

Abstract
A defining characteristic of type 1 diabetes mellitus (T1DM) pathophysiology is pancreatic β-cell death and dysfunction, resulting in insufficient insulin secretion to properly control blood glucose levels. Treatments that promote β-cell replication and survival, thus reversing the loss of β-cell mass, while also preserving β-cell function, could lead to a real cure for T1DM. The α-subunit of the heterotrimeric Gz protein, Gαz, is a tonic negative regulator of adenylate cyclase and downstream cAMP production. cAMP is one of a few identified signaling molecules that can simultaneously have a positive impact on pancreatic islet β-cell proliferation, survival, and function. The purpose of our study was to determine whether mice lacking Gαz might be protected, at least partially, from β-cell loss and dysfunction after streptozotocin treatment. We also aimed to determine whether Gαz might act in concert with an activator of the cAMP-stimulatory glucagon-like peptide 1 receptor, exendin-4 (Ex4). Without Ex4 treatment, Gαz-null mice still developed hyperglycemia, albeit delayed. The same finding held true for wild-type mice treated with Ex4. With Ex4 treatment, Gαz-null mice were protected from developing severe hyperglycemia. Immunohistological studies performed on pancreas sections and in vitro apoptosis, cytotoxicity, and survival assays demonstrated a clear effect of Gαz signaling on pancreatic β-cell replication and death; β-cell function was also improved in Gαz-null islets. These data support our hypothesis that a combination of therapies targeting both stimulatory and inhibitory pathways will be more effective than either alone at protecting, preserving, and possibly regenerating β-cell mass and function in T1DM.
Type 1 diabetes mellitus (T1DM) occurs when immune-mediated pancreatic β-cell destruction leads to near-absolute endogenous insulin deficiency (1, 2). T1DM manifests clinically when the β-cell mass drops below the threshold required to maintain normal glucose tolerance (1), although most patients with newly diagnosed T1DM still have the capacity to secrete insulin in amounts corresponding to 20%–30% of those of nondiabetics (3). This residual β-cell function is quite important for several reasons, including reducing the need for exogenous insulin, promoting better glycemic control, preserving the counterregulatory response to insulin, and potentially reducing diabetic complications (4–6).
The residual β-cell function observed in both early T1DM and pancreatic islet transplant (PIT) patients indicates the presence of a pool of potentially expandable β-cells. A real cure for T1DM might involve halting immune destruction and expanding any residual β-cell mass by increasing replication, cell size, and protection from apoptosis and cell death. Using immunosuppressives in T1DM patients in combination with insulin has shown some promise in some studies, but immunosuppressives themselves often have negative impacts on β-cell biology (7–9).
A more recent strategy for T1DM combination therapy comes from the obesity- and insulin-resistance-linked type 2 diabetes mellitus (T2DM) field. Out of all of the mechanisms of action of T2DM treatments currently in use, agents that stimulate β-cell cAMP production, including dipeptidyl peptidase 4 (DPP-4) inhibitors and glucagon-like peptide 1 (GLP-1) analogs, are the only ones that can positively impact on β-cell replication, neogenesis, and/or survival in rodent models (10–15). There is also evidence that GLP-1 receptor (GLP-1R) agonism positively impacts on replication and neogenesis in human islets (16). Even more interestingly, GLP-1 treatment can protect both rodent and human β-cells from immune-mediated destruction (17, 18). These in vitro analyses, coupled with the relative tolerability and widespread use of GLP-1 agonists and DPP-4 inhibitor in the T2DM field, have lent credence to testing these compounds in rodent models of T1DM and in human T1DM patients and PIT recipients. These studies show prolonged survival, improved glycemia, and maintenance of graft function for a longer duration (19–21). However, even in T2DM, GLP-1 analogs and DPP-4 inhibitors do not work or fail to be completely effective in a significant proportion of individuals, suggesting the existence of negative regulatory pathways that might be dysfunctional in the diabetic state.
The GLP-1R is a heterotrimeric guanine nucleotide binding protein (G protein)-coupled receptor that is coupled to the G protein, Gs. Of the 4 subfamilies of heterotrimeric G protein α-subunits, only those in the Gαs and Gαi subfamilies can regulate adenylate cyclase function, thus positively (Gαs) and negatively (Gαi) modulating cAMP production and concomitant downstream signaling events through changes in intracellular cAMP concentrations. The Gαi subfamily member, Gαz, has such a slow rate of inactivation that it has been proposed as one of, if not the only, Gαi protein that acts as a tonic negative regulator of cAMP production (22). We have previously demonstrated that Gαz is expressed and functional in the pancreatic islet, acting to negatively regulate insulin secretion (23, 24). Furthermore, Gαz-null mice are completely protected from developing glucose intolerance when subjected to long-term high-fat diet feeding due to a significantly increased β-cell replication level, augmenting β-cell mass to compensate for peripheral insulin resistance (25). This led us to hypothesize that β-cells from mice deficient in Gαz might be at least partially protected from the development of experimental diabetes when subjected to induction of diabetes through multiple low-dose streptozotocin (STZ) injections, a well-accepted model of T1DM. Furthermore, we hypothesized that the Gαz-null mutation might synergize with the GLP-1R agonist, exendin-4 (Ex4), perhaps eliciting a complete protection from the development of experimental diabetes. Finally, we hypothesized that removing an inhibitory constraint on cAMP production might allow for a lower dose of Ex4 than has been used in previous studies. In this work, we followed the development of diabetes in mice for 7 weeks after STZ induction, treated or not with 10-μg/kg Ex4. We collected pancreases to determine the levels of apoptosis and replication during the study. We also performed an ultralow dose STZ induction experiment to elicit a β-cell insult, which allowed us to collect islets and perform glucose-stimulated and incretin-potentiated insulin secretion assays in vitro, determining any protection of the Gαz-null mutation and Ex4 treatment on β-cell function. Finally, we used control and constitutively active Gαz constructs along with primary islet gene expression analyses to explore the cellular and molecular mechanisms responsible for the protective effects of Gαz loss in the β-cell. Together, our results support the Gαz pathway as a potential new target for therapeutics aimed at protecting, preserving, and augmenting functional β-cell mass in T1DM.
Materials and Methods
Antibodies, chemicals, and reagents
Sodium chloride (S9888), potassium chloride (P3911), magnesium sulfate heptahydrate (M9397), potassium phosphate monobasic (P0662), sodium bicarbonate (S6014), HEPES (H3375), calcium chloride dehydrate (C3881), and RIA-grade bovine serum albumin (A7888), STZ (S0130), and Ex4 (E7144) were purchased from Sigma-Aldrich. Antiinsulin ELISA antibodies (insulin + proinsulin antibody, 10R-I136a; insulin + proinsulin antibody, biotinylated, 61E-I136bBT) were from Fitzgerald Industries. The 10-ng/mL insulin standard (8013-K) and assay buffer (AB-PHK) were from Millipore. RPMI 1640 medium (11879–020, no glucose), penicillin/streptomycin (15070–063), and fetal bovine serum (12306C: qualified, heat inactivated, USDA-approved regions) were from Life Technologies. Dextrose (D14–500) was from Fisher Scientific. Rabbit anti-Gαz antibody (ab150434) was from Abcam. Mouse antiactin (8H10D10), goat antirabbit horseradish peroxidase (HRP)-conjugated secondary antibody (7074), and horse antimouse HRP-conjugated secondary antibody (7076) were from Cell Signaling Technology. Guinea pig antiinsulin, rabbit anti-Ki67, antibody diluent with background reduction and serum-free blocking agent were from Dako. Citrate based antigen retrieval solution (H-3300), Cy3-coupled antiguinea pig secondary antibody, FITC-coupled antirabbit secondary antibody, and Vectashield Mounting Medium with DAPI were from Vector Laboratories.
Animals
C57Bl/6 mice containing a genomic insertion of a pGKneor cassette 160 basepairs downstream of the translation start site of the Gαz gene (gene symbol Gnaz) have been described and characterized previously (25). Breeding colonies were housed in a limited access, pathogen-free facility on a 12-hour light, 12-hour dark cycle with ad libitum access to water and breeder chow (5058 PicoLab Mouse Diet 20; LabDiet). Gαz-null and wild-type (WT) control mice were generated by heterozygous matings to produce littermate pairs. Upon weaning, mice were housed 5 or fewer per cage with ad libitum access to lower-fat chow (5053 PicoLab Rodent Diet 20; LabDiet). All procedures were performed according to an approved protocol in accordance with the principles and guidelines established by the University of Wisconsin and Madison Veterans Affairs Hospital Institutional Animal Care and Use Committees.
Multiple low-dose STZ protocol for induction of diabetes
Seven-week study
Ten- to 12-week-old Gαz-null and WT mice were separated into 4 groups based on treatment: 1) citrate buffer control, 2) STZ (50 mg/kg), 3) Ex4 (10 μg/kg), or 4) STZ + Ex4. STZ was made at a concentration of 7.5 mg/mL, protected from light, at least 2 hours before injection to equilibrate the levels of the α- and β-enantiomers of STZ (26). Food was removed from the cages between 7 and 9 am for 4–6 hours before mice were given an ip injection of citrate buffer control, STZ, and/or Ex4. This procedure was repeated daily for 5 days. An initial (d 0) blood glucose measurement was recorded, and blood glucose measurements were taken every following Monday for 7 weeks using a blood glucose meter (AlphaTRAK) and rat/mouse-specific test strips. For mice treated with Ex-4 or citrate control, the treatment was repeated daily for the duration of the study. At the end of the study, mice were fasted for 4–6 hours before performing perfusion fixation of the pancreas and dissection for frozen sectioning or isolation of pancreatic islets for in vitro analysis.
One-week study
Mice were treated as above. Blood glucose measurements were taken daily for 5 days during the week of the STZ induction, and 1 week after the first injection. On Monday, after the first STZ treatment, mice were fasted 4–6 hours before performing perfusion fixation of the pancreas for frozen sectioning.
Pancreas perfusion and fixation
Mice were fasted 4–6 hours before sacrificing by CO2. Immediately after clinical death, mouse pancreases were flushed with PBS, perfused with 4% paraformaldehyde (PFA) in PBS, dissected, and fixed for 2 hours in 4% PFA. Next, pancreases were incubated overnight at 4°C in 30% sucrose and frozen at −80°C in Neg-50 frozen section medium (Thermo Scientific). For all immunohistochemical and immunofluorescence (IF) assays, 10-μm serial sections were cut on positively charged slides, with 18 sections per stop position (3/slide) and 3 stop positions per pancreas separated by at least 200 μm.
β-Cell fractional area
For each pancreas, one slide per stop position was postfixed, quenched of peroxidase activity with 2% H2O2, and immunostained using Vectashield Mounting Medium for Fluorescence with 4′,6-Diamidino-2-Phenylindole, (DAPI) according to the manufacturer's protocol (Vector Laboratories). Guinea pig antiinsulin primary antibody was diluted 1:500 in antibody diluent. After insulin immunostaining, the slides were counterstained with hematoxylin. Slides were imaged using an automated pan-and-stich microscope at ×2.5–×10 (AxioPlan or Evos). β-Cell fractional area was determined by quantifying the percent of insulin-positive pancreas area as a total of the full pancreas area for each section per slide, followed by averaging of the 3 sections, and averaging of the 3 slides per mouse. Images were analyzed using ImageJ (64-bit) software (National Institutes of Health, Bethesda, MD) with shading correction.
Nuclear Ki67 IF
The percent of Ki67-positive β-cell nuclei was determined from sections collected from mice 8 weeks after the initiation of STZ injections as a measure of β-cell replication, essentially as in Ref. 25, except that slides did not need to be deparaffinized. Briefly, antigen retrieval was performed with a heat-activated citrate-based unmasking solution. Rabbit anti-Ki67 was used at a 1:100 dilution in Dako antibody diluent with background reducing components. Guinea pig antiinsulin antibody (Dako) was used at a 1:400 dilution to mark β-cells. At least 2 slides per pancreas with 3 sections per slide, and separated by at least 200 μm, were quantified. Ki67 positivity was determined for each section, the values for all sections per slide averaged, and the values for each pancreas averaged to give each biological replicate. Images were collected using a 3-channel fluorescence inverted microscope at ×10 magnification. Ki67-positive nuclei were counted in Adobe Photoshop CS6. The percentage of total β-cell nuclei in each islet was determined using an image-based nuclei counter plugin for ImageJ (64-bit).
Nuclear terminal deoxynucleotidyl transferase 2′-deoxyuridine 5′-triphosphate nick end labeling (TUNEL) assay
The percent of TUNEL-positive β-cell nuclei was determined from sections collected from mice 1 week after the initiation of STZ injection as a measure of β-cell apoptosis. The TACS 2 TdT in situ apoptosis detection kit, fluorescein, was used according to manufacturer's protocol (Trevigen). Antigen retrieval was performed by incubating sections with 50-μL Cytonin (Trevigen) for 30 minutes at room temperature. After completing the streptavidin-fluorescein coupling, slides were washed in PBS and blocked for 30 minutes in 10% donkey serum. The guinea pig antiinsulin antibody was added at a 1:500 dilution in 1% donkey serum overnight. Slides were washed in PBS containing 0.1% Tween 20 and Cy3-coupled antiguinea pig secondary antibody incubated at a 1:400 dilution in 1% donkey serum at room temperature for 30 minutes in the dark. Next, slides were washed in PBS containing 0.1% Tween 20 and mounted in Vectashield mounting medium with DAPI. Slides were dried for 20–30 minutes and then imaged. At least 2 slides per pancreas with 3 sections per slide, and separated by at least 200 μm, were quantified. TUNEL positivity was determined for each section, the values for all sections per slide averaged, and the values for each pancreas averaged to give each biological replicate. TUNEL-positive nuclei were counted in Adobe Photoshop CS6. The percentage of total β-cell nuclei in each islet was determined using an image-based nuclei counter plugin for ImageJ (64-bit).
Ultra low-dose STZ β-cell insult
Gαz-null and WT mice ages 9–12 weeks were fasted 4–6 hours before given an ip injection daily for 5 days with a citrate control solution or 4 mg/kg of STZ, with or without a concurrent ip injection of Ex4 (10 μg/kg). Blood glucose measurements were taken daily for 5 days and then weekly for 2 weeks. Two weeks after the first STZ injection, islet isolation and glucose-stimulated insulin secretion (GSIS) assays were performed.
Islet insulin secretion assay
On the day of isolation, islets were picked into 100 μL of islet medium in each well of a 96-well V-bottom tissue culture-coated plate (catalog number 3894; Corning Life Sciences) using a P-20 micropipettor according to a protocol optimized in our laboratory (27). Insulin ELISAs were performed essentially as in Ref. 28.
Ins-1 (832/3) culture and adenoviral infection
In vitro studies were performed in the rat insulinoma cell line, Ins-1 (832/3). Cells were cultured in RPMI 1640 medium (Gibco) containing 1-g/L glucose supplemented with 10% heat-inactivated fetal bovine serum, 1mM sodium pyruvate, 2mM L-glutamate, 50μM βη-mercaptoethanol, and 100 U/mL of antibiotic-antimycotic (penicillin/streptomycin/fungizone; Gibco). Cells were seeded in 24-well plates or 96-well plates as described for each assay. The day after, cells were washed with PBS before incubation with Opti-MEM (Gibco) medium containing 3–5 × 107 plaque-forming units of adenovirus/mL at a multiplicity of infection of 30–50. After 2 hours, the medium was changed to antibiotic-free RPMI 1640 medium as described above. After 72 hours, cells were washed and used for experimental measurements.
Western blot analysis
Ins-1 832/3 cells were seeded at a density of 400 000 cells/well in a 24-well plate and adenovirally transduced and cultured as described above. To confirm transduction, Ins-1 (832/3) cells from 24-well plates were lysed with 1× RIPA buffer (Sigma) and sonicated 3 times for 10 seconds. Approximately 30-μg protein was loaded on the SDS-PAGE gel, transferred to Polyvinylidene fluoride membranes, and blotted with rabbit anti-Gαz (1:1000 dilution) and mouse anti-β-actin (1:1000 dilution), and then with the appropriate HRP-linked secondary antibody. Proteins were detected by enhanced chemiluminescence.
cAMP production assay
Cells were seeded at density of 100 000 cells/well in 96-well plates and adenovirally transduced and cultured as described above. On the day of assay, cells were washed and preincubated with modified Krebs-Ringer bicarbonate buffer with or without 200μM 3-isobutyl-1-methylxanthine (IBMX) for 45 minutes. Cells were then treated in modified Krebs-Ringer bicarbonate buffer containing 11.1mM glucose, with or without 200μM IBMX and 10μM forskolin in for 45 minutes. Media were removed and cells harvested essentially as in Ref. 29. Cellular cAMP content was analyzed using the Cayman Chemical cAMP EIA kit (catalog number 581001) according to the manufacturer's instructions.
β-Cell death assays
Ins-1 832/3 cells were seeded at a density of 100 000 cells/well in a 96-well plate and adenovirally transduced and cultured as described above. In some cases, 10μM STZ or 1-ng/mL IL-1β were added to the cells for the final 48 or 24 hours of culture, respectively. Trypan blue viability measurements or Promega ApoTox-Glo assays were performed according to manufacturer's instructions.
Quantitative real-time reverse transcription PCR (qRT-PCR)
Islets from WT and Gαz-null mice were isolated and cultured for 24 hours in normal islet medium or medium supplemented with 10μM equilibrated STZ or 1-ng/mL IL-1β before being collected for isolation of whole RNA using the QIAGEN RNeasy kit according to the manufacturer's instructions. cDNA was then generated with random primers (High-Capacity cDNA Reverse Transcription kit; Applied Biosystems). mRNA measurements of Gαz (gene symbol Gnaz), C/EBP homologous protein (Chop), tribbles homolog 3 (Trib3), Caspase 3 (Casp3), and Caspase 9 (Casp9) were made through SYBR quantitative RT-PCR and normalized to β-actin. Primer sequences can be found in Table 1.
Table 1.
Quantitative RT-PCR Primer Sequences
| Target | Forward | Reverse |
|---|---|---|
| Gnaz (mouse) | AAGGCGTCACAGCCATCATCTTCT | TGAGCGAGGTGTTGATGAACCAGT |
| Chop (mouse) | ACAGAGCCAGAATAACAGCCGGAA | TCTGCTTTCAGGTGTGGTGGTGTA |
| Trib3 (mouse) | CGTCGCTTTGTCTTCAGCAACTGT | TCCCACAGAGAGTCATCTGATCCA |
| Casp3 (mouse) | ATGGACAACAACGAAACCTCCGTG | CAGAGTCCATCGACTTGCTTCCAT |
| Casp9 (mouse) | CACGGCTTTGATGGAGATGG | CTGGCAGCCATGAGAGAGGA |
| β-Actin (human/mouse/rat) | TCAAGATCATTGCTCCTCCTGAGC | TTGCTGATCCACATCTGCTGGAAG |
Statistical analysis
Data are expressed as mean ± SEM unless otherwise noted. Data were compared one- or two-way ANOVA as appropriate and as described in the figure legends. P < .05 was considered statistically significant. Statistical analyses were performed with GraphPad Prism version 6 (GraphPad Software).
Results
Gαz-null mice treated with Ex4 are protected from STZ-induced hyperglycemia
Groups of 10- to 12 week-old WT and Gαz-null mice were subjected to the multiple low-dose STZ protocol with and without the addition of daily 10-μg/kg Ex4. Weights and blood glucose levels were taken weekly over a 7-week period. A group of WT mice injected with citrate buffer alone was included to show expected weights and blood glucose levels in the absence of STZ induction.
Although the Gαz-null mice trended towards being lighter, there were no statistically significant differences among the mean starting weights of mice in any of the 4 treatment groups (24.1 ± 1.7 g for WT; 27.2 ± 1.1 g for WT + Ex4; 23.3 ± 3.0 for Gαz-null and 23.8 ± 1.7 g for Gαz-null + Ex4; mean ± SD; n = 5–10/group; P > .05 for all comparisons). The change in weight from baseline did not statistically differ among the groups during most the study, although all 4 of the STZ-induced groups showed a trend towards decreased weight in the first few weeks as compared with the control animals (Figure 1A). In the absence of Ex4, weights in the WT and Gαz-null groups normalized after about 4 weeks, whereas both Ex4-treated groups trended towards maintaining this depressed body weight, a result that was statistically significant in the WT group at week 7 (Figure 1A, open orange circles). This is consistent with the known effect of Ex4 on body weight.
Figure 1.
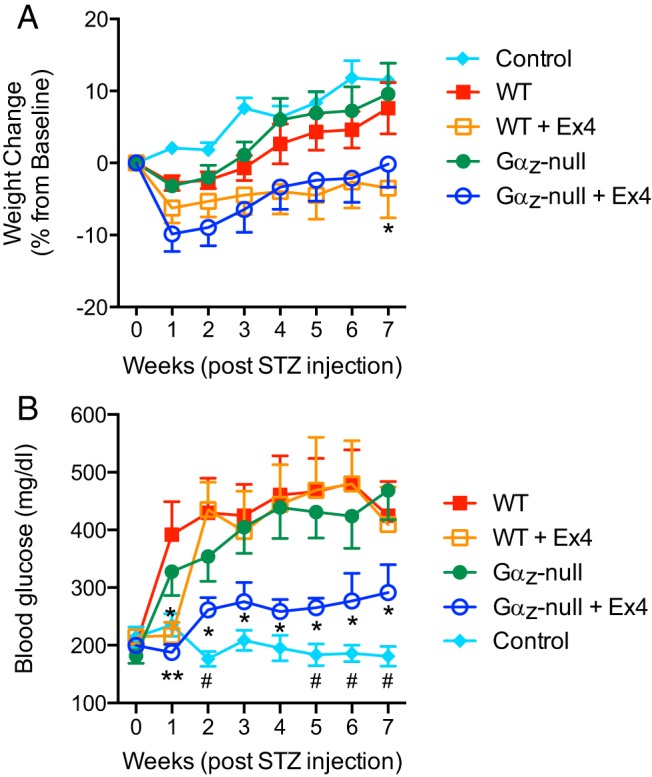
Ex4-treated, Gαz-null mice are protected from developing hyperglycemia after STZ induction. Mice were subjected to the multiple low-dose STZ induction of diabetes protocol, with weights and blood glucose levels recorded weekly for 7 weeks. A, Change in baseline body weights in the 4 groups of experimental mice (WT and Gαz-null, with and without Ex4 treatment) and the citrate buffer control group over the 7 week study. *, P < .05 vs WT. B, Blood glucose levels over the 7 week study. In both A and B, data are shown as the mean ± SEM; n = 5–7 mice per group. Data for each of the experimental groups was compared by two-way paired ANOVA with Dunnet's multiple comparisons test post hoc. *, P < .05 and **, P < .01 vs WT; #, P < .05 for Gαz-null + Ex4 vs control.
There were no statistically significant differences among the initial blood glucose levels of mice in any of the 4 treatment groups (Figure 1B, wk 0). In order to test the progression of diabetes in mice treated with STZ, fasting blood glucose measurements were taken each Monday for 7 weeks after the start of STZ injections. As expected, WT mice injected with STZ developed severe diabetes as measured by blood glucose more than 300 mg/dL 1 week after the first STZ injection (Figure 1B, red filled squares). The Gαz-null mutation alone was enough to slow the onset of severe hyperglycemia (Figure 1B, green filled circles), as was the treatment of WT mice with Ex4 (Figure 1B, orange open squares). The combination of the Gαz-null mutation with Ex4 treatment, however, was sufficient to fully protect mice from developing severe hyperglycemia during the entire 7-week study period, with mean blood glucose levels remaining below 300 mg/dL (Figure 1B, blue open circles). Blood glucose in the Gαz-null mice treated with Ex4 were elevated, however, as compared with WT control mice injected with a citrate buffer control solution (Figure 1B, turquoise diamonds), and this difference was statistically significant at weeks 2, 5, 6, and 7 after STZ injection.
Gαz-null mice treated with Ex4 exhibit increased β-cell proliferation and fractional area
At the end of the 7-week study, mouse pancreases were dissected, fixed in PFA, flash frozen and sectioned for immunostaining to elucidate the physiological mechanisms of diabetes protection in the Gαz-null mouse islets. Nuclear Ki67 was used as a marker for actively replicating cells (see Figure 2, A–D, for representative images: insulin, red; Ki67, green; and DAPI, blue). Upon quantification of Ki67-positive β-cells, islets from WT mice treated with Ex4 and Gαz-null mice alone showed significantly increased β-cell replication as compared with the WT STZ islets (Figure 2E). The mean Ki67 positivity was highest in the Gαz-null mouse islets treated with Ex4, but this difference was not significantly higher than those of the former 2 groups.
Figure 2.
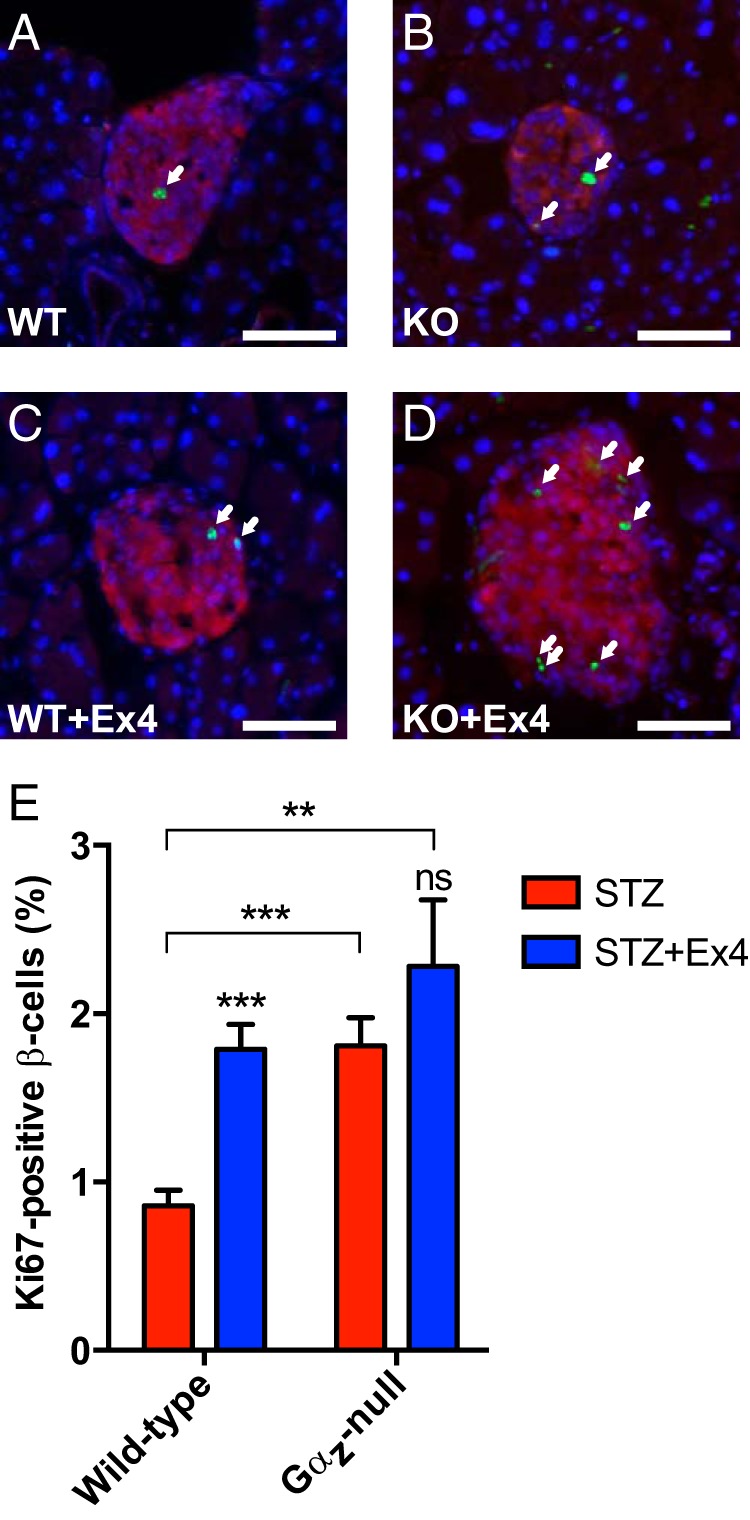
Gαz-null β-cells have an increased replication level independent of Ex4 treatment. A–D, Representative islet imaged from each experimental group subjected to IF analysis for insulin (red) and Ki67 (green), with a DAPI nuclear counterstain (blue). Ki67-positive β-cell nuclei are indicated by white arrows. In all 4 images, the scale bar represents 100 μm. E, Quantification of Ki67-positive β-cell nuclei calculated from all islets counted on 3 sections of each mouse pancreas separated by at least 200 μm. Data are shown as the mean ± SEM; n = 5–7 mice per group. Data for each of the 4 experimental groups was compared by two-way paired ANOVA with Dunnet's multiple comparisons test post hoc. **, P < .01 and ***, P < .001.
Likewise, the overall fractional area of the β-cells within the pancreas sectional area as a whole was measured in mice from each of the treatment groups, with the highest mean fractional area in the Gαz-null islets, regardless of Ex4 treatment (see Figure 3, A–D, for representative images). Pancreases from Gαz-null mice treated with Ex4 had significantly increased β-cell fractional area as compared with WT mice treated with Ex4 (Figure 3E, blue squares). However, all treatment groups had a lower mean β-cell fractional area as compared with WT control mice injected with citrate vehicle (Figure 3E, cyan diamonds), and this difference between the means was statically significant in all groups.
Figure 3.

Gαz-null mice treated with Ex4 have increased β-cell fractional area. A–D, Representative pancreas sections from each experimental group subjected to immunohistochemical analysis for insulin (brown) and counterstained with hematoxylin (purple). In all 4 images, the scale bar indicates 1 mm. A 4-fold enlarged section is shown in an inset in each image, with black arrows indicating the insulin-positive area that was quantified. E, Quantification of β-cell fractional area calculated from all islets counted on 3 sections of each mouse pancreas separated by at least 200 μm. Data are shown as the mean ± SEM; n = 5–7 mice per group. Data were compared by one-way ANOVA with Dunnet's multiple comparisons test post hoc. a, P < .05 vs control; b, P < .05 vs control and P < .05 vs WT STZ + Ex4.
Gαz-null mice treated with Ex4 exhibit decreased β-cell apoptosis
To determine the impact of the Gαz-null mutation on β-cell death, which occurs early after STZ induction, duplicate groups of mice were subjected to multiple low-dose STZ injections with or without the addition of Ex4. Blood glucose levels were recorded daily until the day of pancreas collection (7 d after the start of STZ; equivalent to wk 1 time point in Figure 1B). As above, the Gαz-null mice treated with Ex4 had significantly reduced blood glucose levels as compared with the WT STZ group (Figure 4A, blue open circles vs red filled squares). Pancreata were dissected, fixed, flash-frozen, and sectioned for IF imaging. A fluorescent TUNEL probe was used as a marker for nuclei with fragmented DNA, a marker of both apoptosis and necrosis (see Figure 4, B–E, for representative images). Ex4 treatment significantly reduced β-cell death in the WT group, but as above with β-cell replication, the Gαz-null mutation itself had a significant effect on reducing β-cell death. The treatment of Gαz-null mice with Ex4 had no additional effect on β-cell death (Figure 4F).
Figure 4.
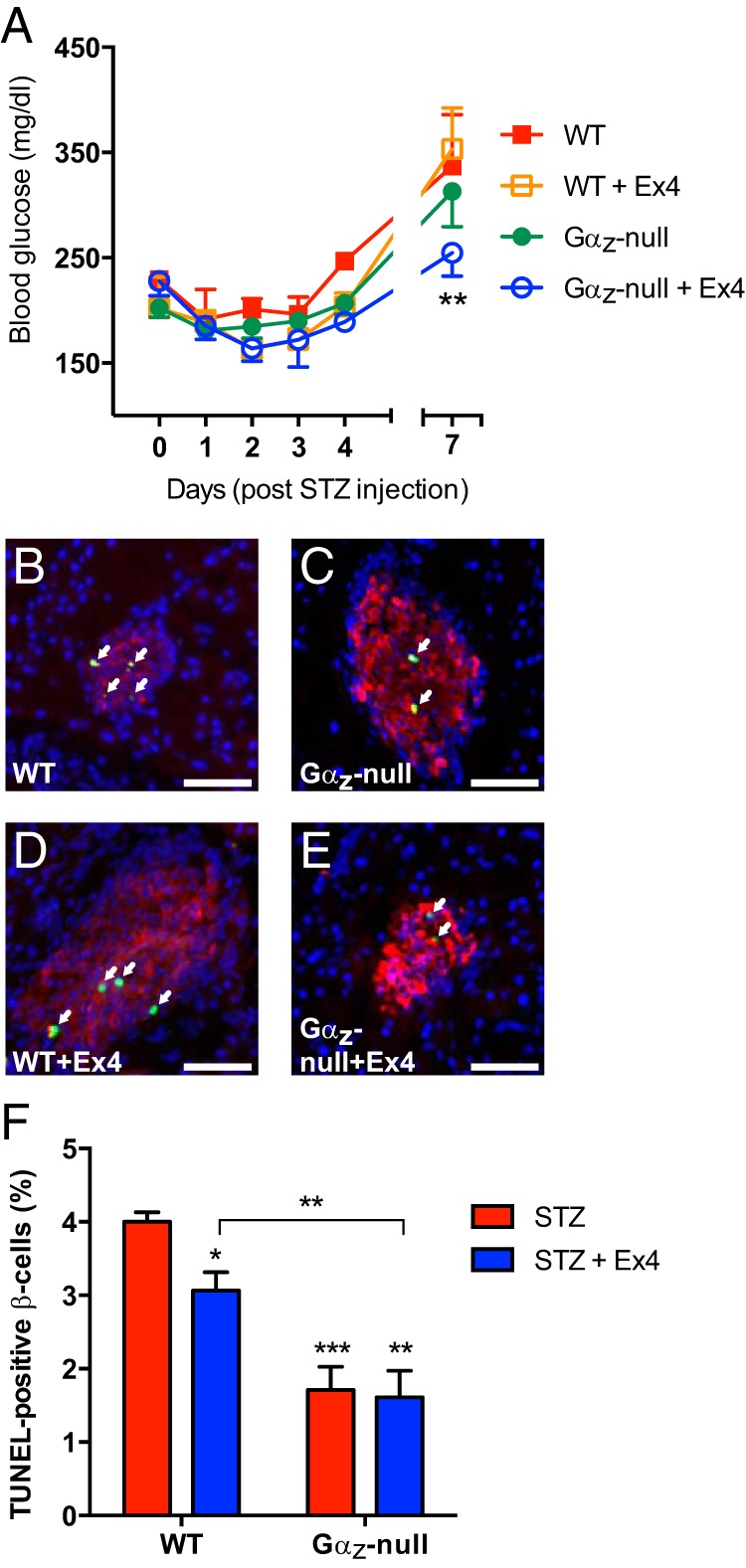
Gαz-null mice, treated or not with Ex4, have decreased β-cell death after STZ induction. A, Blood glucose levels from each group of mice subjected to multiple low-dose STZ induction of diabetes and sacrificed 2 weeks after the initiation of STZ injection. B–E, Representative islet images from pancreas sections of each group subjected to IF analysis for insulin (red) and TUNEL assay (green), with a DAPI nuclear counterstain (blue). TUNEL-positive β-cell nuclei are indicated by white arrows. In all 4 images, the scale bar represents 100 μm. F, Quantification of TUNEL-positive β-cell nuclei calculated from all islets counted on 3 sections of each mouse pancreas separated by at least 200 μm. Data are shown as the mean ± SEM; n = 5–7 mice per group. Data were compared by one-way ANOVA with Dunnet's multiple comparisons test post hoc. *, P < .05; **, P < .01; and ***, P < .001.
Gαz signaling inhibits β-cell survival by several different mechanisms
Having confirmed that loss of Gαz could protect mouse islets from β-cell death, we aimed to determine whether activated Gαz would promote death in the Ins-1 (832/3) β-cell line. First, we confirmed that we could express WT and constitutively active Gαz in the cAMP-responsive Ins-1 (832/3) cells and obtain the expected effect on cellular signaling using a protocol previously optimized in the related Ins-1 (832/13) cell line (24). We used adenoviral expression constructs to overexpress WT or constitutively active Gαz (Gαz(WT) or Gαz(QL), respectively) or a green fluorescent protein (GFP) control virus to monitor infection efficiency. Western blot analysis showed significant overexpression of with both WT and constitutively active Gαz as compared with GFP control (Figure 5A), with more than 90% infection efficiency (data not shown). Next, we performed cAMP production assays 48 hours after infection. Gαz is a known negative regulator of adenylate cyclase, and its activation should result in a significant decrease in cAMP production (24). When normalized to a control condition in which the phosphodiesterase inhibitor IBMX was not added, mean cAMP production was blunted (although this change was not statistically significant) with overexpression of Gαz(WT) (which is estimated to be approximately 30% active at any one time) (22) and was significantly reduced (∼50%) with constitutively active Gαz(QL) (Figure 5B), confirming the Gαz constructs function as expected.
Figure 5.
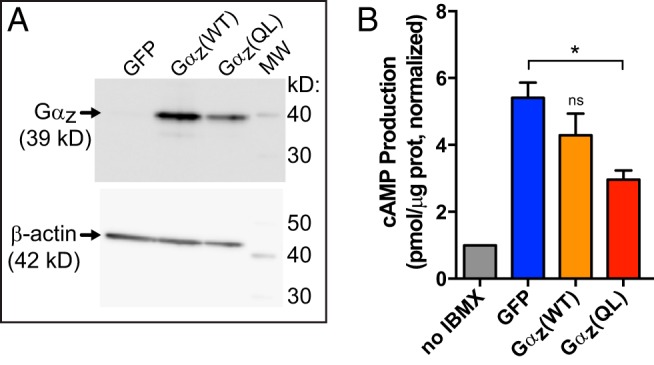
Functional characterization of WT and constitutively active Gαz constructs in the Ins-1 (832/3) cell line. A, Representative Western blot analysis of Ins-1 (832/3) cell lysates adenovirally transduced with Gαz variants, both WT and constitutively active (QL), or a GFP control adenovirus. Both Gαz(WT) and Gαz(QL) are highly overexpressed as compared with endogenous Gαz. β-Actin was used as a loading control. B, Gαz(WT) and Gαz(QL) function as expected to reduce cAMP production in Ins-1 (832/3) cells. Data are combined from 3 independent experiments, each normalized to the basal cAMP production when the phosphodiesterase inhibitor, IBMX, was not included. In the 3 experimental conditions, 10μM forskolin was added to stimulate cAMP production. Data are shown as the mean ± SEM; n = 3 independent experiments/group. Data were compared by one-way ANOVA with Dunnet's multiple comparisons test post hoc. *, P < .05.
As a first determination of the effect of Gαz on β-cell survival, we harvested Ins-1 (832/3) cells 72 hours after infection to determine the number of cells per well in each condition and the percent of those cells that remained viable. This was performed by trypan blue uptake, a marker of compromised plasma membrane integrity occurring late in the cell death process. Both Gαz(WT) and Gαz(QL) reduce the total number of Ins-1 (832/3) cells, with Gαz(QL) having the greater effect. There was about a 25% reduction in total cell number (Figure 6A, green bars) and percent viable cells (Figure 6A, orange bars) with Gαz(WT), and a 40% reduction in total cell number and percent viable cells with Gαz(QL) (Figure 6A, right).
Figure 6.

Gαz negatively regulates insulinoma cell survival through multiple mechanisms. A, Total Ins-1 (832/3) cells (green bars) and viable Ins-1 (832/3) cells (orange bars) remaining 72 hours after transduction with adenoviruses expressing the indicated proteins. Data are shown as the mean ± SEM; n = 6 independent experiments per group. Data were compared by one-way ANOVA with Dunnet's multiple comparisons test post hoc. **, P < .01 and ****, P < .0001 vs GFP. B–D, Ins-1 (832/3) cells were transduced with adenoviruses expressing the indicated proteins and treated with vector control (blue bars), 10μM STZ (pink bars), or 1-ng/mL IL-1β (red bars). Nonvirally transduced cells were used as a control for effects of viral expression alone (black and gray bars in B–D). ApoToxGlo assays (Promega) were performed to determine viability (B), cytotoxicity (C), and cleaved caspase (D). In all panels, data are shown as the mean ± SEM; n = at least 9 separate determinations combined from 4 separate assays. Virally transduced results were compared by one-way ANOVA with Tukey's multiple comparisons test post hoc. “No virus” results were compared by t test. *, P < .05; **, P < .01; ***, P < .001; and ****, P < .0001 as compared with the no virus or LacZ control, as appropriate, unless otherwise indicated. a, P < .05; b, P < .01; c, P < .001; d, P < .0001; and e, not significant as compared with the appropriate virus alone (ie, Gαz(WT) or Gαz(QL)).
Next, we aimed to determine which cellular mechanisms might be responsible for the effect of Gαz on β-cell survival. We tested viability, cytotoxicity, and apoptosis using the ApoToxGlo assay (Promega). Because this assay incorporates a fluorescent component, and having just validated our virus transduction efficiency of the Ins-1 (832/3) line, we switched to a LacZ-expressing negative control adenovirus. Nonvirally transduced cells were used as a control for any effect of adenoviral infection. Expression of Gαz(WT) or Gαz(QL) significantly reduced β-cell viability (Figure 6B, blue bars); increased β-cell cytotoxicity (Figure 6C, blue bars), and increased β-cell apoptosis (Figure 6D, blue bars) as compared with LacZ-expressing cells, with constitutively active Gαz(QL) having the greater effect in all cases. Gαz(QL) expression alone reduced Ins-1 cell viability by approximately 30% and increased cytotoxicity and apoptosis by approximately 400%.
Next, we aimed to determine the effect of activated Gαz on known negative effectors of β-cell survival: STZ and IL-1β. First, we treated control or LacZ-, Gαz(WT)-, or Gαz(QL)-expressing Ins-1 (832/3) cells with 10μM STZ for 48 hours or 1-ng/mL IL-1β for 24 hours. STZ had little to no effect on insulinoma cell viability in either the uninfected or LacZ-expressing cells, whereas cells expressing either Gαz(WT) or Gαz(QL) exhibited an approximate 40% decrease in viability (Figure 6A, medium gray and pink bars). In analyzing cytotoxicity, STZ treatment did not significantly impact β-cell cytotoxicity in LacZ-expressing cells, but it significantly augmented Gαz(WT)-mediated cytotoxicity (Figure 6C, pink bars). STZ did not increase cytotoxicity in Gαz(QL)-expressing cells. Finally, STZ did not significantly increase apoptosis in LacZ-expressing cells, and it did not further increase apoptosis in Gαz(WT)- or Gαz(QL)-expressing cells (Figure 6D, pink bars). Control cells that were not transduced with adenovirus were still responsive to STZ treatment by increasing cytotoxicity and apoptosis (Figure 6, C and D, medium gray bars).
In contrast to STZ, IL-1β significantly reduced Ins-1 (832/3) cell viability, by approximately 50% in all cases, with no additional impact of Gαz-coexpression (Figure 6B, red bars). The combined effects of IL-1β and Gαz expression on Ins-1 (832/3) cell cytotoxicity and apoptosis were almost identical to those mediated by STZ, with 1-ng/mL IL-1β significantly augmenting only Gαz(WT)-mediated cytotoxicity (Figure 6C, red bars) and having no additional effect on apoptosis in any of the virally expressing cells (Figure 6D, red bars). As with STZ treatment, IL-1β had no effect on cytotoxicity or apoptosis in LacZ-expressing cells, whereas control cells that were not transduced with adenovirus were still responsive to IL-1β by increasing cytotoxicity and apoptosis (Figure 6, C and D, medium gray bars).
Because of the strong effect of Gαz on apoptosis and the known role of apoptosis in multiple low-dose STZ induction, we aimed to elucidate putative molecular mechanisms responsible for its effects using islets isolated from WT and Gαz-null mice and the same STZ and IL-1β treatments described above. mRNA for Gnaz, the gene encoding Gαz, was significantly elevated in WT mouse islets by STZ treatment (Figure 7, orange vs red bars), although no significant changes were observed in any of the endoplasmic reticulum (ER) stress or proapoptotic genes analyzed (data not shown). As expected, Gnaz mRNA was significantly reduced in both the Gαz-null control and STZ-treated groups, likely reflecting background qRT-PCR amplification (Figure 7, blue and green bars).
Figure 7.
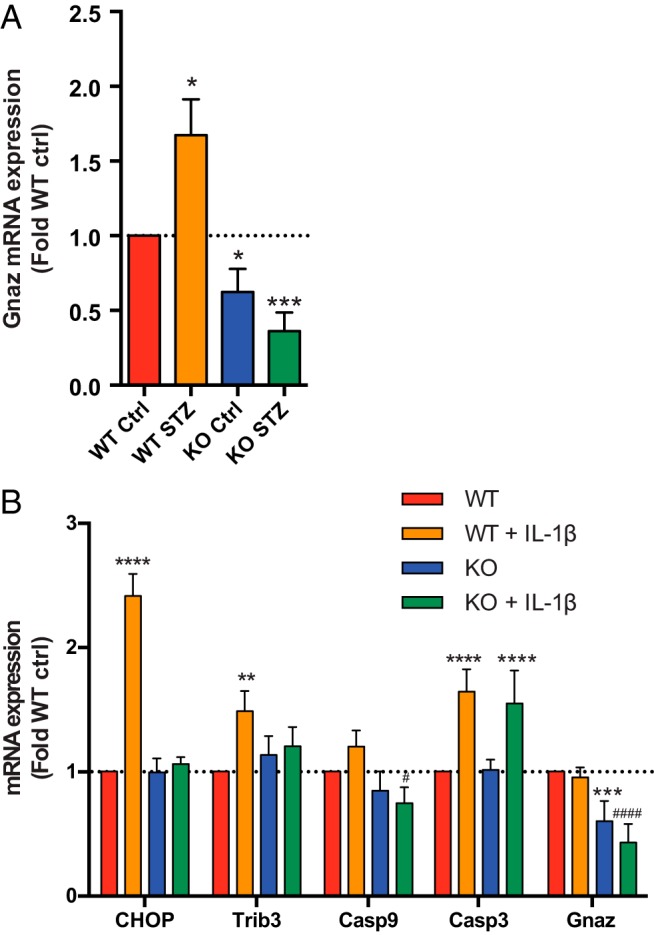
Gαz-null islets have altered changes in gene expression with STZ or IL-1β treatment. A, Islets isolated from WT or Gαz-null (GzKO) mice were treated or not with STZ and subjected to mRNA expression analysis for the Gαz gene (Gnaz). Gαz expression is elevated with STZ treatment in WT islets, but not in Gαz-null islets. *, P < .05 vs WT Ctrl and ###, P < .001 vs WT STZ. B, Islets isolated from WT or GzKO mice were treated or not with IL-1β and subjected to mRNA expression analysis for Gnaz and a number of ER stress/proapoptotic genes. Chop, Trib3, Casp3, and Casp9 are all elevated in WT islets treated with IL-1β but not Gαz-null islets. **, P < .01 and ****, P < .0001 vs WT or KO, as appropriate; #, P < .05 and ####, P < .0001 vs WT + IL-1β.
As STZ induces massive β-cell death in vivo through recruitment of proinflammatory immune cells to the islet, a process that would be absent in islets treated with STZ ex vivo, we repeated these experiments with IL-1β (Figure 7B). Interestingly, Gαz expression was unchanged by IL-1β treatment (Figure 7B, right). Also in contrast to STZ treatment, the ER stress/proapoptotic genes Chop, Trib3, Casp3, and Casp9 expression were all elevated in WT islets treated with IL-1β, the former 3 significantly. In all cases but for Casp3, however, this elevation was ameliorated by loss of islet Gαz expression (Figure 7B, green bars).
The Gαz-null mutation preserves β-cell function under insult
With the traditional β-cell ablation of the STZ induction protocol, isolating pancreatic islets from severely diabetic WT mice is not possible. Therefore, we performed an ultralow-dose STZ protocol (10-fold less than the low-dose protocol above) in groups of WT and Gαz-null mice, to provide a β-cell insult without islet destruction (a pilot experiment was performed with groups of STZ-injected, Ex4-treated mice as well, but the results were not significantly different from STZ alone; therefore, for the full experiment only data from control and STZ-injected mice is shown). Whether injected with STZ or citrate control, the mean blood glucose levels of all groups of mice was not elevated over the 3-week protocol (Figure 8A), suggesting that any decreases in β-cell mass were not sufficient to alter glucose tolerance. Two weeks after the initial STZ injection (d 14), islets were isolated from all 4 groups of mice and GSIS assays were performed. Even though the blood glucose levels of WT mice subjected to STZ-induced β-cell insult were normal, the β-cell function was reduced by about 50% in high glucose as compared with mice receiving a control solution (Figure 8B, left, red bars). In contrast, islets isolated from Gαz-null mice injected with the control solution had elevated GSIS as compared with WT mice treated with control solution, and the ultralow-dose STZ protocol had little effect on reducing this GSIS response, which was still elevated over WT control-treated mouse islets (Figure 8B, right, red bars). Next, the potentiation of GSIS in response to Ex4 was quantified in the STZ-treated mouse islets. Islets isolated from both genotypes of STZ-treated mice had more than a doubling in GSIS with Ex4 treatment, but due to their elevated baseline GSIS response, the absolute percent insulin secreted with Ex4 was significantly higher in the Gαz-null STZ-treated mouse islets (Figure 8B, blue vs red bars). This appeared to be due to a primary effect on the insulin secretion process itself, as insulin content did not differ among any of the groups (Figure 8C). Taken together, our data support an additive effect on insulin secretion with the Gαz-null mutation and Ex4 stimulation. This result, combined with the effects of Gαz loss itself on β-cell replication and survival, appears to explain the relatively complete protection of Gαz-null, Ex4-treated mice from STZ-induced diabetes.
Figure 8.
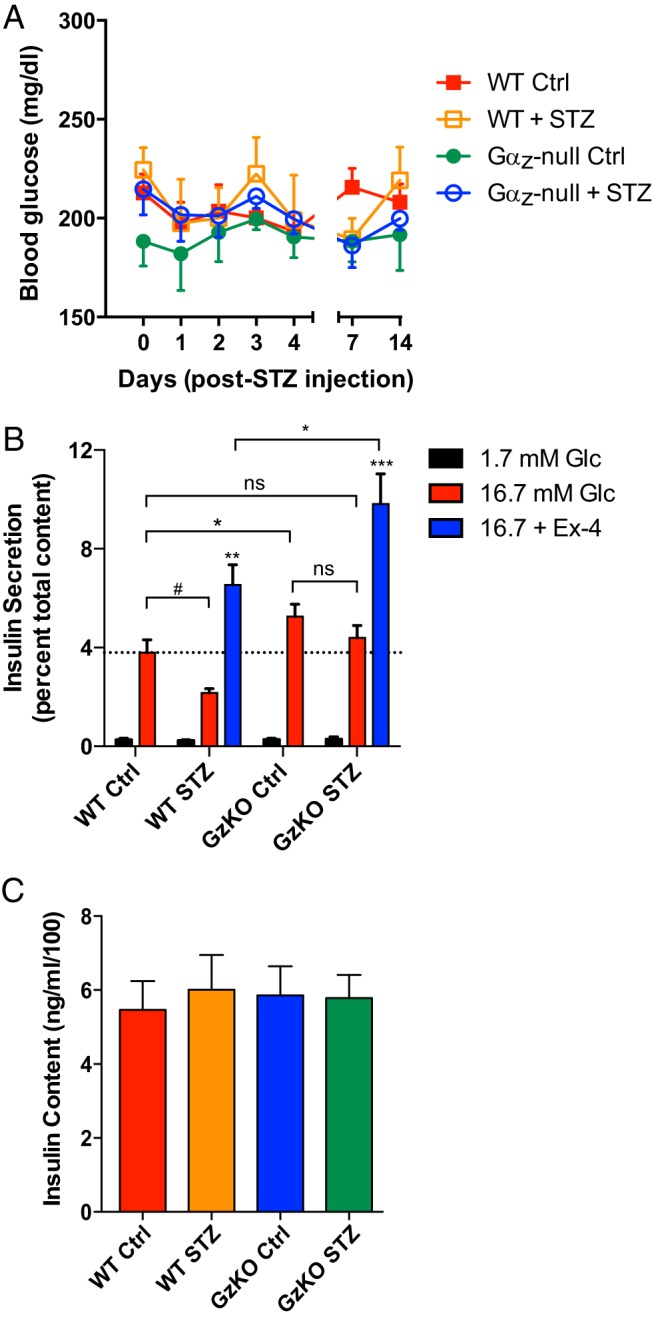
Gαz-null islets have augmented function even in the face of β-cell insult. A, Mice were injected with either citrate control solution or a 10-fold reduced dose of STZ in order to provide a β-cell insult. A, Blood glucose levels recorded during the 3-week time period indicate no mice became hyperglycemic. B, Islets isolated from the 4 groups of mice were analyzed for their glucose-stimulated (and in some cases Ex4-potentiated) insulin secretion response. C, Insulin content measured from the 4 groups of islets indicates no gross differences among the groups. In all 3 panels, data are shown as the mean ± SEM; n = 3 mice/group. Data were compared by one-way ANOVA with Dunnet's multiple comparisons test post hoc. #, P = .06; *, P < .05; **, P < .01; ***, P < .00; ns, not significant.
Discussion
In this study, we confirm that mice lacking the heterotrimeric G protein α-subunit, Gαz, are protected from developing severe hyperglycemia with multiple low-dose STZ induction (a well-accepted model for T1DM) when treated concurrently with the GLP-1R agonist, Ex4. Gαz is a unique Gα-subunit whose tissue distribution outside of the brain is quite limited (30). In the β-cell, Gαz is a tonic negative regulator of GSIS (23, 24), mediated at least in part through the prostaglandin E receptor 3 (EP3) (24, 25). In support of Gαz as a tonic inhibitor of insulin secretion, lean mice genetically deficient for Gαz have a stronger GSIS response and a more effective clearance of glucose from the blood (23). Interestingly, Gαz-null mice fed a high-fat diet undergo a dramatic increase in β-cell replication and mass are fully resistant to developing glucose intolerance (25). Furthermore, antagonizing the dysfunctionally up-regulated EP3 receptor in islets isolated from severely hyperglycemic BTBR-LeptinOb mice restores proper β-cell function; in particular, the responsiveness of the islets to Ex4 (28). These results in T2DM models served as the rationale for testing the effect of Gαz loss, with or without concurrent Ex4 treatment, on STZ-induced diabetes. The fact that GLP-1 agonists themselves had been shown to be at partially protective against the development of hyperglycemia in chemically induced and genetic models of T1DM (31–33) lent support to our hypothesis that relieving signaling through an inhibitory constraint on β-cell function and mass might make a GLP-1 agonist fully protective against severe hyperglycemia.
Other studies have found genetic or chemical alteration of specific protein levels or activities protect from STZ-induced diabetes, but most of these proteins are transcription factors or cell cycle-regulatory genes (such as FoxM1, p21cipYan, p16INK4a, Hnf4g, Mig6, and c-Abl) (34–38). Few proteins that could be direct pharmaceutical targets have been discovered to protect from STZ-induced diabetes. Notable exceptions are a combination of epidermal growth factor and gastrin, which was shown to promote β-cell regeneration and reduce hyperglycemia in chemical and genetic models of T1DM and PIT (39–42). Haploinsufficiency in Mig6, a negative regulator of epidermal growth factor receptor signaling up-regulated by inflammatory cytokines, was similarly protective against chemically induced diabetes (43). The GLP-1R itself, however, is probably the most well-studied pharmacological target for preventing hyperglycemia during diabetes induction, with very promising results. A GLP-1/gastrin combination protected from hyperglycemia in the Non-obese diabetic strain as well as a PIT model (44). Ex4 was shown to ameliorate STZ-induced diabetes in a β-cell GLP-1R-dependent manner (31, 32). In contrast to these previous works, however, we did not find a protective effect of GLP-1 agonist treatment alone on STZ-induced hyperglycemia (Figure 1B). It is our belief differences in the GLP-1 analog dose used are responsible for these discrepancies. The dose that we chose (10 μg/kg) is up to 20 times less than that used in the above-referenced studies among others (100–200 μg/kg). Supporting this, administration of 10-μg/kg Ex4 was insufficient to protect rats from developing hyperglycemia after STZ treatment (whereas interestingly, renal injury was still significantly improved) (45). We feel our study design with a subtherapeutic dose of Ex4 is ideal for studying a putative combination therapy. Being able to reduce the dose of GLP-1 analog used reduces the potential for negative outcomes, including the still controversial yet serious risk of pancreatitis and pancreatic and thyroid cancers (46, 47).
GLP-1 agonists such as Ex4 have long been known to promote β-cell replication and survival (10–15, 32). Supporting these results, we demonstrate that at even at 10 μg/kg, Ex4 promotes β-cell replication to the same extent as deletion of Gαz (Figure 2), supporting their actions through a common pathway. Both Ex4 treatment and Gαz loss have been shown to promote islet cAMP production, a known stimulator of β-cell proliferation. In contrast to results of some of these studies, including a paper by Li et al (32), which used a very similar STZ induction and Ex4 injection protocol and timeline, we found a much smaller (although still statistically significant) effect Ex4 on promoting β-cell survival (Figure 4). We propose this is due to the 10-fold lower dose of Ex4 than that used in this previous study. To summarize, in our hands, most the protective effect on reducing β-cell apoptosis was due solely to loss of Gαz (Figure 4). Synergy between active Gαz and known regulators of β-cell death directly relevant to our study, STZ and IL-1β (Figures 6 and 7), helps to explain why loss of this inhibitory G protein may be able to so strongly preserve β-cell replication and survival.
In our previous work, we have shown that relieving inhibition on cAMP production by a dysfunctionally up-regulated EP3 signaling pathway works in concert with Ex4 to promote GSIS from islets isolated from hyperglycemic BTBR-Ob mice, a model of type 2 diabetes (28). In BTBR-Ob islets, up-regulation of this prostaglandin E2/EP3/Gαz signaling pathway is due to increased EP3 receptor expression and endogenous ligand production occurring in T2D state. In the current work, we propose that up-regulation of Gαz expression in the β-cell itself may be responsible for increased signaling through this inhibitory pathway (Figure 7). Although islet Gαz mRNA expression was only up-regulated by STZ treatment and not IL-1β treatment (Figure 7B), physiologically, increased IL-1β would be come after the initial insult given by STZ from invading immune cells (48).
With IL-1β treatment, however, up-regulation of the early ER stress/proapoptotic genes, Chop and Trib3, were completely blunted by loss of Gαz. CHOP is a canonical marker of chronic ER stress leading to apoptosis (49). Trib3 is directly up-regulated by the ATF4/Chop pathway and required for CHOP-dependent apoptosis (50), and is also an inhibitor of protein kinase B/Akt signaling (51). Supporting a role for Gαz in promoting the intrinsic, ER stress-initiated apoptosis pathway, expression of Casp9 was significantly lower in IL-1β-treated Gαz-null islets as compared with IL-1β-treated WT islets (although the up-regulation of Casp9 expression by IL-1β in WT islets was not statistically significant). Proinflammatory cytokines can activate both the intrinsic and extrinsic apoptosis pathways (52), suggesting a specific role for Gαz in regulating the intrinsic pathway. Expression of Casp3, which mediates both the intrinsic and extrinsic apoptosis pathways, was unaffected by loss of Gαz; this is not unexpected, as Casp3 acts as a convergent effector for both apoptotic pathways (53). It is also possible that we saw no difference in Casp3 expression because the catalytic activity of the protein requires proteolytic cleavage to reveal the active site, which we were not able to capture using our qRT-PCR method (54). It will be interesting to further delineate the molecular mechanisms by which Gαz regulates an initial β-cell insult and the resulting cellular response to the immune reaction.
Finally, although we have not specifically targeted a receptor in this work, the deletion of Gαz is the proof-of-concept that blocking a Gz-coupled receptor would have a positive impact on diabetes protection. A number of clinical trials are testing the impact of stable GLP-1 analogs on T1DM progression or islet transplantation outcomes, with very promising results thus far (55–58). It is possible, however, that relieving an inhibitory constraint on β-cell growth, proliferation, and function could help to amplify these positive results. The possibility of using a putative Gαz-coupled receptor antagonist in concert with a well-accepted T2D diabetes therapeutic such as exenatide or liraglutide makes our work quite appealing. Furthermore, the tissue expression of Gαz is the most limited out of all of the nonsensory cell Gαi subunits (30), meaning that a peripherally restricted Gαz inhibitor could likely be designed that was essentially β-cell specific. It is our hope that the current work will spur research and development into EP3-specific antagonists with appropriate pharmacological properties for use in vivo so that these ideas can be tested in preclinical models.
Acknowledgments
We thank all of the talented and dedicated undergraduate assistants in the laboratory for their technical assistance, in particular Haley Wienkes, who prepared cDNA samples for this manuscript.
This work was supported by the JDRF Grant 17–2011-608 (to M.E.K.), the National Institutes of Health/National Institute of Diabetes and Digestive and Kidney Diseases Grant R01 DK102598 (to M.E.K.), the American Diabetes Association Grant 1–14-BS-115 (to M.E.K.), and the University of Wisconsin-Madison (M.E.K.). A.L.B. was funded in part through a Summer Undergraduate Research Fellowship from the American Society of Pharmacology and Experimental Therapeutics. M.T.C. was funded in part through a Minority Undergraduate Internship Award from the American Diabetes Association. H.K.B. was funded in part through a Hilldale Undergraduate/Faculty Research Award from the University of Wisconsin-Madison. This material is the result of work supported in part with the resources and use of facilities at the William S. Middleton Memorial Veterans Hospital, Madison, WI.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- Casp3
- Caspase 3
- Casp9
- Caspase 9
- Chop
- C/EBP homologous protein
- DAPI
- 4′,6-Diamidino-2-Phenylindole
- DPP-4
- dipeptidyl peptidase 4
- EP3
- prostaglandin E receptor 3
- ER
- endoplasmic reticulum
- Ex4
- exendin-4
- GFP
- green fluorescent protein
- GLP-1
- glucagon-like peptide 1
- GLP-1R
- GLP-1 receptor
- GSIS
- glucose-stimulated insulin secretion
- HRP
- horseradish peroxidase
- IBMX
- 3-isobutyl-1-methylxanthine
- IF
- immunofluorescence
- PFA
- paraformaldehyde
- PIT
- pancreatic islet transplant
- qRT-PCR
- quantitative real-time reverse transcription PCR
- STZ
- streptozotocin
- T1DM
- type 1 diabetes mellitus
- T2DM
- type 2 diabetes mellitus
- Trib3
- tribbles homolog 3
- TUNEL
- terminal deoxynucleotidyl transferase dUTP nick end labeling
- WT
- wild type.
References
- 1. Mathis D, Vence L, Benoist C. β-Cell death during progression to diabetes. Nature. 2001;414:792–798. [DOI] [PubMed] [Google Scholar]
- 2. Devendra D, Liu E, Eisenbarth GS. Type 1 diabetes: recent developments. BMJ. 2004;328:750–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Madsbad S, Krarup T, Regeur L, Faber OK, Binder C. Insulin secretory reserve in insulin dependent patients at time of diagnosis and the first 180 days of insulin treatment. Acta Endocrinol (Copenh). 1980;95:359–363. [DOI] [PubMed] [Google Scholar]
- 4. Madsbad S. Prevalence of residual B cell function and its metabolic consequences in type 1 (insulin-dependent) diabetes. Diabetologia. 1983;24:141–147. [DOI] [PubMed] [Google Scholar]
- 5. Palmer JP, Fleming GA, Greenbaum CJ, et al. C-peptide is the appropriate outcome measure for type 1 diabetes clinical trials to preserve β-cell function: report of an ADA workshop, 21–22 October 2001. Diabetes. 2004;53:250–264. [DOI] [PubMed] [Google Scholar]
- 6. Steffes MW, Sibley S, Jackson M, Thomas W. β-Cell function and the development of diabetes-related complications in the diabetes control and complications trial. Diabetes Care. 2003;26:832–836. [DOI] [PubMed] [Google Scholar]
- 7. Johnson JD, Ao Z, Ao P, et al. Different effects of FK506, rapamycin, and mycophenolate mofetil on glucose-stimulated insulin release and apoptosis in human islets. Cell Transplant. 2009;18:833–845. [DOI] [PubMed] [Google Scholar]
- 8. Martin F, Bedoya FJ. Effects of cyclosporine A on cyclic AMP generation and GTP-binding proteins in isolated islets. Biochem Pharmacol. 1992;44:359–364. [DOI] [PubMed] [Google Scholar]
- 9. Rostambeigi N, Lanza IR, Dzeja PP, et al. Unique cellular and mitochondrial defects mediate FK506-induced islet β-cell dysfunction. Transplantation. 2011;91:615–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Xu G, Stoffers DA, Habener JF, Bonner-Weir S. Exendin-4 stimulates both β-cell replication and neogenesis, resulting in increased β-cell mass and improved glucose tolerance in diabetic rats. Diabetes. 1999;48:2270–2276. [DOI] [PubMed] [Google Scholar]
- 11. Wang Q, Brubaker PL. Glucagon-like peptide-1 treatment delays the onset of diabetes in 8 week-old db/db mice. Diabetologia. 2002;45:1263–1273. [DOI] [PubMed] [Google Scholar]
- 12. Sturis J, Gotfredsen CF, Rømer J, et al. GLP-1 derivative liraglutide in rats with β-cell deficiencies: influence of metabolic state on β-cell mass dynamics. Br J Pharmacol. 2003;140:123–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Perfetti R, Zhou J, Doyle ME, Egan JM. Glucagon-like peptide-1 induces cell proliferation and pancreatic-duodenum homeobox-1 expression and increases endocrine cell mass in the pancreas of old, glucose-intolerant rats. Endocrinology. 2000;141:4600–4605. [DOI] [PubMed] [Google Scholar]
- 14. Gedulin BR, Nikoulina SE, Smith PA, et al. Exenatide (exendin-4) improves insulin sensitivity and β-cell mass in insulin-resistant obese fa/fa Zucker rats independent of glycemia and body weight. Endocrinology. 2005;146:2069–2076. [DOI] [PubMed] [Google Scholar]
- 15. Farilla L, Hui H, Bertolotto C, et al. Glucagon-like peptide-1 promotes islet cell growth and inhibits apoptosis in Zucker diabetic rats. Endocrinology. 2002;143:4397–4408. [DOI] [PubMed] [Google Scholar]
- 16. Farilla L, Bulotta A, Hirshberg B, et al. Glucagon-like peptide 1 inhibits cell apoptosis and improves glucose responsiveness of freshly isolated human islets. Endocrinology. 2003;144:5149–5158. [DOI] [PubMed] [Google Scholar]
- 17. Sano H, Terasaki J, Mishiba Y, Imagawa A, Hanafusa T. Exendin-4, a glucagon-like peptide-1 receptor agonist, suppresses pancreatic β-cell destruction induced by encephalomyocarditis virus. Biochem Biophys Res Commun. 2011;404:756–761. [DOI] [PubMed] [Google Scholar]
- 18. Pugazhenthi U, Velmurugan K, Tran A, Mahaffey G, Pugazhenthi S. Anti-inflammatory action of exendin-4 in human islets is enhanced by phosphodiesterase inhibitors: potential therapeutic benefits in diabetic patients. Diabetologia. 2010;53:2357–2368. [DOI] [PubMed] [Google Scholar]
- 19. Yanay O, Moralejo D, Kernan K, et al. Prolonged survival and improved glycemia in BioBreeding diabetic rats after early sustained exposure to glucagon-like peptide 1. J Gene Med. 2010;12:538–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kielgast U, Holst JJ, Madsbad S. Treatment of type 1 diabetic patients with glucagon-like peptide-1 (GLP-1) and GLP-1R agonists. Curr Diabetes Rev. 2009;5:266–275. [DOI] [PubMed] [Google Scholar]
- 21. Faradji RN, Froud T, Messinger S, et al. Long-term metabolic and hormonal effects of exenatide on islet transplant recipients with allograft dysfunction. Cell Transplant. 2009;18:1247–1259. [DOI] [PubMed] [Google Scholar]
- 22. Casey PJ, Fong HK, Simon MI, Gilman AG. Gz, a guanine nucleotide-binding protein with unique biochemical properties. J Biol Chem. 1990;265:2383–2390. [PubMed] [Google Scholar]
- 23. Kimple ME, Joseph JW, Bailey CL, et al. Gαz negatively regulates insulin secretion and glucose clearance. J Biol Chem. 2008;283:4560–4567. [DOI] [PubMed] [Google Scholar]
- 24. Kimple ME, Nixon AB, Kelly P, et al. A role for G(z) in pancreatic islet β-cell biology. J Biol Chem. 2005;280:31708–31713. [DOI] [PubMed] [Google Scholar]
- 25. Kimple ME, Moss JB, Brar HK, et al. Deletion of GαZ protein protects against diet-induced glucose intolerance via expansion of β-cell mass. J Biol Chem. 2012;287:20344–20355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. de la Garza-Rodea AS, Knaän-Shanzer S, den Hartigh JD, Verhaegen AP, van Bekkum DW. Anomer-equilibrated streptozotocin solution for the induction of experimental diabetes in mice (Mus musculus). J Am Assoc Lab Anim Sci. 2010;49:40–44. [PMC free article] [PubMed] [Google Scholar]
- 27. Truchan NA, Brar HK, Gallagher SJ, Neuman JC, Kimple ME. A single-islet microplate assay to measure mouse and human islet insulin secretion. Islets. 2015;7:e1076607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kimple ME, Keller MP, Rabaglia MR, et al. Prostaglandin E2 receptor, EP3, is induced in diabetic islets and negatively regulates glucose- and hormone-stimulated insulin secretion. Diabetes. 2013;62:1904–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Neuman JC, Truchan NA, Joseph JW, Kimple ME. A method for mouse pancreatic islet isolation and intracellular cAMP determination. J Vis Exp. 2014;e50374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kimple ME, Hultman RC, Casey PJ. Signaling through Gz. In: Bradshaw RA, Dennis E, eds. Handbook of Cell Signaling. 2nd ed Vol 2 New York, NY: Elsevier; 2010:1649–1653. [Google Scholar]
- 31. Maida A, Hansotia T, Longuet C, Seino Y, Drucker DJ. Differential importance of glucose-dependent insulinotropic polypeptide vs glucagon-like peptide 1 receptor signaling for β cell survival in mice. Gastroenterology. 2009;137:2146–2157. [DOI] [PubMed] [Google Scholar]
- 32. Li Y, Hansotia T, Yusta B, Ris F, Halban PA, Drucker DJ. Glucagon-like peptide-1 receptor signaling modulates β cell apoptosis. J Biol Chem. 2003;278:471–478. [DOI] [PubMed] [Google Scholar]
- 33. Smith EP, An Z, Wagner C, et al. The role of β cell glucagon-like peptide-1 signaling in glucose regulation and response to diabetes drugs. Cell Metab. 2014;19:1050–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Golson ML, Maulis MF, Dunn JC, et al. Activated FoxM1 attenuates streptozotocin-mediated β-cell death. Mol Endocrinol. 2014;28:1435–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yang J, Zhang W, Jiang W, et al. P21cip-overexpression in the mouse β cells leads to the improved recovery from streptozotocin-induced diabetes. PLoS One. 2009;4:e8344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Krishnamurthy J, Ramsey MR, Ligon KL, et al. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature. 2006;443:453–457. [DOI] [PubMed] [Google Scholar]
- 37. Baraille F, Ayari S, Carrière V, et al. Glucose tolerance is improved in mice invalidated for the nuclear receptor HNF-4γ: a critical role for enteroendocrine cell lineage. Diabetes. 2015;64:2744–2756. [DOI] [PubMed] [Google Scholar]
- 38. Karunakaran U, Park SJ, Jun do Y, et al. Non-receptor tyrosine kinase inhibitors enhances β-cell survival by suppressing the PKCδ signal transduction pathway in streptozotocin-induced β-cell apoptosis. Cell Signal. 2015;27:1066–1074. [DOI] [PubMed] [Google Scholar]
- 39. Rooman I, Bouwens L. Combined gastrin and epidermal growth factor treatment induces islet regeneration and restores normoglycaemia in C57Bl6/J mice treated with alloxan. Diabetologia. 2004;47:259–265. [DOI] [PubMed] [Google Scholar]
- 40. Suarez-Pinzon WL, Lakey JR, Brand SJ, Rabinovitch A. Combination therapy with epidermal growth factor and gastrin induces neogenesis of human islet β-cells from pancreatic duct cells and an increase in functional β-cell mass. J Clin Endocrinol Metab. 2005;90:3401–3409. [DOI] [PubMed] [Google Scholar]
- 41. Suarez-Pinzon WL, Yan Y, Power R, Brand SJ, Rabinovitch A. Combination therapy with epidermal growth factor and gastrin increases β-cell mass and reverses hyperglycemia in diabetic NOD mice. Diabetes. 2005;54:2596–2601. [DOI] [PubMed] [Google Scholar]
- 42. Suarez-Pinzon WL, Rabinovitch A. Combination therapy with epidermal growth factor and gastrin delays autoimmune diabetes recurrence in nonobese diabetic mice transplanted with syngeneic islets. Transplant Proc. 2008;40:529–532. [DOI] [PubMed] [Google Scholar]
- 43. Chen YC, Colvin ES, Griffin KE, Maier BF, Fueger PT. Mig6 haploinsufficiency protects mice against streptozotocin-induced diabetes. Diabetologia. 2014;57:2066–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Suarez-Pinzon WL, Power RF, Yan Y, Wasserfall C, Atkinson M, Rabinovitch A. Combination therapy with glucagon-like peptide-1 and gastrin restores normoglycemia in diabetic NOD mice. Diabetes. 2008;57:3281–3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kodera R, Shikata K, Kataoka HU, et al. Glucagon-like peptide-1 receptor agonist ameliorates renal injury through its anti-inflammatory action without lowering blood glucose level in a rat model of type 1 diabetes. Diabetologia. 2011;54:965–978. [DOI] [PubMed] [Google Scholar]
- 46. Elashoff M, Matveyenko AV, Gier B, Elashoff R, Butler PC. Pancreatitis, pancreatic, and thyroid cancer with glucagon-like peptide-1-based therapies. Gastroenterology. 2011;141:150–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gier B, Matveyenko AV, Kirakossian D, Dawson D, Dry SM, Butler PC. Chronic GLP-1 receptor activation by exendin-4 induces expansion of pancreatic duct glands in rats and accelerates formation of dysplastic lesions and chronic pancreatitis in the Kras(G12D) mouse model. Diabetes. 2012;61:1250–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Luki ML, Stosi-Grujici S, Shahin A. Effector mechanisms in low-dose streptozotocin-induced diabetes. Dev Immunol. 1998;6:119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tait SW, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol. 2010;11:621–632. [DOI] [PubMed] [Google Scholar]
- 50. Ohoka N, Yoshii S, Hattori T, Onozaki K, Hayashi H. TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. EMBO J. 2005;24:1243–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Du K, Herzig S, Kulkarni RN, Montminy M. TRB3: a tribbles homolog that inhibits Akt/PKB activation by insulin in liver. Science. 2003;300:1574–1577. [DOI] [PubMed] [Google Scholar]
- 52. Chandrasekar B, Vemula K, Surabhi RM, et al. Activation of intrinsic and extrinsic proapoptotic signaling pathways in interleukin-18-mediated human cardiac endothelial cell death. J Biol Chem. 2004;279:20221–20233. [DOI] [PubMed] [Google Scholar]
- 53. Riedl SJ, Shi Y. Molecular mechanisms of caspase regulation during apoptosis. Nat Rev Mol Cell Biol. 2004;5:897–907. [DOI] [PubMed] [Google Scholar]
- 54. Budihardjo I, Oliver H, Lutter M, Luo X, Wang X. Biochemical pathways of caspase activation during apoptosis. Annu Rev Cell Dev Biol. 1999;15:269–290. [DOI] [PubMed] [Google Scholar]
- 55. Traina AN, Lull ME, Hui AC, Zahorian TM, Lyons-Patterson J. Once-weekly exenatide as adjunct treatment of type 1 diabetes mellitus in patients receiving continuous subcutaneous insulin infusion therapy. Can J Diabetes. 2014;38:269–272. [DOI] [PubMed] [Google Scholar]
- 56. Qi M, Kinzer K, Danielson KK, et al. Five-year follow-up of patients with type 1 diabetes transplanted with allogeneic islets: the UIC experience. Acta Diabetol. 2014;51:833–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kielgast U, Krarup T, Holst JJ, Madsbad S. Four weeks of treatment with liraglutide reduces insulin dose without loss of glycemic control in type 1 diabetic patients with and without residual β-cell function. Diabetes Care. 2011;34:1463–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Varanasi A, Bellini N, Rawal D, et al. Liraglutide as additional treatment for type 1 diabetes. Eur J Endocrinol. 2011;165:77–84. [DOI] [PubMed] [Google Scholar]