

Abstract
Fructose consumption has been on the rise for the last two decades and is starting to be recognized as being responsible for metabolic diseases. Metabolic disorders pose a particular threat for brain conditions characterized by energy dysfunction, such as traumatic brain injury. Traumatic brain injury patients experience sudden abnormalities in the control of brain metabolism and cognitive function, which may worsen the prospect of brain plasticity and function. The mechanisms involved are poorly understood. Here we report that fructose consumption disrupts hippocampal energy homeostasis as evidenced by a decline in functional mitochondria bioenergetics (oxygen consumption rate and cytochrome C oxidase activity) and an aggravation of the effects of traumatic brain injury on molecular systems engaged in cell energy homeostasis (sirtuin 1, peroxisome proliferator-activated receptor gamma coactivator-1alpha) and synaptic plasticity (brain-derived neurotrophic factor, tropomyosin receptor kinase B, cyclic adenosine monophosphate response element binding, synaptophysin signaling). Fructose also worsened the effects of traumatic brain injury on spatial memory, which disruption was associated with a decrease in hippocampal insulin receptor signaling. Additionally, fructose consumption and traumatic brain injury promoted plasma membrane lipid peroxidation, measured by elevated protein and phenotypic expression of 4-hydroxynonenal. These data imply that high fructose consumption exacerbates the pathology of brain trauma by further disrupting energy metabolism and brain plasticity, highlighting the impact of diet on the resilience to neurological disorders.
Keywords: Cognition, energy homeostasis, metabolic syndrome, mitochondria, plasticity, traumatic brain injury
Introduction
The rise in consumption of high caloric foods has triggered a metabolic epidemic such that the number of diabetic and prediabetic persons in the U.S. is now estimated at over 40% of the population.1 A high fructose diet can induce several parameters of metabolic disease such as reduced sensitivity to insulin and increased risk factors for cardiometabolic disease in humans and rodents.2 Furthermore, the deleterious effects of fructose extend to the brain where neuronal signaling, which is crucial for supporting brain plasticity and function, is compromised.2 These data raise the question of whether fructose-induced metabolic dysfunction can reduce brain resilience against neurological disorders. Indeed, a new line of studies in humans indicates an association between metabolic disease and disturbances in cognition, emotional health, and reduced quality of life.3
We have embarked in studies to determine whether fructose consumption reduces the capacity of the brain to cope with the pathology of traumatic brain injury (TBI). TBI is a globally prevalent disorder,4,5 which is devastatingly difficult to treat, mainly due to the multifactorial aspect of the pathology.6 A particular aspect of the TBI pathology is the inability of the brain to metabolize energy.7,8 TBI patients experience sudden abnormalities in the control of brain glucose metabolism,9,10 which can increase the risk of secondary brain damage.11,12 Overload of an already disrupted brain metabolic regulation13 may exacerbate the pathobiology, as suggested by clinical reports that metabolic dysfunctions are predictors of outcome in TBI patients,14,15 and can increase incidence of long-term neurological and psychiatric disorders.16,17 The magnitude of the problem is even bigger considering that the incidence of TBI and associated cognitive disorders is on the rise,18 as is the prevalence of metabolic disease.19
The current study is centered on molecular systems that are at the critical interface between cell metabolism and synaptic plasticity; thereby having a strong impact on cognitive function. Reduced sensitivity to the action of insulin is considered a predictor of poor clinical outcome in TBI patients.15 The action of insulin has been associated with mitochondrial function regulation20,21 which suggests that insulin can influence a range of cellular processes. We assessed the effects of fructose consumption and TBI on the development of brain insulin resistance by analyzing changes in insulin signaling, such as insulin receptor (InR), and insulin receptor substrate-1 (IRS-1). Recent studies showing that a high fructose diet disrupts insulin signaling in the brain2 indicate that insulin is part of the pathway by which fructose impacts neuronal function. Insulin activates regulators of mitochondrial biogenesis, such as the peroxisome proliferator-activated receptor gamma coactivator-1alpha (PGC-1α), a member of a family of transcription coactivators,22 which in conjunction with the mitochondrial transcription factor A (TFAM),23,24 and sirtuin 1 (SIRT1) regulate cellular energy metabolism. PGC-1α can interact with brain-derived neurotrophic factor (BDNF)25 and reactive oxygen species (ROS) in the regulation of brain plasticity.26 We also determined the effects of fructose and TBI on mitochondrial functions by assessing the oxygen consumption rate (OCR). In addition, BDNF signaling is crucial for maintaining energy metabolism and cognitive function. Oxidative damage can interfere with plasticity and can be estimated by assessing the end by-product of lipid peroxidation 4-hydroxynonenal (4HNE).27 Cytochrome c oxidase (CO) is a mitochondrial protein that provides an index of mitochondrial function/mass and plays an important role in mitochondrial oxidative phosphorylation. In the current study, we sought to determine whether fructose exacerbates the TBI pathology by disrupting the interaction between cell energy homeostasis and synaptic plasticity underlying cognitive function.
Materials and methods
Animals
Male Sprague–Dawley rats (Charles River Laboratories, Inc., Wilmington, MA) approximately two months old were housed in polyacrylic cages and maintained under standard housing conditions (room temperature 22–24℃) with 12 h light/dark cycle. After acclimatization for one week on standard rat chow, rats were trained on the Barnes maze test for five days to learn the task, and then randomly assigned to either regular (con) or fructose (FRU; 15% w/v) drinking water throughout the study. They were kept in individual cages and had free access to food and drinking water. At six weeks of drinking intervention, all animals were tested for glucose tolerance test (GTT) and then subjected to either sham or fluid percussion injury (FPI) at seven weeks of drinking intervention. Memory retention was tested by Barnes maze after one week of injury (i.e. at eight weeks of drinking intervention) and rats were sacrificed immediately by decapitation. Body weight, food intake, water intake, and calorie intake were measured throughout the study. There were four experimental groups (n = 7/group) formed by the end of the experiment, based on their drinking intervention (con versus FRU) and injury (sham versus FPI): (I) regular water plus sham (con/sham), (II) regular water plus fluid percussion injury (con/FPI), (III) fructose water plus sham (FRU/sham), (IV) fructose water plus fluid percussion injury (FRU/FPI). A separate cohort of animals (kept on regular and fructose drinking water) was sacrificed after 12 h of injury (n = 5/group) for mitochondrial studies. All experiments outlined in this manuscript conform to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health. The animal care and experimental protocols were approved by the University of California at Los Angeles Chancellor’s Animal Research Committee (ID: ARC 2001-164). The suffering and number of animals used were minimized.
Blood analysis
At six weeks of intervention, blood was collected from rat-tail vein after overnight fasting and then centrifuged at 3000g for 15 min at 4℃ to obtain serum samples. Serum triglyceride was assayed enzymatically by ACE triglycerides reagent (Alfa Wassermann, NJ, USA) using VetACE chemistry analyzer (AlfaWassermann, NJ, USA).
GTT
After overnight fasting, blood glucose was measured with a glucose meter (Bayer’s Contour, NJ, USA) using a small drop of blood from tail vein. Following fasting glucose measurement, rats were injected with 50% d-glucose (dextrose; 2 g/kg body weight) intraperitoneally and glucose level was measured at 15, 30, 60, 90, and 120 min to assess glucose clearance.
FPI
FPI was performed as previously described by Sharma et al.28 In brief, animals were anesthetized by using a Laboratory Animal Anesthesia System (VetEquip Inc., CA, USA) that provides a mixture of isoflurane and oxygen. Animals were maintained in a deep anesthetic state during surgery with 2–5% isoflurane mixed with 100% O2 at a flow rate of 0.4 l/min via nose cone. 3.0 mm-diameter craniotomy was made over the left parietal cortex, 3.0 mm posterior to bregma, and 6.0 mm lateral (left) to the midline with a high-speed drill (Dremel, WI, USA). A plastic injury cap was placed over the craniotomy with dental cement. When the dental cement hardened, the cap was filled with 0.9% saline solution. Anesthesia was discontinued and the injury cap was attached to the fluid percussion device. At the first sign of hind-limb withdrawal to a paw pinch, a moderate fluid percussion pulse (2.7 atm) was administered to the epidural space. Immediately upon responding to a paw pinch, anesthesia was restored and the skull was sutured. Neomycin was applied on the suture and the rats were placed in a heated recovery chamber before being returned to their cages. Sham animals underwent an identical preparation with the exception of the lesion.
Barnes maze test
All rats were tested with the Barnes maze before and after experimental exposure to assess spatial learning and memory.29 In brief, our maze was manufactured from acrylic plastic to form a disk 1.5 cm thick and 115 cm in diameter, with 18 evenly spaced 7 cm holes at its edges. Four overhead lamps to provide an aversive stimulus brightly illuminated the disk. Animals were trained to locate a dark escape chamber hidden underneath a hole positioned around the perimeter of a disk. All trials were recorded simultaneously by a video camera installed directly overhead at the center of the maze. Animals were trained with two trials per day for five consecutive days before being subjected to the experimental conditions. A trial was started by placing the animal in the center of the maze covered under a cylindrical start chamber; after a 10 s delay, the start chamber was raised. A training session ended after the animal had entered the escape chamber or when a predetermined time (5 min) had elapsed, whichever came first. Memory retention was tested during Barnes maze trials performed one week after injury (i.e. at eight weeks of drinking intervention). All surfaces were routinely cleaned before and after each trial to eliminate possible olfactory cues from preceding animals.
Tissue collection
After the memory test the animals were killed immediately by decapitation and the fresh brains were dissected out, frozen in dry ice, and stored at −70℃ until use.
Immunoblotting
The hippocampal tissues from the left hemisphere were homogenized in a lysis buffer containing 137 mM NaCl, 20 mM Tris–HCl pH 8.0, 1% NP40, 10% glycerol, 1 mM phenylmethylsulfonylfluoride, 10 µg/ml aprotinin, 0.1 mM benzethonium chloride, 0.5 mM sodium vanadate. The homogenates were then centrifuged, the supernatants were collected, and total protein concentration was determined according to MicroBCA procedure (Pierce, IL, USA), using bovine serum albumin (BSA) as standard.
Briefly, protein samples were separated by electrophoresis on a 10% polyacrylamide gel and electrotransferred to a PVDF membrane (Millipore, MA, USA). Nonspecific binding sites were blocked in Tris-buffered saline (TBS), pH 7.6, containing 5% nonfat dry milk. Membranes were rinsed in buffer (0.05% Tween-20 in TBS) and then incubated with antiactin or anti-BDNF, anti-pTrkB, anti-TrkB, anti-pInR, anti-InR, anti-mtTFA (TFAM), antisynaptophysin (SYP), anti-GAP43 (1:500; Santa Cruz Biotechnology, CA, USA), anti-PGC-1α, anti-pCREB, anti-cyclic AMP response element binding protein (CREB), anti-SIRT1, anti-4HNE, anti-pIRS-1, anti-IRS-1 (1:1000, Millipore, MA, USA) followed by anti-rabbit or anti-goat or anti-mouse IgG horseradish peroxidase-conjugate (1:10,000; Santa Cruz Biotechnology, CA, USA). After rinsing with buffer, the immunocomplexes were visualized by chemiluminescence using the ECL kit (Amersham Pharmacia Biotech Inc., NJ, USA) according to the manufacturer’s instructions. The film signals were digitally scanned and then quantified using ImageJ software, normalized for actin levels.
Immunofluorescence
Additional rats (n = 4) from con/sham and FRU/FPI group were anesthetized with isoflurane, then intracardially perfused with PBS (pH 7.4) followed by 4% paraformaldehyde in phosphate buffer (pH 7.4) and 30% sucrose in paraformaldehyde. Tissues were flash frozen on dry ice and stored at −80℃ until time of use. Serial coronal brain sections (40 µm) were cut on a cryostat and collected into 0.01 M phosphate buffered saline (PBS). After washing, free-floating sections were heated to 80℃ for 30 min in 10 mM sodium citrate buffer (pH 8.5) to promote antigen binding and allowed to return to room temperature. Nonspecific binding was blocked by incubating the sections with 0.01 M PBS solution containing 0.5% BSA and 0.5% Triton X-100 for 2 h at room temperature.
Sections were incubated with either: rabbit polyclonal primary anti-4HNE (1:250; Abcam, Cambridge, MA, USA), rabbit polyclonal anti-pTrkB receptor (Y816) primary antibody at a dilution of 0.3 µg/ml (a kind gift from Dr Moses Chao), or rabbit polyclonal anti-pIRS1 (1:1000; Upstate, Lake Placid, NY, USA) in 0.01 M PBS solution containing 0.2% BSA and 0.6% Triton X-100 at 4℃ overnight. After thorough washing with 0.01 M PBS, sections were incubated with rabbit secondary antibody (Cy3; 1:250; Jackson Immunoresearch; West Grove, PA, USA) for 1.5 h at room temperature. Sections were mounted using Prolong Gold antifade reagent with Dapi (Life technologies, New York, NY, USA). The staining was visualized under Zeiss microscope (Zeiss Imager.Z1; Gottingen, DE) using the Axiovision software (Carl Zeiss Vision, version 4.6). Images were collected using a black and white camera (Axiocam MRm, Zeiss, Gottingen, DE) and pseudocolored green (pTrkB), red (pIRS1), purple (4HNE), or blue (Dapi).
Mitochondrial isolation and bioenergetics analysis
After 12 h of FPI, mitochondria were isolated from the left hemisphere hippocampal tissues using differential centrifugation, nitrogen disruption, and a Ficoll gradient according to the method described by Sauerbeck et al.30 The hippocampal tissues were homogenized in a Potter–Elvehjem homogenizer containing ice-cold isolation buffer (215 mM mannitol, 75 mM sucrose, 0.1% BSA, 1 mM EGTA, and 20 mM HEPES at pH 7.2). Homogenates were then centrifuged at 1300g for 3 min at 4℃. The resultant supernatant was placed in a fresh tube and the pellet was resuspended in isolation buffer and centrifuged at 1300 × g for 3 min at 4℃ to remove cellular debris and nuclei. The pellet was discarded, and the supernatants were combined in a separate tube and centrifuged at 13,000 g for 10 min at 4℃. The crude mitochondrial pellets were resuspended in isolation buffer and then subjected to the nitrogen decompression to release synaptic mitochondria, using a nitrogen cell disruption vessel (Parr Instrument Company, IL, USA) cooled to 4℃, under a pressure of 1200 psi for 10 min. After nitrogen disruption, the mitochondria were placed atop a discontinuous Ficoll gradient (7.5%, 10%), and centrifuged at 100,000 g for 30 min at 4℃. Following the Ficoll purification, the mitochondrial pellet was resuspended in isolation buffer without EGTA and centrifuged at 10,000 g for 10 min at 4℃ to remove residual Ficoll from the purified mitochondrial sample. The final mitochondrial pellet was resuspended in isolation buffer without EGTA and protein concentrations were determined using the Bradford assay reagent (Biorad, CA, USA).
Mitochondrial functions were measured using an XF24 Extracellular Flux Analyzer (Seahorse Bioscience, MA, USA). Briefly, isolated mitochondria were placed into the V7 plate (Seahorse Bioscience) and centrifuged to attach the surface as described by Rogers et al.31 To assess the mitochondrial functions, OCR was measured in the basal state in presence of succinate (complex II substrate) and rotenone (complex I inhibitor) substrates. Following basal respiration, the changes in OCR were measured after sequential injection of ADP, oligomycin (ATP synthase inhibitor), carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP; uncoupler), and antimycin-A (complex III inhibitor). The ATP synthase inhibitor oligomycin allows the detection of ATP-linked respiration by decreasing OCR, thus indicating the extent of respiration by which mitochondria generate ATP. The uncoupler FCCP reveals maximal mitochondrial respiratory capacity, while antimycin-A blocks mitochondrial respiration at complex III.
CO activity
The activity of CO activity was determined by oxidation of reduced cytochrome c in hippocampal mitochondria using CO activity colorimetric assay kit (BioVision Inc., CA, USA) according to the manufacturer’s instructions.
Statistical analysis
The results are represented as mean ± standard error of the mean. Results for body weight, food intake, water intake, total caloric intake, and glucose levels were analyzed by repeated measures ANOVA. Data for triglyceride levels and area under the curve (AUC) were analyzed by Student’s (unpaired) t-test. Data for protein expression and CO activity are represented as percentage of the con/sham group. Results for mitochondrial respiration are expressed as percentage of basal OCR. Statistical analysis was performed by two-way ANOVA ((drinking solution: con versus FRU) and (injury: sham versus FPI)) and post hoc analyses were conducted using Bonferroni’s multiple comparison tests to determine the significance of difference among various groups. A level of 5% probability was considered as statistically significant. Pearson correlation analysis was performed on individual samples to evaluate the association between variables.
Results
Effect of fructose and TBI on cognitive function
Animals were tested for spatial learning in the Barnes maze for five days before being exposed to the fructose. All animals showed similar latencies on last day (day 5), which corroborated that all rats were in a similar baseline cognitive condition prior to the interventions (data not shown). After eight weeks of fructose consumption, the Barnes Maze test was performed to evaluate memory retention. Two-way ANOVA analysis showed a significant effect of drinking solution (F1,24 = 28.66, p < 0.01) and injury (F1,24 = 21.14, p < 0.01) on escape latency. Post hoc analyses showed a marked increase in escape latency due to fructose or TBI. In turn, there was an additive effect of TBI and fructose to increase escape latency (Figure 1).
Figure 1.
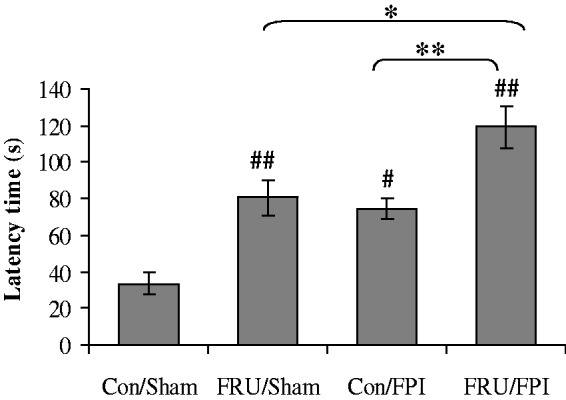
Effect of fructose and TBI on memory retention. Comparison of latency times in the Barnes Maze in regular (con) or fructose-fed (FRU) rats, subjected to either sham or fluid percussion injury (FPI). Data are expressed as latency in seconds (mean ± SEM). #P < 0.05, ##P < 0.01 versus con/sham; **P < 0.01 versus con/FPI; ANOVA (two-way) with Bonferroni’s comparisons post hoc test.
Influence of fructose and TBI on BDNF-related plasticity
The analysis revealed a significant effect of drinking solution (F1,24 = 13.36, p < 0.01) and injury (F1,24 = 70.38, p < 0.01) on BDNF levels. TBI reduced BDNF levels in sham animals exposed to either water or fructose and there was an additive effect of fructose and TBI to reduce BDNF (Figure 2(a)). Fructose exposure per se did not alter BDNF levels. With regards to the phosphorylation of the BDNF receptor TrkB, a significant effect of drinking solution (F1,24 = 35.22, p < 0.01) and injury (F1,24 = 51.74, p < 0.01) as well as the interaction between drinking solution versus injury (F1,24 = 6.07, p < 0.05) was observed. Exposure to fructose reduced the phosphorylation of TrkB, with the greatest reduction in animals that were also subjected to TBI. In turn, TBI decreased TrkB phosphorylation in control and fructose-fed rats to a similar extent (Figure 2(b)). Fructose and FPI together showed a qualitative decrease in the immunofluorescent labeling of pTrkB in CA3 subregion.
Figure 2.

Fructose and TBI affects molecules associated with synaptic plasticity. (a) Levels of BDNF, (b) TrkB phosphorylation, representative immunofluorescent staining of pTrkB (green) with DAPI nuclear stain (blue) (b, right), (c) phosphorylation of CREB, (d) levels of synaptophysin (SYP), and (e) GAP43 in regular (con) or fructose-fed (FRU) rats, subjected to either sham or fluid percussion injury (FPI). Intensity of immunofluorescent pTrkB was reduced in the hippocampal CA3 region of FRU/FPI compared with con/sham (b, right). Data are expressed as percentage of con/sham (mean ± SEM). #P < 0.05, ##P < 0.01 versus con/sham; **P < 0.01 versus con/FPI; ANOVA (two-way) with Bonferroni’s comparison post hoc test.
There was a significant effect of drinking solution and injury (F1,24 = 24.37, p < 0.01) on the phosphorylation of CREB (F1,24 = 31.93, p < 0.01). Treatment with fructose and TBI both reduced CREB phosphorylation to a similar extent and there was an additive effect when both conditions were present (Figure 2(c)).
For markers of synaptic plasticity and axonal growth, we observed a significant effect of drinking solution (F1,24 = 21.98, p < 0.01; F1,24 = 105.64, p < 0.01) and injury (F1,24 = 41.64, p < 0.01; F1,24 = 187.40, p < 0.01) on levels of SYP and GAP-43, respectively. Fructose reduced the levels of SYP and GAP-43 in both sham and TBI animals. In turn, TBI decreased the levels of SYP and GAP-43 and this effect was greater in rats that were also fed fructose (Figure 2(d) and (e)).
Effects of fructose and TBI on insulin signaling in the brain
There was a significant effect of drinking solution on phosphorylation of insulin receptor (InR; F1,24 = 16.03, p < 0.01). Drinking solution (F1,24 = 53.49, p < 0.01) and injury (F1,24 = 19.57, p < 0.01) significantly affected the phosphorylation of IRS-1. Treatment with fructose reduced phosphorylation of InR (Figure 3(a)) and IRS-1 (Figure 3(b)) in both sham and TBI animals. In addition, fructose and TBI together showed a qualitative reduction in IRS-1 immunoreactivity within the pyramidal cells of CA3 subregion as compared to control. There was a negative correlation between InR phosphorylation and latency in the Barnes Maze (r = −0.870, p < 0.01; Figure 3(c)) in sham dataset (con/sham and FRU/sham), indicating that fructose reduced hippocampal insulin signaling. However, there was no correlation between latency and InR phosphorylation in the TBI dataset (con/FPI and FRU/FPI; r = −0.199, p > 0.05).
Figure 3.

Influence of fructose and TBI on insulin signaling in the brain. Phosphorylation of (a) insulin receptor (InR), (b) IRS-1, representative immunofluorescent staining of pIRS1 (red) with DAPI nuclear stain (blue) (b, right), and (c) correlation analysis of InR phosphorylation with latency time in Barnes maze test in sham datasets (con/sham and FRU/sham) and FPI datasets (con/FPI and FRU/FPI). Intensity of immunofluorescent pIRS1 was reduced in the hippocampal CA3 region of FRU/FPI compared with con/sham (b, right). Data are expressed as percentage of con/sham (mean ± SEM). #P < 0.05, ##P < 0.01 versus con/sham; **P < 0.01 versus con/FPI; ANOVA (two-way) with Bonferroni’s comparisons post hoc test.
Effect of fructose and TBI on mitochondrial function
We determined the effects of fructose and TBI on mitochondrial function by assessing the OCR under basal condition and in response to the sequential treatment with APD, oligomycin (ATP synthase inhibitor), carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP; uncoupler), and antimycin-A (complex III inhibitor) (Figure 4(a)). There was a significant effect of injury (F1,16 = 10.57, p < 0.05; F1,16 = 4.54, p < 0.05) and an interaction between injury and drinking solution (F1,16 = 9.43, p < 0.05; F1,16 = 10.06, p < 0.05) on OCR in response to the oligomycin and FCCP treatment, respectively. Animals subjected to fructose and/or FPI had reduced ATP-linked respiration and maximal mitochondrial respiratory capacity as evidenced by reduced mitochondrial OCRs in oligomycin and reduced FCCP response, respectively (Figure 4(b)). There was a significant effect of drinking solution (F1,16 = 12.05, p < 0.05) and injury (F1,16 = 7.54, p < 0.05) on CO activity. The activity of CO was reduced to a similar level in fructose and/or FPI group as compared to control (Figure 4(c)), indicating that these interventions compromise mitochondrial functions at complex IV.
Figure 4.

Mitochondrial function in animals subjected to fructose and TBI. Bioenergetics analysis in mitochondria isolated from hippocampal tissues of regular (con) or fructose (FRU) drinking rats, which were subjected to either sham or fluid percussion injury (FPI). (a) Oxygen consumption rate (OCR) in presence of succinate and rotenone (basal respiration) and in response to the sequential treatment with ADP, oligomycin (ATP synthase inhibitor), carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP; uncoupler), and antimycin-A (complex III inhibitor) using XF24 Extracellular Flux Analyzer; (b) respiration due to ATP turnover was calculated as response to the oligomycin, while maximal mitochondrial respiratory capacity was deduced from the response to treatment with FCCP; and (c) cytochrome C oxidase activity. Data are expressed as percentage of basal OCR (mean ± SEM) of five independent experiments. #P < 0.05 versus con/sham; ANOVA (two-way) with Bonferroni’s comparisons post hoc test.
Fructose aggravates the effects of TBI on cell energy regulators
There was a significant effect of drinking solution (F1,24 = 78.1,7, p < 0.01) on PGC-1α. Fructose-induced MetS and TBI negatively influenced the levels of PGC-1α. In turn, PGC-1α level was further reduced in the fructose-fed FPI group compared to the FPI group alone (Figure 5(a)). The level of PGC-1α varied in proportion to the phosphorylation of CREB (r = 0.599, p < 0.01), suggesting an association between pCREB and PGC-1α (Figure 5(b)). We also observed that the levels of PGC-1α were negatively correlated with latency time in Barnes maze test (r = −0.5208, p < 0.01; Figure 5(c)).
Figure 5.

Fructose and TBI affects cell energy regulators. (a) Levels of PGC-1α, (b, c) correlation analysis of PGC-1α with CREB phosphorylation and latency time in Barnes maze test, (d) TFAM, and (e) SIRT1 in regular (con) or fructose (FRU) drinking rats, subjected to either sham or fluid percussion injury (FPI). Data are expressed as percentage of con/sham (mean ± SEM). #P < 0.05, ##P < 0.01 versus con/sham; **P < 0.01 versus con/FPI; ANOVA (two-way) with Bonferroni’s comparisons post hoc test.
A significant effect of drinking solution (F1,24 = 59.16, p < 0.01) and injury (F1,24 = 29.33, p < 0.01) was observed on the TFAM. Fructose treatment reduced levels of TFAM in both sham and TBI rats. In turn, a significant reduction in TFAM was found in fructose-fed animals subjected to TBI as compared to the TBI alone (Figure 5(d)). There was a significant effect of drinking solution (F1,24 = 44.68, p < 0.01) and injury (F1,24 = 48.26, p < 0.01) on levels of SIRT1. Fructose reduced SIRT1 levels in both sham and TBI groups. However, the effect of fructose was more pronounced after TBI, (Figure 5(e)).
Fructose aggravates the effect of TBI on lipid peroxidation
Two-way ANOVA analysis revealed a significant effect of drinking solution (F1,24 = 16.52, p < 0.01) and injury (F1,24 = 17.43, p < 0.01) on 4HNE levels. Post hoc analyses showed that fructose or TBI increased the levels of 4HNE. In turn, fructose32 worsened the effect of TBI on lipid peroxidation (Figure 6). There was a marked qualitative increase in the immunofluorescent labeling of 4HNE in fructose-fed TBI group as compared to control, which was especially evident in the CA3 subfield.
Figure 6.

Fructose aggravates the effect of TBI on lipid peroxidation. 4HNE immunoblotting and immunofluorescent staining of 4HNE (purple) counterstained with DAPI (blue) in hippocampal coronal sections from regular (con) or fructose (FRU) drinking rats, subjected to either sham or fluid percussion injury (FPI) (scale bar: 20 µm). Data are expressed as percentage of con/sham (mean ± SEM). ##P < 0.01 versus con/sham; **P < 0.01 versus con/FPI; ANOVA (two-way) with Bonferroni’s comparisons post hoc test.
Effect of fructose on peripheral metabolic markers
During the initial six weeks of the intervention, animals on fructose drank more fluid and had reduced food intake compared to animals fed water. Body weights and total caloric intake were similar between groups (supplementary Figure 1 (a) to (d)). Animals fed fructose had impaired insulin sensitivity, as measured by consistently increased blood glucose in the GTT (supplementary Figure 1(e)) corresponding to an elevated AUC (t26 = 6.038; p < 0.01; Figure 1(e)). Animals fed fructose solution had elevated levels of serum triglycerides (t26 = 5.025; p < 0.01; Supplementary Figure 1(f)) in comparison to controls.
Discussion
Herein we report that overconsumption of dietary fructose for a duration sufficient to disrupt peripheral metabolism exacerbates cognitive dysfunction following TBI, while reducing levels of proteins related to brain plasticity and cell energy metabolism. An increasing body of evidence indicates that diet-induced metabolic disease poses a threat for brain function and can increase the risk for neurological and psychiatric disorders.33 Our current dataset provides the framework for a potential mechanism by which dietary fructose may disturb cognition during TBI by disrupting oxidative metabolism, thereby interfering with the activation of systems that support synaptic plasticity. For example, our results show that both fructose and TBI reduce mitochondrial oxidative metabolism. In addition, animals on a high fructose diet or animals with TBI both had reduced markers of cell energy metabolism (PGC-1α, TFAM, and SIRT1), and markers of neuronal plasticity (SYP, GAP43, BDNF-TrkB signaling) as well as elevated markers of lipid peroxidation (4HNE). These data piece together to reveal the compelling possibility that diet is a predictor of resiliency to cognitive impairment due to injury.
We previously reported that metabolic syndrome (MetS), as established by increased serum triglyceride levels and low glucose tolerance following high fructose feeding, reduces hippocampal InR signaling.2 Our current data expand upon this work to show that fructose reduces InR signaling in the hippocampus and potentiates the effects of TBI on behavioral dysfunction and plasticity. The InR has a role in cognitive function such that reduced activity of this receptor in the hippocampus impairs long-term potentiation consistent with poor recognition memory.34 Our data similarly show that the detrimental effects of fructose on hippocampal InR signaling were commensurable to poor performance in the Barnes maze. In addition, fructose consumption reduced phosphorylation of IRS-1 in TBI animals suggesting that impaired insulin signaling can compromise the outcome of TBI. TBI alone did not alter the phosphorylation of InR and IRS-1, which indicates that fructose was the primary stimulus for the alterations in hippocampal insulin signaling.
The effects of fructose and TBI appeared to concert the actions of key elements in the BDNF signaling cascade. The BDNF/trkB signaling pathway is well known to mediate hippocampal-dependent learning and memory and synaptic plasticity. Disruption in BDNF function has been implicated in the pathophysiology of several neuropsychological disorders such as depression35 and schizophrenia.36 Both BDNF/trkB37 and InR38 pathways have been reported to act via PI3K/Akt/mTOR signaling, which is an essential pathway for synaptic plasticity and cognition. As an indicator of changes in synaptic plasticity, we assessed the levels of SYP, a marker of synaptic growth, and the growth associated protein 43 (GAP-43), which is expressed at high levels during neuronal growth and is associated with axonal sprouting.39,40 SYP is involved in calcium binding, channel formation, exocytosis, and synaptic vesicle recycling via endocytosis.41 Our results showed that fructose and TBI each individually reduced the levels of SYP, and that fructose potentiated the reduction caused by TBI. A similar pattern was observed for the growth-associated protein GAP-43. GAP-43 is an intracellular membrane-associated phosphoprotein, present in growth cones and presynaptic terminals important for formation of neuronal connections, and synaptic remodeling following traumatic insult.39,40 Taken together these data strongly suggest that fructose exacerbates the effects of TBI on neuronal growth and synaptic plasticity.
In addition to growth and plasticity, it is now becoming well understood that BDNF can influence the brain by engaging elements of cell energy metabolism.42 CREB is an important step in BDNF signaling and a point of convergence of many signaling pathways regulating synaptic activity and learning and memory.43 Through TrkB receptor, BDNF leads to the activation of CREB, which is a potent activator of PGC-1α.44 Therefore, the marked reduction in the levels of phosphorylated CREB in rats after dietary fructose and TBI interventions emphasizes the interactive action of metabolic and plasticity signals. The interaction between CREB and PGC-1α is reflected by our results showing that phosphorylation of CREB changes in proportion to levels of PGC-1α. When fructose and TBI were combined, we observed a significant and additive depression of pCREB and a similar pattern for molecules directly related to synaptic plasticity (SYP, GAP43) and cellular energy metabolism (PGC-1α, TFAM, and SIRT1). Therefore, it is likely that fructose and TBI affect cognitive performance by engaging the action of metabolic regulators such as PGC-1α.
Mitochondrial abnormalities are getting recognition as a common feature for neurological disorders,45 and a failure in mitochondrial function is as a major sequel of TBI46 and suggests that metabolic disorders can exacerbate the pathobiology of TBI. We observed reduced mitochondrial function in both fructose and TBI conditions as evidenced by a decreased mitochondrial respiratory capacity linked with ATP turnover. This loss in the capacity of the mitochondria to consume oxygen has been shown in most brain injuries32,47 and might be due to oxidative modification of the proteins involved in mitochondrial bioenergetics following TBI.48 It can also be inferred that diminished mitochondrial respiratory capacity in fructose-induced MetS and TBI would work as a maladaptive mechanism to escalate the secondary cascade of events.
We also observed a significant decline in CO activity after fructose and TBI interventions. CO is a protein subunit of the terminal and highly regulated enzyme complex IV of the electron transport chain in mitochondria. CO plays a key role in controlling ATP production as it helps to establish a transmembrane difference of proton electrochemical potential in the electron transport chain of mitochondria. The action of fructose was not effective to lower mitochondrial respiration or CO activity in TBI rats. These results may be explained by a loss bioenergetics capacity due to a reduction in mitochondrial threshold. There are reports suggesting that proatherogenic risk factors such as smoking, hypercholesterolemia, and obesity are associated with increased mitochondrial damage, lowering the threshold for mitochondrial bioenergetics dysfunction.49 Accordingly, fructose pretreatment might have affected the mitochondrial respiration capacity up to a threshold that no further down-regulation can be seen after TBI.
According to our results, fructose and TBI compromised molecular systems important for the maintenance of mitochondrial homeostasis, notably PGC-1α. PGC-1α is a transcriptional coactivator that exerts its action by interacting with the nuclear receptor peroxisome proliferator-activated receptor-gamma. PGC-1α provides a link between physiological stimuli and the regulation of mitochondrial biogenesis and regulates several factors involved in energy homeostasis such as SIRT1. Interestingly, it has been reported that SIRT1 (mammalian Sir2 homolog) modulates synaptic plasticity and memory formation via posttranscriptional regulation of CREB.50 We found a decrease in SIRT1 in the hippocampus of fructose and TBI rats, reflecting a disturbance in the energy management system. The increases in PGC-1α in inverse proportion to the latency in Barnes maze test suggest that PGC-1α is involved in cognitive function. We observed similar reductions in levels of the TFAM, which is a key activator of mitochondrial transcription and genome replication. TFAM was reduced in animals subjected to either fructose or TBI. The combined exposure of fructose and TBI resulted in a greater depression of these metabolic markers (PGC-1α, SIRT1, TFAM). The action of PGC-1α is also related to control of oxidative stress.26 Overproduction of ROS, a by-product of mitochondrial electron transport chain, has been implicated in acute brain injuries such as ischemia.26 Our results showed that fructose exacerbated the effects of TBI on the levels of 4HNE, as indicative of increased lipid peroxidation and neuronal damage. It could be argued that the high oxygen supply necessitated for the delivery of anesthesia could influence the outcome of the brain injury by altering cerebral metabolism and ROS levels. However, all groups were exposed to supraphysiological oxygen for a similar length of time, which seems to indicate that fructose was the primary effector for the elevated 4-HNE and reduced markers of metabolism and plasticity in the TBI animals exposed to fructose. Taken together, these results further support the possibility that disruption in energy homeostasis reduces neuronal resilience and predisposes the brain for worse outcome after TBI.
Conclusions
MetS as a result of fructose consumption exacerbates the deleterious effects of TBI on cognitive function. We propose a framework for a potential model in which fructose and TBI affect systems related to cell energy metabolism, which converge on intracellular pathways that regulate brain plasticity and cognitive function (Figure 7). Our results show the harmful impact of metabolic disease on the outcome of TBI with regards to mitochondrial biogenesis, synaptic plasticity, and cognitive function. These data suggest that fructose consumption reduces neural resilience and may predispose the brain toward cognitive dysfunction and lifelong susceptibility to neurological disorders.
Figure 7.
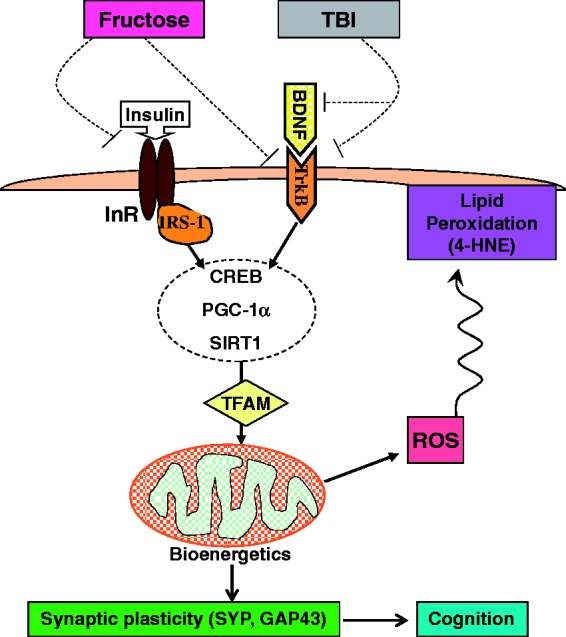
Hypothetical mechanism by which dietary fructose aggravates the pathology of traumatic brain injury (TBI) by disrupting the interaction between cell energy metabolism and synaptic plasticity. It is well understood that TBI reduces the capacity of brain cells to metabolize energy.7,8 Our current data show that TBI affects the BDNF system while fructose interferes with signaling of both insulin receptor and BDNF receptor (dashed lines). Therefore, fructose may present an additional challenge to cellular energy metabolism and TBI outcome. The actions of fructose and TBI converge and inhibit pathways associated with managements of cell energy metabolism and plasticity, such as cAMP response element-binding protein (CREB) and peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), and SIRT1. Based on the interactive action of PGC-1α and mitochondrial transcription factor A (TFAM) on the regulation of mitochondrial biogenesis,23,24 both PGC-1α and TFAM may convey the effects of fructose and TBI on decreasing oxidative phosphorylation and bioenergetics. SIRT1 modulates synaptic plasticity and memory formation via posttranscriptional regulation of CREB50; therefore, the interaction among SIRT1, PGC-1α, and TFAM may be important to regulate cognitive function. The loss in energy homeostasis results in ROS, and the harmful by-product of lipid peroxidation 4-hydroxynonenal (4HNE), thereby compromised plasma membrane function. The lipid peroxidation of the plasma membrane may further exacerbate synaptic plasticity and cognition. Overall, our data suggest a model whereby a high fructose diet and TBI disrupt the interplay between energy metabolism and synaptic plasticity with profound consequences for brain function.
Supplementary Material
Acknowledgments
We also acknowledge support of Brain Injury Center, and Letten Foundation. We acknowledge the assistance of Yumei Zhuang in western blot assays and Frances Hong in the diet experiments.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by National Institutes of Health Grant NS050465 (F.G-P.), and instrument grant NIH NCRR S10RR026744 (K.R.).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
RA: Study concept, study design, molecular and behavioral data acquisition, data analysis, data interpretation, drafting, revising and final approval of the article. EN: Immunohistochemical data acquisition and analysis, revising and final approval of the article. LV: Mitochondria data acquisition and analysis, revising and final approval of the article. ZY: Study design, data analysis, revising and final approval of the version. KR: Bioenergetic data interpretation, revising and final approval of the article. FG-P: Study concept, study design, data interpretation, drafting, revising and approval of the article.
References
- 1.Cowie CC, Rust KF, Ford ES, et al. Full accounting of diabetes and pre-diabetes in the U.S. population in 1988–1994 and 2005–2006. Diabetes Care 2009; 32: 287–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Agrawal R, Gomez-Pinilla F. ‘Metabolic syndrome’ in the brain: deficiency in omega-3 fatty acid exacerbates dysfunctions in insulin receptor signalling and cognition. J Physiol 2012; 590: 2485–2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Creavin ST, Gallacher J, Bayer A, et al. Metabolic syndrome, diabetes, poor cognition, and dementia in the Caerphilly prospective study. J Alzheimers Dis 2012; 28: 931–939. [DOI] [PubMed] [Google Scholar]
- 4.Tagliaferri F, Compagnone C, Korsic M, et al. A systematic review of brain injury epidemiology in Europe. Acta Neurochir (Wien) 2006; 148: 255–268. discussion 268. [DOI] [PubMed] [Google Scholar]
- 5.Feigin VL, Theadom A, Barker-Collo S, et al. Incidence of traumatic brain injury in New Zealand: a population-based study. Lancet Neurol 2013; 12: 53–64. [DOI] [PubMed] [Google Scholar]
- 6.Masel BE, DeWitt DS. Traumatic brain injury: a disease process, not an event. J Neurotrauma 2010; 27: 1529–1540. [DOI] [PubMed] [Google Scholar]
- 7.Vespa P, Bergsneider M, Hattori N, et al. Metabolic crisis without brain ischemia is common after traumatic brain injury: a combined microdialysis and positron emission tomography study. J Cereb Blood Flow Metab 2005; 25: 763–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lakshmanan R, Loo JA, Drake T, et al. Metabolic crisis after traumatic brain injury is associated with a novel microdialysis proteome. Neurocrit Care 2010; 12: 324–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kato T, Nakayama N, Yasokawa Y, et al. Statistical image analysis of cerebral glucose metabolism in patients with cognitive impairment following diffuse traumatic brain injury. J Neurotrauma 2007; 24: 919–926. [DOI] [PubMed] [Google Scholar]
- 10.Eakins J. Blood glucose control in the trauma patient. J Diabetes Sci Technol 2009; 3: 1373–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu-DeRyke X, Collingridge DS, Orme J, et al. Clinical impact of early hyperglycemia during acute phase of traumatic brain injury. Neurocrit Care 2009; 11: 151–157. [DOI] [PubMed] [Google Scholar]
- 12.Griesdale DE, Tremblay MH, McEwen J, et al. Glucose control and mortality in patients with severe traumatic brain injury. Neurocrit Care 2009; 11: 311–316. [DOI] [PubMed] [Google Scholar]
- 13.Stein NR, McArthur DL, Etchepare M, et al. Early cerebral metabolic crisis after TBI influences outcome despite adequate hemodynamic resuscitation. Neurocrit Care 2012; 17: 49–57. [DOI] [PubMed] [Google Scholar]
- 14.Ley EJ, Srour MK, Clond MA, et al. Diabetic patients with traumatic brain injury: insulin deficiency is associated with increased mortality. J Trauma 2011; 70: 1141–1144. [DOI] [PubMed] [Google Scholar]
- 15.Mowery NT, Gunter OL, Guillamondegui O, et al. Stress insulin resistance is a marker for mortality in traumatic brain injury. J Trauma 2009; 66: 145–151. discussion 151–153. [DOI] [PubMed] [Google Scholar]
- 16.Vagnozzi R, Signoretti S, Cristofori L, et al. Assessment of metabolic brain damage and recovery following mild traumatic brain injury: a multicentre, proton magnetic resonance spectroscopic study in concussed patients. Brain 2010; 133: 3232–3242. [DOI] [PubMed] [Google Scholar]
- 17.Casey PA, McKenna MC, Fiskum G, et al. Early and sustained alterations in cerebral metabolism after traumatic brain injury in immature rats. J Neurotrauma 2008; 25: 603–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roozenbeek B, Maas AI, Menon DK. Changing patterns in the epidemiology of traumatic brain injury. Nat Rev Neurol 2013; 9: 231–236. [DOI] [PubMed] [Google Scholar]
- 19.Padwal RS. Obesity, diabetes, and the metabolic syndrome: the global scourge. Can J Cardiol 2014; 30: 467–472. [DOI] [PubMed] [Google Scholar]
- 20.Szendroedi J, Phielix E, Roden M. The role of mitochondria in insulin resistance and type 2 diabetes mellitus. Nat Rev Endocrinol 2012; 8: 92–103. [DOI] [PubMed] [Google Scholar]
- 21.Cheng Z, Tseng Y, White MF. Insulin signaling meets mitochondria in metabolism. Trends Endocrinol Metab 2010; 21: 589–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ventura-Clapier R, Garnier A, Veksler V. Transcriptional control of mitochondrial biogenesis: the central role of PGC-1alpha. Cardiovasc Res 2008; 79: 208–217. [DOI] [PubMed] [Google Scholar]
- 23.Campbell CT, Kolesar JE, Kaufman BA. Mitochondrial transcription factor A regulates mitochondrial transcription initiation, DNA packaging, and genome copy number. Biochim Biophys Acta 2012; 1819: 921–929. [DOI] [PubMed] [Google Scholar]
- 24.Ekstrand MI, Falkenberg M, Rantanen A, et al. Mitochondrial transcription factor A regulates mtDNA copy number in mammals. Hum Mol Genet 2004; 13: 935–944. [DOI] [PubMed] [Google Scholar]
- 25.Cheng A, Wan R, Yang JL, et al. Involvement of PGC-1α in the formation and maintenance of neuronal dendritic spines. Nat Commun 2012; 3: 1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen SD, Yang DI, Lin TK, et al. Roles of oxidative stress, apoptosis, PGC-1α and mitochondrial biogenesis in cerebral ischemia. Int J Mol Sci 2011; 12: 7199–7215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Singh IN, Gilmer LK, Miller DM, et al. Phenelzine mitochondrial functional preservation and neuroprotection after traumatic brain injury related to scavenging of the lipid peroxidation-derived aldehyde 4-hydroxy-2-nonenal. J Cereb Blood Flow Metab 2013; 33: 593–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sharma S, Ying Z, Gomez-Pinilla F. A pyrazole curcumin derivative restores membrane homeostasis disrupted after brain trauma. Exp Neurol 2010; 226: 191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barnes CA. Memory deficits associated with senescence: a neurophysiological and behavioral study in the rat. J Comp Physiol Psychol 1979; 93: 74–104. [DOI] [PubMed] [Google Scholar]
- 30.Sauerbeck A, Pandya J, Singh I, et al. Analysis of regional brain mitochondrial bioenergetics and susceptibility to mitochondrial inhibition utilizing a microplate based system. J Neurosci Methods 2011; 198: 36–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rogers GW, Brand MD, Petrosyan S, et al. High throughput microplate respiratory measurements using minimal quantities of isolated mitochondria. PLoS One 2011; 6: e21746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lifshitz J, Sullivan PG, Hovda DA, et al. Mitochondrial damage and dysfunction in traumatic brain injury. Mitochondrion 2004; 4: 705–713. [DOI] [PubMed] [Google Scholar]
- 33.Farooqui AA, Farooqui T, Panza F, et al. Metabolic syndrome as a risk factor for neurological disorders. Cell Mol Life Sci 2012; 69: 741–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nisticò R, Cavallucci V, Piccinin S, et al. Insulin receptor β-subunit haploinsufficiency impairs hippocampal late-phase LTP and recognition memory. Neuromolecular Med 2012; 14: 262–269. [DOI] [PubMed] [Google Scholar]
- 35.Dwivedi Y. Brain-derived neurotrophic factor: role in depression and suicide. Neuropsychiatr Dis Treat 2009; 5: 433–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Angelucci F, Brenè S, Mathé AA. BDNF in schizophrenia, depression and corresponding animal models. Mol Psychiatry 2005; 10: 345–352. [DOI] [PubMed] [Google Scholar]
- 37.Chen TJ, Wang DC, Chen SS. Amyloid-beta interrupts the PI3K-Akt-mTOR signaling pathway that could be involved in brain-derived neurotrophic factor-induced Arc expression in rat cortical neurons. J Neurosci Res 2009; 87: 2297–2307. [DOI] [PubMed] [Google Scholar]
- 38.Lee CC, Huang CC, Wu MY, et al. Insulin stimulates postsynaptic density-95 protein translation via the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway. J Biol Chem 2005; 280: 18543–18550. [DOI] [PubMed] [Google Scholar]
- 39.Schirmer L, Merkler D, König FB, et al. Neuroaxonal regeneration is more pronounced in early multiple sclerosis than in traumatic brain injury lesions. Brain Pathol 2013; 23: 2–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grasselli G, Mandolesi G, Strata P, et al. Impaired sprouting and axonal atrophy in cerebellar climbing fibres following in vivo silencing of the growth-associated protein GAP-43. PLoS One 2011; 6: e20791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Glantz LA, Gilmore JH, Hamer RM, et al. Synaptophysin and postsynaptic density protein 95 in the human prefrontal cortex from mid-gestation into early adulthood. Neuroscience 2007; 149: 582–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gomez-Pinilla F. Brain foods: the effects of nutrients on brain function. Nat Rev Neurosci 2008; 9: 568–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Alonso M, Vianna MR, Izquierdo I, et al. Signaling mechanisms mediating BDNF modulation of memory formation in vivo in the hippocampus. Cell Mol Neurobiol 2002; 22: 663–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Herzig S, Long F, Jhala US, et al. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature 2001; 413: 179–183. [DOI] [PubMed] [Google Scholar]
- 45.Mattson MP, Gleichmann M, Cheng A. Mitochondria in neuroplasticity and neurological disorders. Neuron 2008; 60: 748–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Singh IN, Sullivan PG, Deng Y, et al. Time course of post-traumatic mitochondrial oxidative damage and dysfunction in a mouse model of focal traumatic brain injury: implications for neuroprotective therapy. J Cereb Blood Flow Metab 2006; 26: 1407–1418. [DOI] [PubMed] [Google Scholar]
- 47.Sullivan PG, Rabchevsky AG, Waldmeier PC, et al. Mitochondrial permeability transition in CNS trauma: cause or effect of neuronal cell death? J Neurosci Res 2005; 79: 231–239. [DOI] [PubMed] [Google Scholar]
- 48.Opii WO, Nukala VN, Sultana R, et al. Proteomic identification of oxidized mitochondrial proteins following experimental traumatic brain injury. J Neurotrauma 2007; 24: 772–789. [DOI] [PubMed] [Google Scholar]
- 49.Gutierrez J, Ballinger SW, Darley-Usmar VM, et al. Free radicals, mitochondria, and oxidized lipids: the emerging role in signal transduction in vascular cells. Circ Res 2006; 99: 924–932. [DOI] [PubMed] [Google Scholar]
- 50.Gao J, Wang WY, Mao YW, et al. A novel pathway regulates memory and plasticity via SIRT1 and miR-134. Nature 2010; 466: 1105–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.