

Abstract
Background
X linked intellectual disability (XLID) syndromes account for a substantial number of males with ID. Much progress has been made in identifying the genetic cause in many of the syndromes described 20–40 years ago. Next generation sequencing (NGS) has contributed to the rapid discovery of XLID genes and identifying novel mutations in known XLID genes for many of these syndromes.
Methods
2 NGS approaches were employed to identify mutations in X linked genes in families with XLID disorders. 1 involved exome sequencing of genes on the X chromosome using the Agilent SureSelect Human X Chromosome Kit. The second approach was to conduct targeted NGS sequencing of 90 known XLID genes.
Results
We identified the same mutation, a c.12928 G>C transversion in the HUWE1 gene, which gives rise to a p.G4310R missense mutation in 2 XLID disorders: Juberg-Marsidi syndrome (JMS) and Brooks syndrome. Although the original families with these disorders were considered separate entities, they indeed overlap clinically. A third family was also found to have a novel HUWE1 mutation.
Conclusions
As we identified a HUWE1 mutation in an affected male from the original family reported by Juberg and Marsidi, it is evident the syndrome does not result from a mutation in ATRX as reported in the literature. Additionally, our data indicate that JMS and Brooks syndromes are allelic having the same HUWE1 mutation.
Strengths and limitations of this study.
Using the power of next generation sequencing, we have linked Juberg-Marsidi syndrome (JMS) and Brooks syndrome as allelic conditions.
This study provides better organisation to the field of X linked disorders by providing evidence that JMS is not caused by mutation in the ATRX gene.
We provide clinical descriptions and comparisons to support our conclusions.
The reported missense mutations have detrimental effects on HUWE1 functional activity.
Based on the limited number of patients/families, we are unable to make specific genotype–phenotype correlations at this time.
Introduction
In the two decades since the discovery of the trinucleotide repeat expansion in FMR1 responsible for fragile X syndrome, substantial progress has been made in defining the other X linked intellectual disability (XLID) syndromes at the molecular level.1–3 Mutations in over 100 X-chromosome genes have been assigned at least provisional roles in these syndromes.2 4 Some syndromes, initially believed to be separate conditions, have now been lumped together while others that appeared to be the same have been separated on the basis of molecular findings.2 3 5 6
Juberg-Marsidi syndrome (JMS) has heretofore been considered by some observers to be allelic to α-thalassemia intellectual disability (ATRX syndrome) while Brooks syndrome (aka Brooks-Wisniewski-Brown syndrome) has been considered to be distinct from JMS.7–10 A missense mutation in ATRX has been previously reported in a family considered to have JMS.8 On clinical re-evaluation of the original family reported by Juberg and Marsidi,7 we considered the findings in the sole affected male still alive to be inconsistent with ATRX syndrome. While there are some similarities between JMS and ATRX syndrome, there are distinctive differences.7 Specifically, the surviving male in the Juberg-Marsidi family had bifrontal narrowing, hypotelorism, prominent nose with overhanging columella, and thin lips, all facial manifestations very different than those of ATRX syndrome. Subsequently, we identified in this male, a novel missense alteration in HUWE1, a gene located at Xp11.23 that encodes an E3 ubiquitin ligase that regulates numerous proteins, such as key cellular factors p53 and Mcl1.11 12 The identical missense alteration in HUWE1 was also identified and shown to properly segregate in another XLID syndrome reported by Brooks et al.10 The clinical phenotypes in the two conditions previously considered to be unrelated show close similarity. Here, we update the clinical and molecular findings in these two families and report an additional family with another missense mutations in HUWE1.
Methods
Informed consent
The authors note that we have obtained proper informed consent from the participants (or parental consent for participants under 18 years of age) for use of their personal information, including photos, for publication purposes. Copies of these consents have been shared with the office of the Editor.
Families
Clinical update: K9149 (original JMS family)
Clinical findings in four affected males in three generations included generalised growth retardation, severe intellectual disability, tall forehead, epicanthus, short palpebral fissures, pale retina, flat nasal bridge, large or cupped ears, small penis with undescended testes and deafness.7 The two brothers in generation II died in infancy or childhood.
The pedigree and clinical findings were updated in 2006. The nephew (III-2 in figure 1A), age 27 years, was found to have moderate hearing loss of unknown type but believed to be related to recurrent otitis media, severe growth impairment (all measurements below the third centile) and intellectual disability (IQ<20). Craniofacial findings included facial asymmetry, bifrontal narrowing, furrowing of the forehead, prominent brow giving the eyes and midface a sunken appearance, hypotelorism, upslanting palpebral fissures, prominent nose with overhanging columella, long philtrum and thin lips with a beak at the midpoint of the upper lip (figure 1). Additional features were brachydactyly, decreased musculature of the distal legs, foot deformation (right foot everted, left foot with midfoot varus), scoliosis, flexion of the hips, genu vara, soft loose skin over the hands and hyperextensible joints. Sexual hair and penis appeared normal but the testes were not palpable. He had no speech, but uttered sounds when under stress and used gestures to indicate his desires (table 1).
Figure 1.

Pedigrees of families with HUWE1 mutations. (A) Pedigree of K9149. Photographs show microcephaly, cupped ears and thin upper lip in infant (II-6); and 27-year-old nephew (III-2) from original family reported by Juberg and Marsidi.7 (B) Pedigree of family K9223. Photographs show microcephaly, cupped ears, blepharophimosis, deeply set eyes, depressed nasal bridge and bulbous nasal tip in affected male (III-1) from original family reported by Brooks et al10 at age 4 months and 4 years. (C) Pedigree of family 3. The affected boys' mother (I-2) and one sister (II-2) have mild learning disabilities. Photographs show microcephaly, frontal hair upsweep, deep-set eyes, broad nasal tip, wide mouth, thin upper lip and prominent jaw in brothers, ages 10 (II-5) and 13 years (II-4). The older brother also has cupped ears, thick eyebrows and coarse face. +, presence of mutation; −, normal;  , proband;
, proband;  , affected;
, affected;  , carrier.
, carrier.
Table 1.
Comparison of findings in families with missense mutations and duplications of HUWE1*
| Juberg and Marsidi7 | Brooks et al10 | Family 3 | Froyen et al22 |
Isrie et al24 | Mattei et al9 | ||||
|---|---|---|---|---|---|---|---|---|---|
| K9149 | K9223 | A323 | UK444 | UK106 | Dup HUWE1 | ATRX mutation | |||
| Affected individuals | 4M | 3M | 2M | 4M | 3M | 3M | 24M | 2M | 7M |
| Mutation | p.G4310R | p.G4310R | p.R4063Q | p.R4013W | p.R4187C | p.R2981H | Dup | p.R4013W | p.R1272Q |
| Low birth weight (<3c) | 4/4 | 3/3 | 2/2 | 0/1 | – | – | 0/12 | – | 2/2 |
| Low birth length (<3c) | 4/4 | – | 2/2 | – | – | – | 0/1 | – | 1/2 |
| Short stature (<3c) | 4/4 | 3/3 | 2/2 | – | 1/1 | – | 0/19 | 0/2 | – |
| Microcephaly (<3c) | 4/4 | 3/3 | 2/2 | 0/4 | 0/1 | 0/1 | 1/19 | 0/2 | 1/2 |
| Macrocephaly (>97c) | 0/4 | 0/3 | 0/2 | 2/4 | 0/1 | 0/1 | 0/19 | 0/2 | 0/2 |
| Strabismus | 2/3 | 2/3 | – | – | 1/1 | – | 1/11 | 0/2 | – |
| Epicanthus | 2/2 | 3/3 | – | – | – | – | 3/8 | 0/2 | 1/1 |
| Deep-set eyes | 1/1 | 3/3 | 2/2 | – | – | – | 0/8 | 1/2 | – |
| Blepharophimosis/small pf | 4/4 | 3/3 | – | – | – | – | 3/8 | 0/2 | 1/1 |
| Cupped ears | 1/3 | 2/3 | 2/2 | 0/4 | – | 1/2 | 3/8 | 0/2 | 0/1 |
| Prominent nose | 1/3 | 3/3 | 2/2 | 0/3 | – | – | 3/8 | 0/2 | 0/1 |
| Thin lip(s) | 2/3 | 3/3 | 2/2 | 1/3 | – | – | 0/9 | – | 0/1 |
| Undescended testes | 4/4 | – | 0/2 | 0/1 | – | – | 0/8 | – | 2/3 |
| Deafness | 4/4 | 1/3 | 0/2 | 1/4 | – | – | 0/20 | – | 1/3 |
| Seizures | 1/4 | 0/3 | 0/2 | 0/4 | – | – | 0/8 | – | – |
| Hypotonia | 1/4 | 0/3 | 0/2 | 1/1 | – | – | 5/11 | – | 1/1 |
| Severe ID (IQ<50) | 4/4 | 3/3 | 2/2 | 4/4 | – | – | 12/17 | 2/2 | 3/3 |
| Absent/limited speech | 3/3 | 3/3 | 1/2 | 1/3 | 0/1 | 2/2 | 5/8 | – | – |
| Other findings | Bifrontal narrowing, hypotelorism, contractures | Bifrontal narrowing, short philtrum, contractures, clumsiness | Contractures | Tapered fingers | Clumsiness | Contractures | Hypertelorism, down-slanting palpebral fissures | Non-attentive gaze, GI disturbances | |
*The de novo p.V950A mutation in one of two brothers with autism reported by Nava et al25 is not included because of lack of clinical details.
<3c, less than 3rd centile; >97c, greater than 97th centile; dash (–), indicates information not available; GI, gastrointestinal; pf, palpebral fissures.
All females, including two obligate and four non-obligate carriers identified by gene testing, had normal growth, craniofacial appearance and intellectual function. Marked skewing (>90:10) of X chromosome inactivation in all carrier females provided a plausible reason for absence of clinical manifestations. One carrier subsequently had an affected son. He had growth retardation when delivered at 38 weeks with a weight of 1510 g, head circumference of 30 cm and length of 42 cm, all measurements less than the third centile for 38 weeks. He had hypotelorism, small appearing eyes, flat nasal bridge, right cleft lip and palate, supravalvar aortic stenosis, small hands and feet, externally rotated feet and undescended testes. Ultrasound examination of the brain showed a small cyst (1.0×1.6 cm) within each lateral ventricle. He failed the newborn hearing test.
Clinical update: K9223 (original Brooks syndrome family)
Clinical findings in two affected brothers and a nephew were generalised intrauterine and postnatal growth retardation, severe intellectual disability, triangular-shaped face, low posterior hairline, bifrontal narrowness, malar flatness, blepharophimosis, optic atrophy, esotropia, nystagmus, bulbous nose, low-set and cupped ears, short philtrum, thin tented upper lip and pectus excavatum (figure 1B).10 They were poorly coordinated and became self-abusive, hyperactive and spastic with large joint contractures.
The pedigree and clinical findings were updated in 2010. The affected males were aged 23, 45 and 52 years. All growth parameters remained well below the third centile. Affected males became ambulatory by about age 3 years, but remained non-verbal or had very limited speech (table 1). Hearing loss was believed to be related to recurrent otitis media. Cranial imaging showed enlarged ventricles and small brain.
Three carrier females had normal intellectual function and showed no phenotypic abnormalities. X-inactivation was markedly skewed (>90:10) in the two carriers who were informative at the AR locus.
Clinical report: family 3
Family 3 consists of two brothers, now aged 10 and 13 years (II-5 and II-4 in figure 1C), who have severe intellectual disability, microcephaly and postnatal growth failure. The brothers have three sisters, one of whom has minor literacy problems. Their mother also has a minor learning difficulty, not present in her three brothers and sister. X-inactivation testing in the mother and the daughter with literacy problems was uninformative.
The two brothers were born at term gestation with weights and lengths at term at or below the third centile, being 2.52 and 2.67 kg and 44 and 47 cm, respectively. Neither was microcephalic at birth but have been less than third centile since early childhood. Early motor milestones were delayed with both boys not walking independently until after age 2 years. The oldest brother had tight Achilles tendons and required serial casting and ankle-foot orthoses. His gait was broad based and ataxic, with reduced extension of hips and knees. The younger brother had better language skills, with over 100 single words, and could follow single step commands; the older brother only had a few single words.
Both brothers had early onset growth failure, with significant short stature. Their heights were more than −4.0 SD from the mean by age 3 years. Dynamic testing excluded growth hormone deficiency (peak growth hormone levels 54.3 and 13.9 mIU/L to glucagon stimulation). Alternative causes of short stature were not identified. Both boys were treated with and responded to growth hormone, with their heights now approaching the third centile. Neither boy had clinical or biochemical evidence of other endocrine abnormalities. Their genitalia were normal and the older brother had normal pubertal development. The youngest brother has astigmatism, but otherwise vision and hearing in both boys is normal.
Both brothers are now markedly microbrachycephalic, with head circumference of 47 cm (less than third centile) and 48 cm (less than third centile), respectively. They have sparse scalp hair, which was more obvious in early infancy. Craniofacial features include deep-set eyes, broad nasal bridge and nasal tip, a wide mouth, a thin upper lip and prognathism (figure 1C and table 1). The older brother's ears are cupped, his eyebrows are thick and his face is now coarse, resembling the pictures of the patient with JMS (figure 1A).
Skeletal surveys in both boys demonstrated only brachycephaly and delayed bone age (prior to treatment with growth hormone). Neuroimaging showed some increased extra-axial space and ventriculomegaly; white matter loss in the parietal, occipital and periventricular regions; a normal pituitary gland and a posterior pituitary bright spot.
Marker analysis
As the clinical evaluation of K9149, the original Juberg-Marsidi family was not consistent with ATRX syndrome and sequencing of ATRX conducted at the Greenwood Genetic Center failed to detect a mutation, marker analysis using 28 X-chromosome markers was performed.13
Sequencing
Genomic DNA from affected male, III-2, in family K9149 was enriched for exons of 718 genes on the X chromosome using the Agilent SureSelect Human X Chromosome Kit and subjected to next generation sequencing (NGS) as previously described.14 DNA from affected male, III-1, was enriched for 90XLID genes using RainDance microdroplet technology (RainDance Technologies, Lexington, Massachusetts, USA). Sequencing of these 90 genes was performed on an Applied Biosystems SOLiD 3 Plus Sequencer.
Preparation of whole cell extracts
Lymphoblastoid cells were collected and washed twice with ice cold phosphate buffered saline (PBS), followed by flash freezing of the cell pellet in liquid N2. Subsequently, the thawed cells were resuspended in one packed cell volume (PCV) of lysis buffer I (10 mM Tris-HCl pH 7.8, 200 mM KCl, 1 mM N-ethylmaleimide (NEM), 0.1 mM MG-132, 1 μM PMSF, 2 μM leupeptin, 3 μM bestatin, 1.5 μM pepstatin) and two PCVs of lysis buffer II (10 mM Tris-HCl pH 7.8, 600 mM KCl, 2 mM EDTA, 40% glycerol, 0.2% NP-40, 1 mM NEM, 0.1 mM MG-132, 1 μM phenylmethylsulfonyl fluoride (PMSF), 2 μM leupeptin, 3 μM bestatin, 1.5 μM pepstatin). The samples were further incubated for 30 min at 4°C, sonicated and centrifuged. The supernatants were either stored at −80°C or used directly for immunoblot analysis.
Immunoblotting
High molecular weight proteins present in the whole cell extract were separated on a 7% Tris-acetate polyacrylamide (PA) gel, while lower molecular weight proteins (p53 and Mcl-1) were separated by a 10% Tris-glycine PA gel. After separation, proteins were transferred to an Immobilon-FL membrane (Millipore), followed by subsequent immunoblotting. Primary antibodies against HUWE1 (Bethyl Laboratories), p53 (Santa Cruz) and Mcl-1 (BD Pharmingen) were detected using infrared (IR) dye-conjugated secondary antibodies (Rockland). The signal was visualised using direct IR fluorescence via the Odyssey Scanner (LI-COR Biosciences).
shRNA knockdown
Cells from male control were lentivirally transduced with plasmids carrying the non-specific control shRNA (pGIPZ-Ns, Open Biosystems) or the mule-targeting shRNA (GIPZ Human HUWE1 shRNA, Clone-ID V3LHS_353153, Open Biosystems). Stable clones were subsequently selected with puromycin, while the selection efficiency was monitored by the percentage of green fluorescent protein (GFP)-positive cells.
Results
Segregation analysis in K9149 of 28 markers spaced along the X chromosome indicated four markers from DX51055 (Xp11.2) to AR (Xq12) were linked to JMS (data not shown). The region did not include the ATRX gene confirming the negative gene sequencing and clinical impression that JMS was not allelic to ATRX. But as the region contained many genes, NGS of exons on the X chromosome was initiated.
NGS of genes on the X chromosome in individual III-2 identified only one unique variant within the ‘linked’ interval, a G>C transversion at nucleotide 12928 (c.12928G>C) in HUWE1. This change results in a missense mutation, p.G4310R. The finding was confirmed by Sanger sequencing. The alteration segregated with the phenotype (figure 1A) as expected since HUWE1 is within the ‘linked’ interval.
NGS of individual III-1 in family K9223 for 90 XLID genes identified the same missense HUWE1 mutation as the only unique change after other single nucleotide polymorphisms were removed from further consideration based on available database and in-house frequency information. Sanger sequencing confirmed the NGS finding and the alteration segregated properly in the family (figure 1B). The Sorting Intolerant From Tolerant (SIFT), PolyPhen-2 and Mutation Taster prediction algorithms indicated that replacing a non-polar amino acid with one that is charged is not expected to be tolerated, consistent with a damaging effect on the protein (see online supplementary table S1).
bmjopen-2015-009537supp_table1.pdf (597.7KB, pdf)
The older brother, II-4, in family 3 was found to have a mutation in HUWE1 (c.12188G>A; p. R4063Q) by an x exome analysis. This mutation alters a highly conserved amino acid in the HECT domain. The mutation arose as a de novo event in the mother and is also present in her other affected son as well as two of her daughters (figure 1C). Neither this alteration nor the shared variant for families 1 and 2 has been reported in the available databases, including the Exome Variant Server database that includes exome data for approximately 6500 individuals.
In order to determine if the p.G4310R mutation affected the HUWE1 function, protein levels were compared between the cells derived from an affected male (figure 1A, III-2) from the original JMS family (K9149) and the cells from a healthy male family member (figure 1A, IV-1). As shown in figure 2, the amount of HUWE1 in cells from the affected male was significantly reduced to approximately 39% of the HUWE1 level in the cells from the healthy relative (figure 1A, IV-1). The levels of two important HUWE1 substrates, Mcl1 and p53, were significantly increased in the patient's cells (figure 3). To confirm that reduced HUWE1 levels directly cause the upregulation in p53 and Mcl1 levels, we performed HUWE1 knock-down experiments in cells from healthy individuals. On HUWE1 knock-down, we observed an accumulation in Mcl1 and p53, thus indicating that the change in HUWE1 status is directly correlated with changes in p53 and Mcl1 levels (see online supplementary figure S1). Taken together, these findings suggest that p. G4310R mutation identified in the affected male from the JMS family (K9149) leads to reduced E3 ligase levels and consequent accumulation of downstream targets p53 and Mcl1.
Figure 2.

E3 ubiquitin ligase HUWE1 protein level is decreased in the affected male, III-2, from family K9149. (A) Immunoblot analysis of the HUWE1 protein level a lymphoblastoid cell line from the affected male (figure 1A, III-2) and in a normal male in the family (figure 1A, IV-1). The star indicates unspecific bands. (B) Quantification of three independent experiments as the one presented in (A); error bars indicate mean±SD; **p<0.01.
Figure 3.

Protein levels of well-known HUWE1 substrates are upregulated in the affected male III-2, from family K9149. Immunoblot analysis of Mcl-1 (A) and p53 (C) protein levels in a normal male in the family (figure 1A, IV-1) and a lymphoblastoid cell line from the affected male (family 1A, III-2). (B) and (D) Quantification of three independent experiments as the ones presented in (A) and (C), respectively; error bars indicate mean±SD; *p<0.05.
bmjopen-2015-009537supp_figure1.pdf (824.1KB, pdf)
In family 3, levels of the HUWE1 p.R4063Q protein were increased, and consequently the levels of p53 substrate were reduced in the two affected brothers compared with their normal father (figure 4). Taken together, altered levels of mutant HUWE1 proteins and as a consequence, the levels of its target substrates suggest that missense changes potentially affect HUWE1 function.
Figure 4.
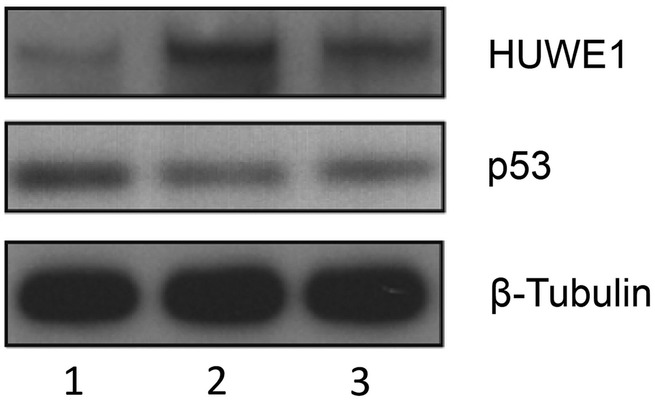
Functional assessment of the p.R4063Q HUWE1 mutation in family 3. HUWE1 protein levels are increased in p.R4063Q lymphoblastoid cell lines (LCLs) derived from the affected brothers and results in moderate reduction in the levels of the p53 protein. Unaffected father I-1 (lane 1), affected sons, II-3 and II-4 (lanes 2 and 3). Total LCL protein lysates were western blotted and probed for HUWE1, p53 and B-tubulin (loading control).
Discussion
HUWE1 plays a role in various essential physiological processes by regulating the stability of multiple proteins. Initially, HUWE1 was shown to bind the tumour suppressor p53, thus inhibiting transactivation.11 15 Further HUWE1 was shown through ubiquitylation to contribute to regulation of the antiapoptotic protein Mcl1.12 Additional substrates of HUWE1 include proteins such as histones,16 transcription factor Miz1,17 DNA polymerases β and λ,18 19 topoisomerase TopBP1,20 and others.
Among the 200 named XLID syndromes, a number of entities have been found to be allelic using molecular technologies.2 3 5 6 Prominent among these are PQBP1-associated XLID (Renpenning, Sutherland-Haan, Hamel Cerebro-Palato-Cardiac, Golabi-Ito-Hall and Porteous syndromes), ATRX-associated XLID (ATRX syndrome, Carpenter-Waziri, Holmes-Gang, Chudley-Lowry and XLID-Hypotonia-Arch Fingerprints syndromes) and ARX-associated XLID (Partington, West, Lissencephaly and Abnormal Genitalia, and Hydranencephaly with Abnormal Genitalia syndromes).
In 1983, Mattei et al9 reported an XLID family with seven affected males which they considered to have JMS. Villard et al8 found a missense mutation in the helicase domain of ATRX in the family, and since that time JMS has been called an ATRX-associated XLID syndrome in much of the literature. Our clinical re-evaluation of the surviving male in the original JMS family showed findings that were not consistent with ATRX-associated XLID, specifically bifrontal narrowing, hypotelorism, prominent nose and thin lips, prompting us to question their conclusion. Sequencing of the ATRX gene in this male failed to find a pathogenic alteration (data not shown). Further, review of the males reported by Mattei et al9 shows them to have clinical findings very different from JMS and more consistent with ATRX-associated XLID (table 1).
The availability of NGS provided an opportunity to sequence 718 X-chromosome genes simultaneously in the surviving affected member of the Juberg-Marsidi family. This effort identified a missense mutation in HUWE1 (c.12928G>C; p.G4310R) which was considered to be likely disease causing (Mutation Taster), not tolerated (SIFT) and probably damaging (PolyPhen-2; figure 5 and online supplementary table S1). Functional studies performed on a cell line from the surviving affected male in the original family with JMS showed significantly decreased HUWE1 protein levels and consequently increased levels of two well-known substrates, p53 and Mcl-1, as compared with a normal family member (figures 3 and 4).
Figure 5.

Domains and missense mutations found in HUWE1 with the relative boundaries of each domain indicated. The known missense alterations are shown below the DUF4414 and HECT domains. The p.G4310R mutation reported in this paper is in red. The WWE domain is named for its conserved residues and is predicted to mediate specific protein–protein interactions in ubiquitin and ADP-ribose conjugation systems. The C-terminal catalytic HECT belongs to a subclass of ubiquitin-protein E3 ligases. DUF913 belongs to a family found in various ubiquitin protein ligases and DUF4414 is in a family of domains frequently found in DNA-binding proteins of the URE-B1 type. DUF, domains of unknown function.
This molecular finding clearly establishes JMS as an XLID disorder caused by a mutation in HUWE1 rather than in ATRX. The same mutation was also found in the family reported by Brooks et al10 using a 90-gene XLID panel (see online supplementary table). Comparisons of the phenotype of Juberg-Marsidi and Brooks syndromes convinced us that they were examples of the same syndrome clinically. We have been unable to link the two kindreds through ancestry or common area of residence. A third family was also found to have a slightly more proximal missense mutation (p.R4063Q) which altered a highly conserved arginine in the HECT domain, which was likewise predicted to be deleterious (see online supplementary table S1). HUWE1 p.R4063Q protein had increased stability and p53 levels reduced in the lymphoblast cell lines derived from the affected males in this family. Vandewalle et al21 recently reported that an extra dose of HUWE1 affects axon branching of dorsal cluster neurons of Drosophila melanogaster through the Wnt/3-catenin pathway.
It appears that the phenotypic spectrum resulting from HUWE1 mutations is quite broad table 1. The Juberg-Marsidi-Brooks phenotype appears most severe with marked growth impairment, somatic manifestations, absent or limited speech and delayed ambulation. Two other families (families A323 and UK444 of Froyen et al22) are less severely affected, having normal or large head circumference, minimal facial dysmorphism and milder intellectual disability.23 It is of interest that carrier females in families with less severe phenotypic expression may have mild expression, not being protected by skewed inactivation of their affected X chromosome. The location of the mutations in the reported cases does not suggest an obvious explanation for the phenotypic variability. The mutation in Juberg-Marsidi-Brooks syndrome is located in the HECT domain (the catalytic domain of the HUWE1 ubiquitin protein ligase) as are the mutations in the third family reported here and in the more mildly affected UK444 family.22 The other two missense mutations in HUWE1, the p.R2981H mutation in a severely affected family and the p.R4013W mutation in a family with a less severe phenotype, are located between the WWE and HECT domains.22 Further cases with HUWE1 mutations will be necessary to determine if a genotype–phenotype correlation can be made.
Acknowledgments
The authors would like to thank the families who were patient while they searched for the genetic cause over many years. Dedicated to the memory of Ethan Francis Schwartz, 1996–1998.
Footnotes
Contributors: MJF, MJB and SS were responsible for sequencing family 2. SSB and CoS contributed clinical information and material for family 2. RES reviewed clinical material for all families and summarised them. MF, MEC, LCA, MAS, AG and LM contributed family 3. MB, BvL conducted the functional HUWE1 studies. SM conducted bioinformatic analysis. CS coordinated sample collection for all families. MC and RK conducted analysis of family 3. RJS evaluated family. PST and FA conducted sequencing of family 1. JG and YYY analysed data for family 3. CES coordinated the study of all families. MF, RES and CES wrote the manuscript.
Funding: This work was supported by the National Health and Medical Research Council of Australia grants APP628952, APP1041920 and APP1008077 to JG; National Institutes of Health grants 2R01HD026202 (NICHD) and 1R01NS73854 (NINDS) to CES and in part by a grant from the South Carolina Department of Disabilities and Special Needs (DDSN). MB was funded by Swiss National Science Foundation (SNSF) MDPhD Fellowship and BvL by SNSF grant 31003A_152621.
Competing interests: None declared.
Patient consent: Obtained.
Ethics approval: South Carolina.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data sharing statement: Raw data or clarification of reported results can be obtained by emailing Michael Friez (friez@ggc.org).
References
- 1.Oberlé I, Rousseau F, Heitz D et al. Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science 1991;252:1097–102. 10.1126/science.252.5009.1097 [DOI] [PubMed] [Google Scholar]
- 2.Lubs HA, Stevenson RE, Schwartz CE. Fragile X and X-linked intellectual disability: four decades of discovery. Am J Hum Genet 2012;90:579–90. 10.1016/j.ajhg.2012.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stevenson R, Schwartz C, Rogers C. Atlas of X-linked intellectual disability syndromes. New York: Oxford University Press, 2012. [Google Scholar]
- 4.Piton A, Redin C, Mandel JL. XLID-causing mutations and associated genes challenged in light of data from large-scale human exome sequencing. Am J Hum Genet 2013;93:368–83. 10.1016/j.ajhg.2013.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stevenson RE. Splitting and lumping in the nosology of XLMR. Am J Med Genet 2000;97:174–82. 10.1002/1096-8628(200023)97:3<174::AID-AJMG1034>3.0.CO;2-4 [DOI] [PubMed] [Google Scholar]
- 6.Kleefstra T, Hamel BC. X-linked mental retardation: further lumping, splitting and emerging phenotypes. Clin Genet 2005;67:451–67. 10.1111/j.1399-0004.2005.00434.x [DOI] [PubMed] [Google Scholar]
- 7.Juberg RC, Marsidi I. A new form of X-linked mental retardation with growth retardation, deafness, and microgenitalism. Am J Hum Genet 1980;32:714–22. [PMC free article] [PubMed] [Google Scholar]
- 8.Villard L, Gecz J, Mattéi JF et al. XNP mutation in a large family with Juberg-Marsidi syndrome. Nat Genet 1996;12:359–60. 10.1038/ng0496-359 [DOI] [PubMed] [Google Scholar]
- 9.Mattei JF, Collignon P, Ayme S et al. X-linked mental retardation, growth retardation, deafness and microgenitalism. A second familial report. Clin Genet 1983;23:70–4. 10.1111/j.1399-0004.1983.tb00439.x [DOI] [PubMed] [Google Scholar]
- 10.Brooks SS, Wisniewski K, Brown WT. New X-linked mental retardation (XLMR) syndrome with distinct facial appearance and growth retardation. Am J Med Genet 1994;51:586–90. 10.1002/ajmg.1320510458 [DOI] [PubMed] [Google Scholar]
- 11.Gu J, Dubner R, Fornace AJ Jr et al. UREB1, a tyrosine phosphorylated nuclear protein, inhibits p53 transactivation. Oncogene 1995;11:2175–8. [PubMed] [Google Scholar]
- 12.Zhong Q, Gao W, Du F et al. Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell 2005;121:1085–95. 10.1016/j.cell.2005.06.009 [DOI] [PubMed] [Google Scholar]
- 13.Ramser J, Abidi FE, Burckle CA et al. A unique exonic splice enhancer mutation in a family with X-linked mental retardation and epilepsy points to a novel role of the renin receptor. Hum Mol Genet 2005;14:1019–27. 10.1093/hmg/ddi094 [DOI] [PubMed] [Google Scholar]
- 14.Takano K, Liu D, Tarpey P et al. An X-linked channelopathy with cardiomegaly due to a CLIC2 mutation enhancing ryanodine receptor channel activity. Hum Mol Genet 2012;21:4497–507. 10.1093/hmg/dds292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gu Z, Pim D, Labrecque S et al. DNA damage induced p53 mediated transcription is inhibited by human papillomavirus type 18 E6. Oncogene 1994;9:629–33. [PubMed] [Google Scholar]
- 16.Liu Z, Oughtred R, Wing SS. Characterization of E3Histone, a novel testis ubiquitin protein ligase which ubiquitinates histones. Mol Cell Biol 2005;25:2819–31. 10.1128/MCB.25.7.2819-2831.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang Y, Do H, Tian X et al. E3 ubiquitin ligase Mule ubiquitinates Miz1 and is required for TNFalpha-induced JNK activation. Proc Natl Acad Sci USA 2010;107:13444–9. 10.1073/pnas.0913690107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Parsons JL, Tait PS, Finch D et al. Ubiquitin ligase ARF-BP1/Mule modulates base excision repair. EMBO J 2009;28:3207–15. 10.1038/emboj.2009.243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Markkanen E, van Loon B, Ferrari E et al. Regulation of oxidative DNA damage repair by DNA polymerase lambda and MutYH by cross-talk of phosphorylation and ubiquitination. Proc Natl Acad Sci USA 2012;109:437–42. 10.1073/pnas.1110449109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Herold S, Hock A, Herkert B et al. Miz1 and HectH9 regulate the stability of the checkpoint protein, TopBP1. EMBO J 2008;27:2851–61. 10.1038/emboj.2008.200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vandewalle J, Langen M, Zschätzsch M et al. Ubiquitin ligase HUWE1 regulates axon branching through the Wnt/beta-catenin pathway in a Drosophila model for intellectual disability. PLoS ONE 2013;8:e81791 10.1371/journal.pone.0081791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Froyen G, Corbett M, Vandewalle J et al. Submicroscopic duplications of the hydroxysteroid dehydrogenase HSD17B10 and the E3 ubiquitin ligase HUWE1 are associated with mental retardation. Am J Hum Genet 2008;82:432–43. 10.1016/j.ajhg.2007.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Turner G, Gedeon A, Mulley J. X-linked mental retardation with heterozygous expression and macrocephaly: pericentromeric gene localization. Am J Med Genet 1994;51:575–80. 10.1002/ajmg.1320510456 [DOI] [PubMed] [Google Scholar]
- 24.Isrie M, Kalscheuer VM, Holvoet M et al. HUWE1 mutation explains phenotypic severity in a case of familial idiopathic intellectual disability. Eur J Med Genet 2013;56:379–82. 10.1016/j.ejmg.2013.05.005 [DOI] [PubMed] [Google Scholar]
- 25.Nava C, Lamari F, Héron D et al. Analysis of the chromosome X exome in patients with autism spectrum disorders identified novel candidate genes, including TMLHE. Transl Psychiatry 2012;2:e179 10.1038/tp.2012.102 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
bmjopen-2015-009537supp_table1.pdf (597.7KB, pdf)
bmjopen-2015-009537supp_figure1.pdf (824.1KB, pdf)