, Sagen JV, et al. Reprogramming the posttranslational code of SRC-3 confers a switch in mammalian systems biology. Proc Natl Acad Sci USA. 2010;107:11122–11127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and β-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–419. [DOI] [PubMed] [Google Scholar]
- 14. Ribar TJ, Epstein PN, Overbeek PA, Means AR. Targeted overexpression of an inactive calmodulin that binds Ca2+ to the mouse pancreatic β-cell results in impaired secretion and chronic hyperglycemia. Endocrinology. 1995;136:106–115. [DOI] [PubMed] [Google Scholar]
- 15. Carter JD, Dula SB, Corbin KL, Wu R, Nunemaker CS. A practical guide to rodent islet isolation and assessment. Biol Proced Online. 2009;11:3–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ferrara CT, Wang P, Neto EC, et al. Genetic networks of liver metabolism revealed by integration of metabolic and transcriptional profiling. PLoS Genet. 2008;4:e1000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Newgard CB, An J, Bain JR, et al. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. 2009;9:311–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. York B, Reineke EL, Sagen JV, et al. Ablation of steroid receptor coactivator-3 resembles the human CACT metabolic myopathy. Cell Metab. 2012;15:752–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. An J, Muoio DM, Shiota M, et al. Hepatic expression of malonyl-CoA decarboxylase reverses muscle, liver and whole-animal insulin resistance. Nat Med. 2004;10:268–274. [DOI] [PubMed] [Google Scholar]
- 20. Benjamini YaH, Y Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol. 1995;57:289–300. [Google Scholar]
- 21. Li C, Wong WH. Model-based analysis of oligonucleotide arrays: expression index computation and outlier detection. Proc Natl Acad Sci USA. 2001;98:31–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lin F, Ribar TJ, Means AR. The Ca2+/calmodulin-dependent protein kinase kinase, CaMKK2, inhibits preadipocyte differentiation. Endocrinology. 2011;152:3668–3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Racioppi L, Noeldner PK, Lin F, Arvai S, Means AR. Calcium/calmodulin-dependent protein kinase kinase 2 regulates macrophage-mediated inflammatory responses. J Biol Chem. 2012;287:11579–11591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lin F, Marcelo KL, Rajapakshe K, et al. The camKK2/camKIV relay is an essential regulator of hepatic cancer. Hepatology. 2015;62:505–520. [DOI] [PMC free article] [PubMed] [Google Scholar]