

Abstract
To promote the tumor growth, angiogenesis, metabolism, and invasion, glioblastoma multiforme (GBM) cells subvert the surrounding microenvironment by influencing the endogenous activity of other brain cells including endothelial cells, macrophages, astrocytes, and microglia. Large number of studies indicates that the intracellular communication between the different cell types of the GBM microenvironment occurs through the functional transfer of oncogenic components such as proteins, non-coding RNAs, DNA and lipids via the release and uptake of extracellular vesicles (EVs). Unlike the communication through the secretion of chemokines and cytokines, the transfer and gene silencing activity of microRNAs through EVs is more complex as the biogenesis and proper packaging of microRNAs is crucial for their uptake by recipient cells. Although the specific mechanism of EV-derived microRNA uptake and processing in recipient cells is largely unknown, the screening, identifying and finally targeting of the EV-associated pro-tumorigenic microRNAs are emerging as new therapeutic strategy to combat the GBM.
Keywords: exosomes, extracellular vesicles, glioblastoma, microRNA
Introduction
Understanding the communication between tumor cells and their surrounding microenvironment is rapidly becoming the new frontier in cancer research. It is now widely accepted that cancer cells, including GBM, exert their dynamic effects over their microenvironment from a normal to a tumor supportive state which permits sustained growth, angiogenesis, and invasion. GBM is the most frequent and malignant primary brain tumor in adults and despite the progress in understanding of GBM pathophysiology, surgical procedures and therapy options, the overall survival of GBM patients has remained poor with a prognosis of only 12–15 months [1, 2]. GBM has been described as a complex, heterogeneous tumor containing not only neoplastic cells but also cancer stem like cells, endothelial cells, microglia, peripheral immune cells, neural precursor cells and reactive extracellular components. As GBM is characterized by extensive heterogeneity and variety of subtypes, a unique target which will eradicate the GBM cells has not been identified to-date. There is increasing evidence that communication between tumor cells and the surrounding components such as stromal cells, immune cells, and extracellular matrix may directly affect various hallmark features of GBM. Extracellular vesicles are now emerging as entities engaged in intratumoral communication through which the horizontal transfer of functional proteins or nucleic acids (mRNA, DNA, microRNA, and long non-coding RNA) from the GBM cells to the normal brain cells occurs. In this review, we will discuss the current knowledge regarding the multifaceted roles of both cellular and EV microRNAs in shaping the GBM microenvironment. We also review the current state of knowledge on EVs as potential biomarkers of the disease.
Biogenesis of microRNAs
MicroRNAs belong to a family of small non-coding RNAs of approximately 22 nt that are involved in post-transcriptional control of gene expression by reducing the expression of target genes by partial complementary binding of their mRNA's 3′ untranslated regions (UTRs) resulting in accelerated mRNA decay and/or blocks their translation into the proteins[3]. It is now established that the aberrant expression and microRNA activity are closely associated with several human disease including cancers[4]. Human genome encodes 2588 microRNAs identified by miRBase (release June 2014), transcribed from either the intergenic (∼ 70%) or intronic regions of protein coding genes by RNA polymerase II [5]. While the intronic primary transcripts (pri-microRNAs) bypass the RNase III processing by Drosha-DGCR8 (DiGeorge syndrome chromosome region 8) complex, the pri-microRNAs from intergenic regions are cleaved by this microprocessor complex into a stem-loop precursor of approximately 70 nt (pre-microRNA) [5-7]. A single pri-microRNA can either produce an individual mature microRNA or contain clusters of several microRNAs that are transcribed from a common primary transcript. The hairpin structured pre-microRNAs are then exported from the nucleus to cytoplasm by exportin-5 (XPO-5) and further processed by DICER 1, a RNase III enzyme [8]. DICER 1 and TAR RNA binding protein (TRBP) complex binds to the double stranded pre-microRNAs and further processes the stem loop pre-microRNAs into a ∼22 nt microRNA duplexes. These unstable microRNA duplexes are soon cleaved into single stranded mature microRNAs and loaded onto an Argonaute protein (AGO1, AGO2, AGO3 or AGO4) to form the microRNA induced silencing complexes (miRISCs) by a TRBP-dependent process. This miRISC complex then binds to the complimentary sequences of target mRNA for post transcriptional gene silencing through mRNA decay or translation inhibition [9]. Considering the complex nature of mRNA-targeting, an individual mRNA can be coordinately repressed by multiple microRNAs and each microRNA may target hundreds of different mRNAs. It will not be thus an exaggeration that an individual microRNA can powerfully regulate the complex biological processes in a well-regulated fashion.
The role of microRNAs in pathophysiology of GBM
Deregulation of microRNA expression has been observed in several cancers, including GBM. In 2005, two separate groups first described the functional role and the altered expression of microRNAs in GBM [10, 11]. A microarray analysis confirmed by Northern blotting of GBM tumor samples, as well as GBM cell lines showed aberrant expression of several microRNAs including miR-221, miR-128, miR-181a, miR-181b, and miR-181c [11], while another study demonstrated that single microRNA (miR-21) is significantly upregulated at 5-100 fold in GBM when compared to normal tissue. Inhibition of miR-21 in GBM cell lines using antisense oligonucleotide regulated cell growth by increasing apoptosis without affecting cell proliferation, which suggested the oncogenic properties of this microRNA [10]. Since then ever increasing number of studies demonstrated both oncogenic or tumor suppressor roles of multiple microRNAs regulating the development and progression of GBM. The hallmark features of GBM such as uncontrolled malignant proliferation, increased angiogenesis and invasion, high rate of necrosis, diffuse infiltration, resistance to apoptosis and genomic instability [12] are caused not only due to the deregulation of certain signaling pathways controlled by protein-coding genes, but also as a consequence of the aberrant microRNA-mRNA interaction [13, 14]. In the next chapters we will describe subtype-specific pattern of certain microRNAs and illustrate the role of specific microRNAs as crucial regulators of signaling pathways related to GBM proliferation, stemness, angiogenesis, and metabolism.
MicroRNAs and GBM subtypes
Large scale genomic analysis of The Cancer Genome Atlas (TCGA) datasets based on the combination of genetic alteration, epigenetic changes and transcriptome modifications, revealed four GBM subtypes. GBM can be thus sub-divided into four major subtypes – proneural, neural, classical and mesenchymal [14-16]. The proneural subtype has better prognosis and TP53 is frequently mutated (in 54% cases) along with common mutation in the isocitrate dehydrogenase 1 and 2 (IDH1/IDH2) and platelet derived growth factor receptor-α (PDGFR-α) genes. The classical subtype demonstrates high rates of epidermal growth factor receptor (EGFR) amplification. The mesenchymal subtype is characterized by frequent mutations in the neurofibromin 1 (NF1) (in 37% of cases), phosphatase and tensin homolog (PTEN) and tumor protein p53 (TP53) genes whereas no distinctive mutation has been associated with neural GBMs. The differential expression of microRNAs between GBM subtypes suggests that deregulation of subsets of microRNAs can transform the tumor cell phenotype. Genomic analysis of microRNA expression profile obtained from the TCGA GBM dataset identified five subgroups in whose microRNA profiles largely overlap with the microRNA profiles of neural precursor from neuronal, oligoneuronal, multipotent, mesenchymal and astrocytic lineages [17]. The microRNA based classification appeared to predict clinical outcomes more accurately than mRNA profile [18]. Some microRNAs have been linked to a specific GBM subtype such as the expression of miR-18 family and miR-218 which are significantly downregulated in mesenchymal subtype, whereas miR-34a has been shown to be preferentially expressed in proneural subset [19-21]. A group of nine microRNAs associated with pathobiology and therapeutic responsiveness of GBM, have been recently shown as a cluster of prognostic biomarkers [22]. Based on TCGA microRNA dataset, miR-128 can also classify more aggressive mesenchymal phenotype as its expression is significantly down regulated in this subtype compared to proneural [23]. Overall, these microRNA-based classifications of GBM subtype should be integrated with the mRNA expression to provide comprehensive information for better disease diagnosis.
MicroRNA and proliferation of GBM cells
Highly elevated cell proliferation in GBM is ascribed mainly to excessive proliferative signals caused by the aberrant activity of several receptor tyrosine kinases (RTK) such as PI3K/AKT and RAS/mitogen-activated protein kinase (MAPK) pathways. Several RTK signaling pathways such as EGFR, PDGFR-α, MET proto-oncogene (MET) and fibroblast growth factor receptor (FGFR) are genetically deregulated in GBM [14, 24]. Several microRNAs such as miR-128 [23], miR-218 [20], miR-181c [25], miR-7 [26], miR-124, miR-137 [27], and miR-34a [28] can regulate cell proliferation of GBM by direct targeting of the oncogenic kinases such as EGFR and PDGFR-α. Using both in vitro and in vivo approaches miR-128 has been shown to target these RTKs and repress the growth of GBM stem cells by enhancing neuronal differentiation [23]. MiR-218 acts as a suppressor of GBM proliferation by targeting multiple RTK pathway molecules such as EGFR, phosphoinositide-specific phospholipase C γ1 (PLCγ1), a-Raf proto-oncogene, serine/threonine kinase (ARAF), and phosphatidylinositol-4-phosphate 3-kinase catalytic subunit type 2 alpha (PIK3C2-α) which are also mediators of the activation of hypoxia inducible factors (HIFs), mainly HIF2α [20]. PI3-Kinase/AKT pathway is the major downstream signaling route of active EGFR increasing the proliferation of tumor cells. Overexpression of miR-181c in GBM cells inhibited the phosphorylation of AKT and reduced the proliferation and invasion of GBM [25]. MiR-7 inhibits both EGFR and AKT pathways reducing GBM cell viability either by direct binding to EGFR mRNA or by targeting insulin receptor substrate (IRS) 1 and 2, major upstream regulators of AKT pathway [26]. As an oncogenic microRNA, miR-148a reduced EGFR trafficking for endosomal and lysosomal degradation by inhibiting MIG6 expression resulting in reduced degradation and elevated expression and activation of EGFR, thus increasing migration and invasion by promoting GSC neurosphere formation [29]. As tumor suppressor microRNAs, miR-124 and miR-137 induce differentiation of adult neural stem cells, oligodendroglioma-derived stem cells and GBM-derived stem cells and inhibit the GBM cell proliferation [27]. The expression of miR-34a and activity of EGFR are inversely correlated in GBM samples and forced expression of miR-34a has been shown to regulate the EGFR activity and expression of cell cycle proteins [28]. As a negative regulator of EGFR expression and activity, all the microRNAs described above are expressed at a very low level in GBM tissue or cell lines. Apart from few upregulated pro-oncogenic microRNAs such as miR-21, miR-10b, and miR-26a in general, the majority of microRNAs deregulated in numerous malignancies are expressed at much lower level in cancer tissues compared with their normal counterpart [30]. Oncogenic miR-21 targets several key factors for both the pro-apoptotic programmed cell death protein 4 [31] and the anti-invasive matrix metalloproteinase regulators RECK (reversion-inducing cysteine-rich protein with kazal motifs) and tissue inhibitor of metalloproteinase 3 [32]. Knock down of miR-21 and induction of tumor necrosis factor-related apoptosis inducing ligand (TRAIL) in GBM cell lines led to significant reduction of tumor growth in vivo by synergistic apoptotic effects via enhanced caspase activity [33]. In GBM levels of sprout homolog 2 (SPRY2) and miR-21 are inversely correlated, and increased level of miR-21 regulates the level of Spry2 mRNA which is also one of key regulators of GBM invasion, by disrupting the negative feedback circuit of Ras/MAPK signaling [34]. MiR-10b, another microRNA frequently studied in GBM, is highly upregulated and a significant correlation has been observed between WHO grades and the expression of this microRNA [35]. This microRNA targets many tumor suppressive genes including TP53 and inhibits the pro-apoptotic signaling/upregulate proliferation by direct targeting Bim, activation protein 2γ, and the cell cycle inhibitors p16 and p21 [36]. The expression of miR-17∼92 cluster (comprising of miR-17-3p, miR-17-5p, miR-18a, miR-19a, miR-19b, miR-20a, and miR-92a) is upregulated in GBM tissue and cell lines [37, 38] and facilitates GBM proliferation by targeting of anti-proliferative genes, such as transforming growth factor (TGF) beta receptor II, SMAD family member 4 (SMAD4) [38, 39]. Another example of pro-oncogenic microRNA in GBM is miR-26a which can promote proliferation both in vitro and in vivo by increasing AKT phosphorylation as a downstream effect of decreasing protein expression of PTEN, retinoblastoma 1, and mitogen-activated protein 3 kinase 2 (MAP3K2)/MAP extracellular signal-regulated kinase kinase 2 (MEKK2) [40]. Overall, these studies suggest that microRNAs functionally integrate into crucial pathways of GBM cell proliferation, and that the deregulation of these microRNAs is accountable for sustaining proliferative signaling in these cells.
MicroRNA and glioblastoma stem cells
Relatively small sub-population of GBM cells retain stem cell properties, that is highly tumorigenic [41-43] and therapy-resistant [44], is known as glioblastoma stem cells (GSC). This specific sub-population of cells has the ability to generate a tumor upon intracranial transplantation that recapitulates the cellular heterogeneity and molecular characteristics present in the parental tumor, indicative of crucial role in gliomagenesis and progression of GBM [41, 43]. Several de-regulated microRNA have been identified to regulate signaling pathway related to the stemness and self-renewal by targeting SOX2, NOTCH, TGF- β and polycomb repressor complex (PRC) dependent signaling pathway. Overexpression of miR-137, microRNA weakly expressed in GSCs, have been reported to significantly decrease the self-renewal of GSCs and other stem cell markers such as OCT4, NANOG, SOX2 and Sonic Hedgehog (SHH) [45]. Expression of SOX2 has inverse relationship with the level of miR-21 in GBM and both in vivo and in vitro studies and in silico analysis of TCGA confirmed that miR-21-dependent suppression of SOX2 could significantly affect the stemness network [46]. Reprogramming transcription factors OCT4 and SOX2 trigger GBM cells to change into stem-like and tumor-propagating cells and recently, miR-148a repression was shown to be required for this microRNA-dependent transcriptional axis involving direct DNA methyltransferase (DNMT) promoter transactivation [47]. Notch-dependent pathway controlling GSC stemness has been shown to be regulated by miR-34a [48] and miR-326 [49]. TGF-β and nuclear factor–kappa β (NF-κβ) have been identified as factors important in maintenance of GSCs and a regulation of TGF-β signaling by miR-34a has been shown via SMAD4 transcription factor dependent pathway [19]. GSC self-renewal is partially regulated by maintaining the cell-population balance between induction of apoptosis/differentiation and those gained through proliferation. MiR-363 and miR-582-5p, two microRNAs highly expressed in GSCs, regulate the self-renewal process by direct targeting of anti-apoptotic factors such as Caspase 3, Caspase 9 and Bim [50]. Our group has demonstrated a crucial link between miR-128, which is significantly downregulated in GBM, with the ability of GSCs to self-renew [51]. We have shown that miR-128 coordinately targets both PRC components PRC1 by targeting B lymphoma Mo-MLV insertion region 1 homolog (BMI1) and PRC2 by targeting suppressor of Zeste 12 Homolog (SUZ12), epigenetic regulators of chromatin status. MiR-128 mediated inhibition of both PRCs blocks their partially redundant function [52] and associated chromatin modifications. Stable overexpression of miR-128 in GSCs changed their molecular landscape by downregulating signaling pathways related to the maintenance of stemness properties. The role of miR-128 in tumorigenicity was also demonstrated by using a genetically engineered mouse model of GBM. Loss of miR-128 expression in neural stem cells precedes the onset of symptoms of tumor in mice and thus indicates that loss of miR-128 is an early event in GBM pathogenesis [52]. Recently, using in silico analysis of TCGA, miR-31 and miR-148a has been identified to be associated with GBM growth and progression. Both in vivo and in vitro studies showed that inhibition of these two microRNAs reduced GSC proliferation, normalized tumor vasculature, suppressed tumor growth and increased animal survival via HIF-lα dependent Notch signaling pathway [53]. GSC self-renewal and maintenance is also supported by some cell adhesion factors and miR-145 has been demonstrated to interact with junctional adhesion molecule – A (JAM-A) to regulate GSC stemness [54]. MiR-134 targeting KRAS and STAT5b, has also been demonstrated to regulate GSC self-renewal and stemness both in vitro and in vivo [55]. These studies thus indicate that several microRNAs have a prime role in the regulation of GSCs by regulating self-renewal, proliferation and differentiation through the regulation of stemness specific genes at molecular level.
MicroRNA and angiogenesis in GBM
Angiogenesis is a complex process of sprouting of endothelial cells into micro-vessels to provide nutrients and oxygen to the tumor cells. The interactions between blood vessels and infiltrating tumor cells facilitate the tumor growth and metastasis [56]. The angiogenesis of GBM occurs through microvascular hyper-proliferation influenced by abundance of growth factors and extracellular matrix with in tumor microenvironment [56]. To verify whether microRNAs can be used as therapeutic inhibitors of angiogenesis-, the involvement of miR-7 in the regulation of the angiogenic process in GBM has been studied [57]. Overexpression of miR-7 in endothelial cells resulted in inhibition of cell viability, tube formation, sprouting and migration in a xenograft model of GBM cell line [57]. MiR-137 was recently shown to regulate the proliferation and angiogenesis of GBM cell line in vivo by directly targeting enhancer of zeste 2 (EZH2) [58]. Hypoxia and HIF-1 have been shown as critical regulator of neo-vascularization. Various HIF-1 target genes have been shown to regulate the angiogenesis by promoting the migratory and mitogenic activities of endothelial cells [59]. Recently miR-218 has been shown to regulate HIF activity through RTK pathway and thus control angiogenesis in mesenchymal GBM subtype [20]. Using subcutaneous xenograft model of GBM, co-culture of endothelial cells with GBM cells overexpressing miR-93 promoted endothelial cells growth, migration and tube formation by targeting the expression of integrin-β8 [60]. GBM cells are known to facilitate angiogenesis through hyper activation of vascular endothelial growth factor (VEGF) [56]. MiR-125b is downregulated in GBM-associated endothelial cells, resulting in overexpression of its target, myc-associated zinc finger protein (MAZ), a transcription factor that regulates the expression of VEGF [61]. Recently our group has showed that overexpression of tumor suppressor miR-1 in GBM cells significantly inhibited the angiogenesis and invasion both in vivo and in vitro through altered secretion of pro-angiogenic factors into the microenvironment [62]. Taken together, these studies demonstrated the relationship between microRNA alterations in GBM cells and the mechanisms of microRNA-mediated regulation of tumor angiogenesis. Further investigations will provide valuable insights into the pivotal roles played by microRNAs in GBM angiogenesis.
MicroRNA and metabolism of GBM
In rapidly proliferating cancer cells like GBM, metabolic alterations are intrinsically involved in tumor growth that adapts the cancer cell to survive in low glucose /low ATP conditions in hypoxic microenvironments. The interference with this metabolic features is termed ‘Warburg effect’ [63]. Our laboratory has focused on the role of miR-451 as a potent regulator of GBM metabolism and invasiveness [64-67]. In a high glucose environment, the expression of miR-451 is maintained at high levels in GBM cells and acts as a potent inhibitor of adenosine monophosphate-activated protein kinase (AMPK) complex by directly targeting calcium binding protein 39, an essential co-factor of liver kinase B1 (LKB1) that activates AMPK [68]. Overexpression of miR-451 sensitized GBM cells to low glucose microenvironment and strongly inhibits their migration in vitro [64, 65]. In a recent study from our group, we have also demonstrated that in a glucose-deprived microenvironment, active AMPK complex phosphorylates OCT1 which is sufficient to reduce the transcription of miR-451 in GBM cells [66]. Octamer binding protein 1 (OCT1) thus acts as transcriptional regulator of miR-451 expression and endogenous OCT1 was recruited to the promoter of miR-451 gene in a glucose-dependent manner [66]. Our findings suggest that the AMPK/OCT1/miR-451/LKB1 loop provides a nutrient-dependent regulatory mechanism so that GBM cells can adapt to altered microenvironmental conditions. The AMPK-regulated inactivation of the OCT1 transcriptional activator of miR-451 facilitates GBM cells leaving low glucose areas. We found miR-451 as unique microRNA that is not deregulated in GBM cells, but which activity is well regulated by microenvironmental niche by promoting or suppressing brain tumor cell phenotypes.
Another metabolic regulator, pyruvate kinase isoform 2 (PKM2) is also a core regulator of GBM cell metabolism and tumor growth. Tumor suppressive miR-326 has been demonstrated to directly target PKM2 which is highly expressed in GBM tissue compared to normal brain suggesting that miR-326 could regulate the GBM metabolism through the downregulation of PKM2 [69]. C-Myc is another critical regulator of tumor cell metabolism including GBM and its expression can be regulated by miR-34c in a mTOR complex 2 dependent forkhead box O3 A (FOXO3A) transcriptional signaling mechanism [70]. Overall, by controlling metabolism of GBM cells these microRNAs contribute to the regulation of migratory and invasive phenotypes of the tumors.
MicroRNA and Immune Regulation in GBM
The GBM immuno-phenotype is significantly altered in comparison to the normal brain, and is characterized by leakage in blood-brain barrier, acidosis due to high rate of glycolytic activity, hypoxia, accumulation of tumor-specific antigens and infiltration of leukocytes/macrophages from the peripheral circulation [71]. GBM tumors mediate profound immune suppression by increasing the secretion of immuno suppressive cytokines such as prostaglandin E, Interleukin (IL)-1, IL-10, and TGF-β secreted by regulatory T cells (Treg) to the microenvironment [72]. Some recent findings show the role of microRNAs in GBM-mediated immune suppression and linked microRNAs as potent candidates for immunotherapeutic agents against GBM [72-80]. Over expression of tumor suppressive miR-124 in Treg cells isolated from GBM samples reduced the immune suppressive properties by reversing a block in T-cell proliferation and inhibiting the signal transducer and activator of transcription 3 (STAT3) driven immune response [74]. The main surface receptor of cytotoxic T lymphocytes (CTL) CTLA4 is considered as one of the immune checkpoints for tumor-mediated immune suppression and an earlier study showed that inhibition of oncogenic miR-222 and miR-339 increased the susceptibility of GBM cells to CTLs by targeting the activity of intracellular adhesion molecule 1 (ICAM-1) [75]. In a very recent scientific report, miR-138 has been demonstrated to target both immune checkpoints CTLA4 and program cell death -1 (PD-1) to inhibit tumor-infiltrating Tregs and subsequently released the brake by these immunosuppressive cells within the GBM microenvironment [78]. In GBM, tumor associated macrophages are the largest population of infiltrating immune cell and after adopting tumor-specific M2 phenotype, they are recruited to the tumor site to mediate the immune suppression [76]. Recently, miR-142 weakly expressed in GBM-specific M2 macrophages has been demonstrated to regulate the GBM immunity by reducing the macrophage infiltration through TGF-β signaling pathway [77].
MiR-17-92 is down-regulated in T-cells derived from GBM patients and it was shown that the over expression of miR-17-92 and chimeric antigen receptor (CAR) in T-cells improves the therapeutic efficacy in a preclinical model of GBM [79]. Recent study from the same group, demonstrated that overexpression of miR-17-92 promoted CD8+ T cell proliferation and enhanced IFN-γ production and cytotoxicity which provided resistance to immunosuppressive effect of TGF-β1 [80]. To develop targeted immunotherapy, the identification of specific mechanisms of microRNA targeting of GBM-specific immune modulators is a promising approach that could lead to augmenting of the activity of tumor infiltrating immune cells in the tumor microenvironment resulting in GBM regression.
Biogenesis and composition of EVs
EVs are membrane-surrounded particles released by most human cells. Based on their subcellular origin, size, and composition, several classes of EVs have been described including exosomes and microvesicles[81]. However, the overlap of size range and the lack of standard isolation protocols make it difficult to obtain a homogeneous population of vesicles.
Exosomes are the smallest vesicles with a diameter ranging between 30 and 100 nm, and with a characteristic cup-shaped morphology. They originate by inward budding of the endosomal membrane leading to the formation of multivesicular bodies (MVBs). Exosomes are then released into the extracellular milieu by the fusion of MVBs with the plasma membrane[82]. Larger microvesicles range from 100 to 1000 nm and originate from the plasma membrane through a mechanism similar to virus budding[83]. In this review we will refer to all the different classes of vesicles as EVs.
EVs transport several bioactive molecules including proteins, DNA, mRNAs and non-coding RNAs (e.g. microRNAs and long non-coding RNAs). The specific composition of EVs varies greatly depending on the cell of origin and a unique tissue/cell type EV signatures were revealed[84]. Conserved sets of vesicular proteins also exist and are now used as markers to distinguish exosome from microvesicles. In fact, the molecules engaged in biogenesis of MVBs, such as Alix and Tsg101, are highly associated with exosomes[82]. Exosomes also contain Rab proteins, the largest family of small GTPases, which regulate docking and fusion of MVBs with the plasma membrane[85]. Microvesicles instead originate from plasma membrane domains specifically enriched in membrane proteins such as integrin-β1 [86].
In the brain, secreted EVs are involved in the cross-talk between neurons, glial and endothelial cells to regulate several physiologic processes such as the formation of blood-brain barrier [87, 88]. An emerging theme in neuro-oncology is that brain cancer cells mold the tumor microenvironment to their advantage in part by the release of substantial quantities of EVs containing cancer-specific molecules, including pro-oncogenic microRNAs. MicroRNAs represent about one third of all small non-coding RNAs identified in GBM EVs[89]. GBM EVs, as well as EVs derived from other tumors, contain microRNA signatures that only partially reflect the microRNA composition of the tumor cell of origin, with several microRNAs that are specifically enriched in the tumor EVs [90-92]. The specific sorting of microRNA into EVs has been described to be driven by the binding of sumoylated heterogeneous nuclear ribonucleoprotein A2/B1 (hnRNPA2B1) to specific EXO motifs present in the sequence of mature microRNAs. Mutation of these EXO motifs or hnRNPA2B1 reduced expression level resulted in impaired selective loading of microRNAs into the EVs [93]. More recently post-transcriptional modification of microRNA has been suggested as an additional mechanism for selective sorting of microRNAs. More specifically, it has been shown that microRNAs with adenylated 3′ end are relatively enriched in the cells, whereas 3′ end uridylated microRNAs are found mostly in EVs[94]. Interestingly, mature miR-451 that is a microRNA highly secreted by GBM cells [95], contains two uracil residues at the 3′ end. This microRNA was shown to be preferentially sorted into EVs without undergoing post-translational modifications, suggesting that the presence of the uracil residues itself is sufficient as a sorting signal [94]. These findings show that the secretion of microRNAs through EVs is not a passive process, and suggest that selective packaging of specific microRNAs into EVs may play an important role in the modulation of the tumor microenvironment.
Interaction of EVs with recipient cells
It has been demonstrated that upon release from their cell of origin, EVs are avidly taken up by recipient cells both in vitro [62, 96] and in vivo [95, 97]. Moreover, it seems that EVs do not interact with just any cell they come into contact with, but rather only with cells that they specifically recognize[98-100]. Hoshino and colleagues have elegantly demonstrated that tumor-derived EVs are internalized by organ-specific cells and that this process played an important role in the preparation of the pre-metastatic niche [101].
Equally important is the nature of EV interaction with the recipient cell. In some cases, it is restricted to the surface and mediated, at least in part, by receptor binding [102]. In such case, EVs may act as signaling complexes by direct stimulation of receptors on the surface of recipient cells. EVs may also directly fuse with the plasma membrane of recipient cells releasing their content into their cytoplasm. This process may also be important in the transfer of receptors between the cells. In fact, GBM cells expressing the oncogenic form of EGF receptor (EGFRvIII) release EVs containing the EGFRvIII that is transferred to other cancer cells expressing wild-type receptor. Such inter-cellular transfer leads to aberrant intra-cellular signaling by the oncogenic receptor and subsequent transformation of the recipient cells [103]. Finally, EVs can be internalized by the target cells through endocytosis [104, 105]. This process requires an initial receptor-mediated binding step followed by the internalization. In GBM EVs this binding seems to be, at least in part, mediated by heparan sulfate proteoglycans[106, 107]. Once endocytosed, EVs may release their content in the cytoplasm by fusion with the membrane of the endocytic compartment [108, 109], or they may be shared between neighbor cells through the formation of inter-cellular connections called nanotubes[110].
Role of EV microRNAs in the modulation of GBM microenvironment
GBM EVs have been shown to be enriched in several oncogenic microRNAs including miR-21, miR-23a, miR-30a, miR-221 and miR-451 [89]. EV-mediated exchange of these oncogenic microRNAs between GBM cells may represent an important mechanism to regulate tumor cell growth, invasion and survival. In parallel, GBM EV microRNAs can be transferred to non-malignant cells in the microenvironment changing their phenotype to promote tumor progression. Van der Vos and colleagues have shown that GBM EVs interact in vivo with microglia/macrophages to deliver functional miR-21 and miR-451. Exposure of microglia/macrophages to GBM EVs resulted in increased level of both microRNAs in the recipient cells with consequent decrease in the levels of the common target c-Myc[95].
GBM EVs can also be internalized by endothelial cell inducing their activation[96]. During GBM progression the formation of a hypoxic environment causes the activation of stress response pathways inside the tumor cells that in turn result in generation of EVs enriched in hypoxia-regulated mRNAs and proteins. Such secreted EVs then mediate a hypoxia-dependent intercellular communication between GBM and endothelial cells that results in strong activation of tumor neovascularization [111]. Thus changes in EVs composition and their effects in the environment overwhelmed by hypoxia may be, at least in part, due to changed microRNA signature in hypoxic GBM cells. In fact, upon hypoxic stress, several microRNAs have been shown to be upregulated, including miR-210, miR-1275, miR-376c, miR-23b, miR-193a and miR-145. Among these, miR-210 is the most up-regulated microRNA during hypoxia. Expression of miR-210 represses glycerol-3-phosphate dehydrogenase 1-like (GPD1L) and HIF3A which, in turn, stabilize HIF1A resulting in increased level of its downstream target VEGF [112]. Interestingly, it has been shown that during hypoxia miR-210 itself can be secreted through EVs to directly affect endothelial cell response [113]. Thus changes in cellular microRNA expression may affect EV composition both at microRNA and proteome/mRNA level.
Our group has recently shown that ectopic expression of the tumor suppressor miR-1 in GBM results in inhibition of tumor growth, invasion and neovascularization. This effect was in part due to alteration of GBM EV cargo, including augmented levels of the mature miR-1 in released EVs. We found that reintroduction of miR-1 resulted in the coordinated targeting of the key oncogenic GBM network composed of ANXA2, MET, JNK and members of the polycomb repressor complex. Analysis of the composition of EVs secreted by miR-1 overexpressing GBM cells also revealed ANXA2 as the most down-regulated protein. Moreover, we demonstrated functional transfer of EV miR-1 to recipient cells resulting in reduced levels of its targets [62]. In summary, our data showed how modulation of microRNA expression may be an alternative strategy for the development of new therapies for the management of GBM.
EV microRNAs as GBM biomarkers
The ability of a single microRNA, or a group of microRNAs, to regulate the expression of multiple genes, both in tumor cells and in the microenvironment, highlight the importance of microRNAs as major component of the inter-cellular communication. MicroRNAs expression in the cancercells have also been shown to reflect the tumor stage, subtype and stress response [4, 112, 114-116]. For these reasons microRNAs can be very promising biomarkers for monitoring GBM recurrence and patient response to chemotherapy. The current option for monitoring GBM over-time based on repeated stereotactic biopsyis not optimal, and it is associated with increased risks [117]. Moreover, biopsies may not reflect the heterogeneity of GBM tumors. EVs have been found in several body fluids including blood [118], urine [119], saliva [120] and cerebrospinal fluid (CSF) [121]. Thus, microRNAs circulating in complex with tumor-originated EVs represent a potential source of biomarkers for early detection and monitoring of tumor response to treatment.
Among the microRNAs, miR-21 is of particular relevance as a biomarker being one of the most upregulated microRNAs in GBM. A meta-analysis performed by Qu and colleagues to assess the diagnostic value of microRNAs showed that miR-21 as the most powerful clinical biomarker in diagnosis of GBM [122]. In fact, levels of EV-bound miR-21 s in CSF have been shown to be a good marker to discriminate between GBM and non-oncologic patients [121]. MiR-21 has been also found to protect GBM cells from TMZ-induced apoptosis by inducing a reduction of the pro-apoptotic protein BAX associated with reduced activity of caspase-3 [123]. This suggests that miR-21 may be useful as a biomarker to predict patient response to chemotherapy. Analysis of microRNAs in the CSF also revealed that levels of miR-10b, together with miR-21, are elevated in patients with GBM and brain metastases, when compared to control patients and patient in remission. On the contrary, members of the miR-200 family were present only in the CSF of patients with metastatic brain tumors but not GBM, allowing discrimination between the different types of brain tumors[124].
Another source of GBM biomarker is patient serum. It has been found that, compared to normal controls, patient with GBM presented with elevated levels of miR-128 [125], miR-320 and miR-574-3p [126] in the serum. Interestingly, both miR-128 and miR-320 have been shown to be tumor suppressor microRNAs [51, 52, 127] with a reduced expression in GBM tissue in comparison to the normal counterpart [51, 128]. Thus additional studies will be required to identify the source of these microRNAs in the blood of GBM patients.
Conclusions
MicroRNAs are important modulators of the tumor pathophysiology in GBM, as well as in other cancers. Recent findings have also shown the role of microRNAs as mediators of the inter-cellular communication within the GBM tumor microenvironment. MicroRNAs are secreted into the extracellular milieu, at least in part by EVs, and then taken up by normal and cancer cells to sustain and promote tumor growth (FIGURE 1). GBM EVs transport several tumor-specific microRNAs; however the function of most of these microRNAs has still to be characterized. Better understanding of the role of secreted microRNAs may unveil novel mechanism of GBM progression and resistance to current treatments, and may provide novel diagnostic and prognostic biomarkers. In turn, these biomarkers can be used for the development of targeted therapies aimed at improving GBM patients' outcomes.
Figure 1. Mechanism of microRNA action and secretion into the tumor microenvironment.
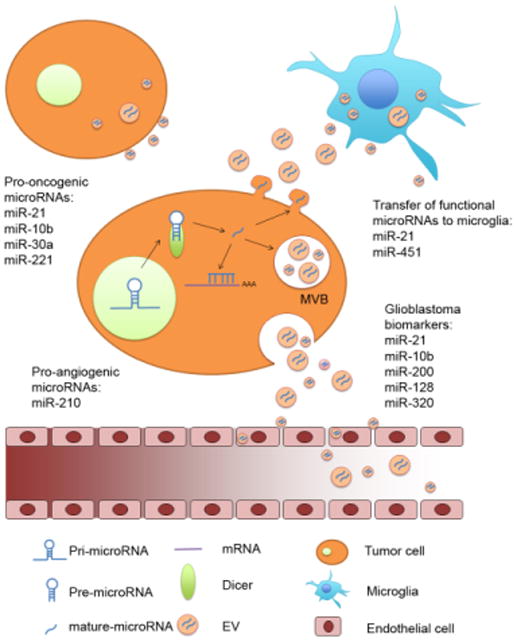
Primary microRNAs are transcribed in the nucleus by DNA polymerase II and then cleaved to generate precursor microRNAs (pre-microRNAs). Pre-microRNAs are then transferred to the cytoplasm were they are further processed by Dicer to form the 17-23 nucleotide long mature microRNAs. Such mature microRNAs can bind the target mRNA to regulate their stability and translation, or they can be secreted through extracellular vesicles (EVs). EVs may generate directly from the cell plasma membrane, or they can be released by fusion of the multi-vesicular bodies (MVBs) with the plasma membrane. After secretion,GBM EV microRNAs are taken up by other tumor cells or normal cells in the microenvironment to change their phenotype and support tumor progression. GBM EV microRNAs are also released into the cerebrospinal fluid and into the bloodstream were they can be collected for detection of tumor biomarkers.
Acknowledgments
Financial Support: NCI 1R01 CA176203-01A1 (JG)
Footnotes
Authors declare no conflict of interest
References
- 1.Stupp R, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10(5):459–66. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 2.Ostrom QT, et al. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2007-2011. Neuro Oncol. 2014;16 Suppl 4:iv1–63. doi: 10.1093/neuonc/nou223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huntzinger E, Izaurralde E. Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat Rev Genet. 2011;12(2):99–110. doi: 10.1038/nrg2936. [DOI] [PubMed] [Google Scholar]
- 4.Piwecka M, et al. Comprehensive analysis of microRNA expression profile in malignant glioma tissues. Mol Oncol. 2015;9(7):1324–40. doi: 10.1016/j.molonc.2015.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee Y, et al. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004;23(20):4051–60. doi: 10.1038/sj.emboj.7600385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Denli AM, et al. Processing of primary microRNAs by the Microprocessor complex. Nature. 2004;432(7014):231–5. doi: 10.1038/nature03049. [DOI] [PubMed] [Google Scholar]
- 7.Gregory RI, et al. The Microprocessor complex mediates the genesis of microRNAs. Nature. 2004;432(7014):235–40. doi: 10.1038/nature03120. [DOI] [PubMed] [Google Scholar]
- 8.Bohnsack MT, Czaplinski K, Gorlich D. Exportin 5 is a RanGTP-dependent dsRNA-binding protein that mediates nuclear export of pre-miRNAs. RNA. 2004;10(2):185–91. doi: 10.1261/rna.5167604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chendrimada TP, et al. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature. 2005;436(7051):740–4. doi: 10.1038/nature03868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chan JA, Krichevsky AM, Kosik KS. MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res. 2005;65(14):6029–33. doi: 10.1158/0008-5472.CAN-05-0137. [DOI] [PubMed] [Google Scholar]
- 11.Ciafre SA, et al. Extensive modulation of a set of microRNAs in primary glioblastoma. Biochem Biophys Res Commun. 2005;334(4):1351–8. doi: 10.1016/j.bbrc.2005.07.030. [DOI] [PubMed] [Google Scholar]
- 12.Furnari FB, et al. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. 2007;21(21):2683–710. doi: 10.1101/gad.1596707. [DOI] [PubMed] [Google Scholar]
- 13.Sumazin P, et al. An extensive microRNA-mediated network of RNA-RNA interactions regulates established oncogenic pathways in glioblastoma. Cell. 2011;147(2):370–81. doi: 10.1016/j.cell.2011.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brennan CW, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155(2):462–77. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Phillips HS, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9(3):157–73. doi: 10.1016/j.ccr.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 16.Verhaak RG, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17(1):98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim TM, et al. A developmental taxonomy of glioblastoma defined and maintained by MicroRNAs. Cancer Res. 2011;71(9):3387–99. doi: 10.1158/0008-5472.CAN-10-4117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tang W, et al. Subtyping glioblastoma by combining miRNA and mRNA expression data using compressed sensing-based approach. EURASIP Journal on Bioinformatics and Systems Biology. 2013;2013(1):2. doi: 10.1186/1687-4153-2013-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Genovese G, et al. microRNA regulatory network inference identifies miR-34a as a novel regulator of TGF-beta signaling in glioblastoma. Cancer Discov. 2012;2(8):736–49. doi: 10.1158/2159-8290.CD-12-0111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mathew LK, et al. miR-218 opposes a critical RTK-HIF pathway in mesenchymal glioblastoma. Proc Natl Acad Sci U S A. 2014;111(1):291–6. doi: 10.1073/pnas.1314341111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang H, et al. Upregulation of miR-181s reverses mesenchymal transition by targeting KPNA4 in glioblastoma. Sci Rep. 2015;5:13072. doi: 10.1038/srep13072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hayes J, et al. Prediction of clinical outcome in glioblastoma using a biologically relevant nine-microRNA signature. Mol Oncol. 2015;9(3):704–14. doi: 10.1016/j.molonc.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Papagiannakopoulos T, et al. Pro-neural miR-128 is a glioma tumor suppressor that targets mitogenic kinases. Oncogene. 2012;31(15):1884–95. doi: 10.1038/onc.2011.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cancer Genome Atlas Research, N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061–8. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang F, et al. MiRNA-181c inhibits EGFR-signaling-dependent MMP9 activation via suppressing Akt phosphorylation in glioblastoma. Tumour Biol. 2014;35(9):8653–8. doi: 10.1007/s13277-014-2131-6. [DOI] [PubMed] [Google Scholar]
- 26.Kefas B, et al. microRNA-7 inhibits the epidermal growth factor receptor and the Akt pathway and is down-regulated in glioblastoma. Cancer Res. 2008;68(10):3566–72. doi: 10.1158/0008-5472.CAN-07-6639. [DOI] [PubMed] [Google Scholar]
- 27.Silber J, et al. miR-124 and miR-137 inhibit proliferation of glioblastoma multiforme cells and induce differentiation of brain tumor stem cells. BMC Medicine. 2008;6(1):1–17. doi: 10.1186/1741-7015-6-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yin D, et al. miR-34a functions as a tumor suppressor modulating EGFR in glioblastoma multiforme. Oncogene. 2013;32(9):1155–63. doi: 10.1038/onc.2012.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim J, et al. microRNA-148a Is a Prognostic oncomiR That Targets MIG6 and BIM to Regulate EGFR and Apoptosis in Glioblastoma. Cancer Research. 2014;74(5):1541–1553. doi: 10.1158/0008-5472.CAN-13-1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lu J, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435(7043):834–8. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 31.Chen Y, et al. MicroRNA-21 down-regulates the expression of tumor suppressor PDCD4 in human glioblastoma cell T98G. Cancer Lett. 2008;272(2):197–205. doi: 10.1016/j.canlet.2008.06.034. [DOI] [PubMed] [Google Scholar]
- 32.Gabriely G, et al. MicroRNA 21 promotes glioma invasion by targeting matrix metalloproteinase regulators. Mol Cell Biol. 2008;28(17):5369–80. doi: 10.1128/MCB.00479-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Corsten MF, et al. MicroRNA-21 Knockdown Disrupts Glioma Growth In vivo and Displays Synergistic Cytotoxicity with Neural Precursor Cell–Delivered S-TRAIL in Human Gliomas. Cancer Research. 2007;67(19):8994–9000. doi: 10.1158/0008-5472.CAN-07-1045. [DOI] [PubMed] [Google Scholar]
- 34.Kwak HJ, et al. Downregulation of Spry2 by miR-21 triggers malignancy in human gliomas. Oncogene. 2011;30(21):2433–42. doi: 10.1038/onc.2010.620. [DOI] [PubMed] [Google Scholar]
- 35.Sasayama T, et al. MicroRNA-10b is overexpressed in malignant glioma and associated with tumor invasive factors, uPAR and RhoC. Int J Cancer. 2009;125(6):1407–13. doi: 10.1002/ijc.24522. [DOI] [PubMed] [Google Scholar]
- 36.Gabriely G, et al. Human glioma growth is controlled by microRNA-10b. Cancer Res. 2011;71(10):3563–72. doi: 10.1158/0008-5472.CAN-10-3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lavon I, et al. Gliomas display a microRNA expression profile reminiscent of neural precursor cells. Neuro Oncol. 2010;12(5):422–33. doi: 10.1093/neuonc/nop061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ernst A, et al. De-repression of CTGF via the miR-17-92 cluster upon differentiation of human glioblastoma spheroid cultures. Oncogene. 2010;29(23):3411–22. doi: 10.1038/onc.2010.83. [DOI] [PubMed] [Google Scholar]
- 39.Dews M, et al. The myc-miR-17∼92 axis blunts TGF{beta} signaling and production of multiple TGF{beta}-dependent antiangiogenic factors. Cancer Res. 2010;70(20):8233–46. doi: 10.1158/0008-5472.CAN-10-2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim H, et al. Integrative genome analysis reveals an oncomir/oncogene cluster regulating glioblastoma survivorship. Proceedings of the National Academy of Sciences. 2010;107(5):2183–2188. doi: 10.1073/pnas.0909896107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Galli R, et al. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004;64(19):7011–21. doi: 10.1158/0008-5472.CAN-04-1364. [DOI] [PubMed] [Google Scholar]
- 42.Singh SK, et al. Identification of human brain tumour initiating cells. Nature. 2004;432(7015):396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 43.Yuan X, et al. Isolation of cancer stem cells from adult glioblastoma multiforme. Oncogene. 2004;23(58):9392–400. doi: 10.1038/sj.onc.1208311. [DOI] [PubMed] [Google Scholar]
- 44.Bao S, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444(7120):756–60. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 45.Bier A, et al. MicroRNA-137 is downregulated in glioblastoma and inhibits the stemness of glioma stem cells by targeting RTVP-1. Oncotarget. 2013;4(5):665–76. doi: 10.18632/oncotarget.928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sathyan P, et al. Mir-21–Sox2 Axis Delineates Glioblastoma Subtypes with Prognostic Impact. The Journal of Neuroscience. 2015;35(45):15097–15112. doi: 10.1523/JNEUROSCI.1265-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lopez-Bertoni H, et al. DNMT-dependent suppression of microRNA regulates the induction of GBM tumor-propagating phenotype by Oct4 and Sox2. Oncogene. 2015;34(30):3994–4004. doi: 10.1038/onc.2014.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li Y, et al. MicroRNA-34a inhibits glioblastoma growth by targeting multiple oncogenes. Cancer Res. 2009;69(19):7569–76. doi: 10.1158/0008-5472.CAN-09-0529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kefas B, et al. The neuronal microRNA miR-326 acts in a feedback loop with notch and has therapeutic potential against brain tumors. J Neurosci. 2009;29(48):15161–8. doi: 10.1523/JNEUROSCI.4966-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Floyd DH, et al. Novel Anti-Apoptotic MicroRNAs 582-5p and 363 Promote Human Glioblastoma Stem Cell Survival via Direct Inhibition of Caspase 3, Caspase 9, and Bim. PLoS One. 2014;9(5):e96239. doi: 10.1371/journal.pone.0096239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Godlewski J, et al. Targeting of the Bmi-1 oncogene/stem cell renewal factor by microRNA-128 inhibits glioma proliferation and self-renewal. Cancer Res. 2008;68(22):9125–30. doi: 10.1158/0008-5472.CAN-08-2629. [DOI] [PubMed] [Google Scholar]
- 52.Peruzzi P, et al. MicroRNA-128 coordinately targets Polycomb Repressor Complexes in glioma stem cells. Neuro Oncol. 2013;15(9):1212–24. doi: 10.1093/neuonc/not055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wong HK, et al. The Cancer Genome Atlas Analysis Predicts MicroRNA for Targeting Cancer Growth and Vascularization in Glioblastoma. Mol Ther. 2015;23(7):1234–47. doi: 10.1038/mt.2015.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Alvarado AG, et al. Coordination of self-renewal in glioblastoma by integration of adhesion and microRNA signaling. Neuro-Oncology. 2015 doi: 10.1093/neuonc/nov196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang Y, et al. Multiple receptor tyrosine kinases converge on microRNA-134 to control KRAS, STAT5B, and glioblastoma. Cell Death Differ. 2014;21(5):720–734. doi: 10.1038/cdd.2013.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gilbertson RJ, Rich JN. Making a tumour's bed: glioblastoma stem cells and the vascular niche. Nat Rev Cancer. 2007;7(10):733–6. doi: 10.1038/nrc2246. [DOI] [PubMed] [Google Scholar]
- 57.Babae N, et al. Systemic miRNA-7 delivery inhibits tumor angiogenesis and growth in murine xenograft glioblastoma. Oncotarget. 2014;5(16):6687–700. doi: 10.18632/oncotarget.2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sun J, et al. MiR-137 inhibits proliferation and angiogenesis of human glioblastoma cells by targeting EZH2. J Neurooncol. 2015;122(3):481–9. doi: 10.1007/s11060-015-1753-x. [DOI] [PubMed] [Google Scholar]
- 59.Ostergaard L, et al. The relationship between tumor blood flow, angiogenesis, tumor hypoxia, and aerobic glycolysis. Cancer Res. 2013;73(18):5618–24. doi: 10.1158/0008-5472.CAN-13-0964. [DOI] [PubMed] [Google Scholar]
- 60.Fang L, et al. MicroRNA miR-93 promotes tumor growth and angiogenesis by targeting integrin-beta8. Oncogene. 2011;30(7):806–21. doi: 10.1038/onc.2010.465. [DOI] [PubMed] [Google Scholar]
- 61.Smits M, et al. Myc-associated zinc finger protein (MAZ) is regulated by miR-125b and mediates VEGF-induced angiogenesis in glioblastoma. FASEB J. 2012;26(6):2639–47. doi: 10.1096/fj.11-202820. [DOI] [PubMed] [Google Scholar]
- 62.Bronisz A, et al. Extracellular vesicles modulate the glioblastoma microenvironment via a tumor suppression signaling network directed by miR-1. Cancer Res. 2014;74(3):738–50. doi: 10.1158/0008-5472.CAN-13-2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309–14. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 64.Godlewski J, et al. MicroRNA-451 regulates LKB1/AMPK signaling and allows adaptation to metabolic stress in glioma cells. Mol Cell. 2010;37(5):620–32. doi: 10.1016/j.molcel.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Godlewski J, et al. microRNA-451: A conditional switch controlling glioma cell proliferation and migration. Cell Cycle. 2010;9(14):2742–8. [PubMed] [Google Scholar]
- 66.Ansari KI, et al. Glucose-based regulation of miR-451/AMPK signaling depends on the OCT1 transcription factor. Cell Rep. 2015;11(6):902–9. doi: 10.1016/j.celrep.2015.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bronisz A, Chiocca EA, Godlewski J. Response to energy depletion: miR-451/AMPK loop. Oncotarget. 2015;6(20):17851–2. doi: 10.18632/oncotarget.4606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hardie DG. AMPK: positive and negative regulation, and its role in whole-body energy homeostasis. Curr Opin Cell Biol. 2015;33:1–7. doi: 10.1016/j.ceb.2014.09.004. [DOI] [PubMed] [Google Scholar]
- 69.Kefas B, et al. Pyruvate kinase M2 is a target of the tumor-suppressive microRNA-326 and regulates the survival of glioma cells. Neuro Oncol. 2010;12(11):1102–12. doi: 10.1093/neuonc/noq080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Masui K, et al. mTOR complex 2 controls glycolytic metabolism in glioblastoma through FoxO acetylation and upregulation of c-Myc. Cell Metab. 2013;18(5):726–39. doi: 10.1016/j.cmet.2013.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Charles NA, et al. The brain tumor microenvironment. Glia. 2011;59(8):1169–80. doi: 10.1002/glia.21136. [DOI] [PubMed] [Google Scholar]
- 72.Nduom EK, Weller M, Heimberger AB. Immunosuppressive mechanisms in glioblastoma. Neuro Oncol. 2015;17 Suppl 7:vii9–vii14. doi: 10.1093/neuonc/nov151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Codo P, et al. MicroRNA-mediated down-regulation of NKG2D ligands contributes to glioma immune escape. Oncotarget. 2014;5(17):7651–62. doi: 10.18632/oncotarget.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wei J, et al. miR-124 Inhibits STAT3 Signaling to Enhance T Cell–Mediated Immune Clearance of Glioma. Cancer Research. 2013;73(13):3913–3926. doi: 10.1158/0008-5472.CAN-12-4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ueda R, et al. Dicer-regulated microRNAs 222 and 339 promote resistance of cancer cells to cytotoxic T-lymphocytes by down-regulation of ICAM-1. Proceedings of the National Academy of Sciences. 2009;106(26):10746–10751. doi: 10.1073/pnas.0811817106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hussain SF, et al. The role of human glioma-infiltrating microglia/macrophages in mediating antitumor immune responses. Neuro Oncol. 2006;8(3):261–79. doi: 10.1215/15228517-2006-008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Xu S, et al. Effect of miR-142-3p on the M2 macrophage and therapeutic efficacy against murine glioblastoma. J Natl Cancer Inst. 2014;106(8) doi: 10.1093/jnci/dju162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wei J, et al. MiR-138 exerts anti-glioma efficacy by targeting immune checkpoints. Neuro Oncol. 2015 doi: 10.1093/neuonc/nov292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ohno M, et al. Expression of miR-17-92 enhances anti-tumor activity of T-cells transduced with the anti-EGFRvIII chimeric antigen receptor in mice bearing human GBM xenografts. J Immunother Cancer. 2013;1:21. doi: 10.1186/2051-1426-1-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kosaka A, et al. Transgene-derived overexpression of miR-17-92 in CD8+ T-cells confers enhanced cytotoxic activity. Biochem Biophys Res Commun. 2015;458(3):549–54. doi: 10.1016/j.bbrc.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Heijnen HF, et al. Activated platelets release two types of membrane vesicles: microvesicles by surface shedding and exosomes derived from exocytosis of multivesicular bodies and alpha-granules. Blood. 1999;94(11):3791–9. [PubMed] [Google Scholar]
- 82.Kowal J, Tkach M, Thery C. Biogenesis and secretion of exosomes. Curr Opin Cell Biol. 2014;29:116–25. doi: 10.1016/j.ceb.2014.05.004. [DOI] [PubMed] [Google Scholar]
- 83.Dainiak N. Surface membrane-associated regulation of cell assembly, differentiation, and growth. Blood. 1991;78(2):264–76. [PubMed] [Google Scholar]
- 84.Mathivanan S, et al. Proteomics analysis of A33 immunoaffinity-purified exosomes released from the human colon tumor cell line LIM1215 reveals a tissue-specific protein signature. Mol Cell Proteomics. 2010;9(2):197–208. doi: 10.1074/mcp.M900152-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ostrowski M, et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat Cell Biol. 2010;12(1):19–30. doi: 10.1038/ncb2000. sup pp 1-13. [DOI] [PubMed] [Google Scholar]
- 86.Dolo V, et al. Selective localization of matrix metalloproteinase 9, beta1 integrins, and human lymphocyte antigen class I molecules on membrane vesicles shed by 8701-BC breast carcinoma cells. Cancer Res. 1998;58(19):4468–74. [PubMed] [Google Scholar]
- 87.Schiera G, et al. Neurons produce FGF2 and VEGF and secrete them at least in part by shedding extracellular vesicles. J Cell Mol Med. 2007;11(6):1384–94. doi: 10.1111/j.1582-4934.2007.00100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Proia P, et al. Astrocytes shed extracellular vesicles that contain fibroblast growth factor-2 and vascular endothelial growth factor. Int J Mol Med. 2008;21(1):63–7. [PubMed] [Google Scholar]
- 89.Li CC, et al. Glioma microvesicles carry selectively packaged coding and non-coding RNAs which alter gene expression in recipient cells. RNA Biol. 2013;10(8):1333–44. doi: 10.4161/rna.25281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hessvik NP, et al. Profiling of microRNAs in exosomes released from PC-3 prostate cancer cells. Biochim Biophys Acta. 2012;1819(11-12):1154–63. doi: 10.1016/j.bbagrm.2012.08.016. [DOI] [PubMed] [Google Scholar]
- 91.Armstrong DA, et al. MicroRNA molecular profiling from matched tumor and bio-fluids in bladder cancer. Mol Cancer. 2015;14(1):194. doi: 10.1186/s12943-015-0466-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Li CCY, et al. Glioma microvesicles carry selectively packaged coding and non-coding RNAs which alter gene expression in recipient cells. RNA Biol. 2013;10(8):1333–1344. doi: 10.4161/rna.25281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Villarroya-Beltri C, et al. Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat Commun. 2013;4:2980. doi: 10.1038/ncomms3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Koppers-Lalic D, et al. Nontemplated nucleotide additions distinguish the small RNA composition in cells from exosomes. Cell Rep. 2014;8(6):1649–58. doi: 10.1016/j.celrep.2014.08.027. [DOI] [PubMed] [Google Scholar]
- 95.van der Vos KE, et al. Directly visualized glioblastoma-derived extracellular vesicles transfer RNA to microglia/macrophages in the brain. Neuro Oncol. 2016;18(1):58–69. doi: 10.1093/neuonc/nov244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Skog J, et al. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol. 2008;10(12):1470–6. doi: 10.1038/ncb1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ridder K, et al. Extracellular vesicle-mediated transfer of functional RNA in the tumor microenvironment. Oncoimmunology. 2015;4(6):e1008371. doi: 10.1080/2162402X.2015.1008371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gasser O, et al. Characterisation and properties of ectosomes released by human polymorphonuclear neutrophils. Exp Cell Res. 2003;285(2):243–57. doi: 10.1016/s0014-4827(03)00055-7. [DOI] [PubMed] [Google Scholar]
- 99.Eken C, et al. Polymorphonuclear neutrophil-derived ectosomes interfere with the maturation of monocyte-derived dendritic cells. J Immunol. 2008;180(2):817–24. doi: 10.4049/jimmunol.180.2.817. [DOI] [PubMed] [Google Scholar]
- 100.Pluskota E, et al. Expression, activation, and function of integrin alphaMbeta2 (Mac-1) on neutrophil-derived microparticles. Blood. 2008;112(6):2327–35. doi: 10.1182/blood-2007-12-127183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hoshino A, et al. Tumour exosome integrins determine organotropic metastasis. Nature. 2015;527(7578):329–35. doi: 10.1038/nature15756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gasser O, Schifferli JA. Activated polymorphonuclear neutrophils disseminate anti-inflammatory microparticles by ectocytosis. Blood. 2004;104(8):2543–8. doi: 10.1182/blood-2004-01-0361. [DOI] [PubMed] [Google Scholar]
- 103.Al-Nedawi K, et al. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat Cell Biol. 2008;10(5):619–24. doi: 10.1038/ncb1725. [DOI] [PubMed] [Google Scholar]
- 104.Nakase I, et al. Active macropinocytosis induction by stimulation of epidermal growth factor receptor and oncogenic Ras expression potentiates cellular uptake efficacy of exosomes. Sci Rep. 2015;5:10300. doi: 10.1038/srep10300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Svensson KJ, et al. Exosome uptake depends on ERK1/2-heat shock protein 27 signaling and lipid Raft-mediated endocytosis negatively regulated by caveolin-1. J Biol Chem. 2013;288(24):17713–24. doi: 10.1074/jbc.M112.445403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Christianson HC, et al. Cancer cell exosomes depend on cell-surface heparan sulfate proteoglycans for their internalization and functional activity. Proc Natl Acad Sci U S A. 2013;110(43):17380–5. doi: 10.1073/pnas.1304266110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Atai NA, et al. Heparin blocks transfer of extracellular vesicles between donor and recipient cells. J Neurooncol. 2013;115(3):343–51. doi: 10.1007/s11060-013-1235-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Montecalvo A, et al. Mechanism of transfer of functional microRNAs between mouse dendritic cells via exosomes. Blood. 2012;119(3):756–66. doi: 10.1182/blood-2011-02-338004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Parolini I, et al. Microenvironmental pH is a key factor for exosome traffic in tumor cells. J Biol Chem. 2009;284(49):34211–22. doi: 10.1074/jbc.M109.041152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Mineo M, et al. Exosomes released by K562 chronic myeloid leukemia cells promote angiogenesis in a Src-dependent fashion. Angiogenesis. 2012;15(1):33–45. doi: 10.1007/s10456-011-9241-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kucharzewska P, et al. Exosomes reflect the hypoxic status of glioma cells and mediate hypoxia-dependent activation of vascular cells during tumor development. Proc Natl Acad Sci U S A. 2013;110(18):7312–7. doi: 10.1073/pnas.1220998110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Agrawal R, et al. Hypoxic signature of microRNAs in glioblastoma: insights from small RNA deep sequencing. BMC Genomics. 2014;15:686. doi: 10.1186/1471-2164-15-686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Tadokoro H, et al. Exosomes derived from hypoxic leukemia cells enhance tube formation in endothelial cells. J Biol Chem. 2013;288(48):34343–51. doi: 10.1074/jbc.M113.480822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Li R, et al. Identification of intrinsic subtype-specific prognostic microRNAs in primary glioblastoma. J Exp Clin Cancer Res. 2014;33:9. doi: 10.1186/1756-9966-33-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Yan W, et al. MicroRNA expression patterns in the malignant progression of gliomas and a 5-microRNA signature for prognosis. Oncotarget. 2014;5(24):12908–15. doi: 10.18632/oncotarget.2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hu J, et al. MiR-215 Is Induced Post-transcriptionally via HIF-Drosha Complex and Mediates Glioma-Initiating Cell Adaptation to Hypoxia by Targeting KDM1B. Cancer Cell. 2016;29(1):49–60. doi: 10.1016/j.ccell.2015.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Chen CC, et al. Stereotactic brain biopsy: Single center retrospective analysis of complications. Clin Neurol Neurosurg. 2009;111(10):835–9. doi: 10.1016/j.clineuro.2009.08.013. [DOI] [PubMed] [Google Scholar]
- 118.Westphal M, Lamszus K. Circulating biomarkers for gliomas. Nat Rev Neurol. 2015;11(10):556–66. doi: 10.1038/nrneurol.2015.171. [DOI] [PubMed] [Google Scholar]
- 119.Pisitkun T, Shen RF, Knepper MA. Identification and proteomic profiling of exosomes in human urine. Proc Natl Acad Sci U S A. 2004;101(36):13368–73. doi: 10.1073/pnas.0403453101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Gallo A, et al. The majority of microRNAs detectable in serum and saliva is concentrated in exosomes. PLoS One. 2012;7(3):e30679. doi: 10.1371/journal.pone.0030679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Akers JC, et al. MiR-21 in the extracellular vesicles (EVs) of cerebrospinal fluid (CSF): a platform for glioblastoma biomarker development. PLoS One. 2013;8(10):e78115. doi: 10.1371/journal.pone.0078115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Qu S, Guan J, Liu Y. Identification of microRNAs as novel biomarkers for glioma detection: a meta-analysis based on 11 articles. J Neurol Sci. 2015;348(1-2):181–7. doi: 10.1016/j.jns.2014.11.036. [DOI] [PubMed] [Google Scholar]
- 123.Shi L, et al. MiR-21 protected human glioblastoma U87MG cells from chemotherapeutic drug temozolomide induced apoptosis by decreasing Bax/Bcl-2 ratio and caspase-3 activity. Brain Res. 2010;1352:255–64. doi: 10.1016/j.brainres.2010.07.009. [DOI] [PubMed] [Google Scholar]
- 124.Teplyuk NM, et al. MicroRNAs in cerebrospinal fluid identify glioblastoma and metastatic brain cancers and reflect disease activity. Neuro Oncol. 2012;14(6):689–700. doi: 10.1093/neuonc/nos074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Roth P, et al. A specific miRNA signature in the peripheral blood of glioblastoma patients. J Neurochem. 2011;118(3):449–57. doi: 10.1111/j.1471-4159.2011.07307.x. [DOI] [PubMed] [Google Scholar]
- 126.Manterola L, et al. A small noncoding RNA signature found in exosomes of GBM patient serum as a diagnostic tool. Neuro Oncol. 2014;16(4):520–7. doi: 10.1093/neuonc/not218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Bronisz A, et al. Reprogramming of the tumour microenvironment by stromal PTEN-regulated miR-320. Nat Cell Biol. 2012;14(2):159–67. doi: 10.1038/ncb2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sun JY, et al. MicroRNA-320 inhibits cell proliferation in glioma by targeting E2F1. Mol Med Rep. 2015;12(2):2355–9. doi: 10.3892/mmr.2015.3657. [DOI] [PubMed] [Google Scholar]