

Abstract

Single-electron transmetalation via photoredox/nickel dual catalysis provides the opportunity for the construction of Csp3–Csp2 bonds through the transfer of alkyl radicals under very mild reaction conditions. A general procedure for the cross-coupling of primary and secondary (bis-catecholato)alkylsilicates with alkenyl halides is presented. The developed method allows not only alkenyl bromides and iodides but also previously underexplored alkenyl chlorides to be employed.
Transition-metal-mediated cross-coupling has been instrumental in facile construction of numerous linkages including C–N, C–O, and C–C bonds.1 Employment of cross-coupling strategies to accomplish C–C bond formation traditionally relies on a three-step catalytic cycle based on two-electron processes. Although such a reaction manifold is effective for Csp2–Csp2 coupling, extension to Csp3–Csp2 bond formation has proved challenging.2 Very recently, several groups, including our own, have reported a means of overcoming this limitation via dual catalysis between a visible-light activated photoredox catalyst and a classic transition metal cross-coupling catalyst.3 By exploiting the propensity of photoredox catalysts to promote single electron transfer (SET) events to generate Csp3 radicals and the propensity of these radicals to undergo single-electron transmetalation with transition metal catalysts, Csp3–Csp2 cross-coupling can be accomplished under remarkably mild conditions.3,4 The simplistic but powerful nature of this paradigm has led to a series of reports on its application in the context of the cross-coupling of α-alkoxy,5 α-amino,6 benzylic,3a and secondary alkyl7 Csp3 radicals typically with aryl electrophiles. These radicals are generated from a variety of radical precursors, including organotrifluoroborates and carboxylic acids, among others.3
Although alkyltrifluoroborate salts have been demonstrated to be exceptional radical precursors,8 they possess several drawbacks (e.g., release of corrosive BF3, high oxidation potential requiring use of an expensive Ir-based photocatalyst, etc.) prompting us to initiate a program to explore alternate radical precursors. As part of this program, our laboratory has successfully highlighted a highly efficient class of coupling partners suitable for a dual catalytic manifold: alkylbis(catecholato)silicates.9 Although recent reports by our group and the groups of Fensterbank and Goddard,10 in addition to the seminal studies of Nishigaichi,11 have demonstrated the use of these synthons as radical precursors, the full reactive potential of these bench stable and easily prepared solids is yet unrealized.
Recently, the MacMillan group reported that carboxylic acids bearing an α-heteroatom and alkenyl halides, namely iodides and bromides, can be effectively cross-coupled via dual catalysis.12 Although our group has shown that a variety of alkyltrifluoroborates function as radical precursors in Ni-photoredox cross-couplings with aryl electrophiles,3a,5−7 integration of these same trifluoroborates with alkenyl halides has yet to prove fruitful. Therefore, we wondered whether alkylsilicates would be suitable as nucleophilic partners in alkenyl dual catalytic cross-coupling (Scheme 1). Indeed, as recently demonstrated,9,10 organosilicates allow easy access to primary and secondary alkyl radicals without necessitating the presence of an α-heteroatom, allowing greater diversity in the structure of the radical precursor and hence substrate scope. In addition, the net-neutral fragmentation of the [Si]–C bond obviates the need for basic additives traditionally required when using carboxylic acids or organotrifluoroborates as radical precursors. Furthermore, the oxidation potentials of alkylsilicates (E° = +0.75 V vs SCE for primary silicates, on average)13 allow [Ru(bpy)3](PF6)2, an easily accessible photocatalyst, to be employed.14 Herein, we report the successful integration of (bis-catecholato)alkylsilicates in photoredox cross-coupling with alkenyl halides.
Scheme 1. Proposed Catalytic Cycle for Photoredox-Ni Dual Catalysis with Alkylsilicates and Alkenyl Halides.

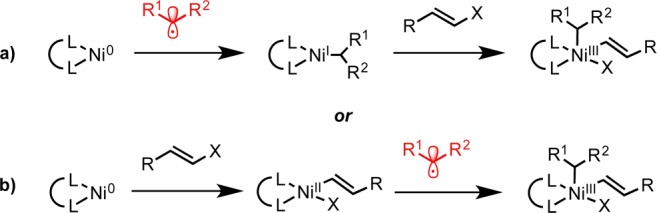
Based on MacMillan’s success with carboxylic acids,12 we posited that an alkyl–Ni(I) complex produced during photoredox cross-coupling involving silicates would be capable of oxidative addition into the alkenyl halide’s C–X bond (Scheme 2a). Alternatively, oxidative addition into this same C–X bond could occur prior to ligation of the alkyl radical by in situ generation of Ni(0) (Scheme 2b).4 Because of the rapid rate of oxidative addition of alkenyl halides to d10 transition metals (e.g., Ni and Pd) compared to aryl halides,15 it may mean that the latter sequence is operative in alkenyl photoredox cross-coupling. Ultimately, both pathways presumably furnish the same reactive Ni(III) intermediate. Following reductive elimination, the resulting Ni(I)–X intermediate is postulated to be reduced by the ground-state Ru(I) photocatalyst, regenerating the Ni(0) and the Ru(II) photocatalyst.4
Scheme 2. Potential Pathways for the Oxidative Addition of Alkenyl Halides onto Ni.
Our initial studies probing the amenability of silicates to this type of cross-coupling proved very fruitful. Thus, our previously established conditions developed for aryl cross-coupling of ammonium alkylsilicates with aryl bromides9 {2 mol % [Ru(bpy)3](PF6)2, 5 mol % [NiCl2(dme)], 5 mol % dtbbpy in DMF (0.1 M)} could be applied to the coupling with alkenyl halides without need for modification (dme: dimethoxyethane; dtbbpy: 4,4′-di-tert-butyl-2,2′-dipyridyl; bpy: 2,2′-dipyridyl). Control experiments confirmed that light,16 a Ru photocatalyst, a Ni catalyst, and the bipyridyl ligand are all necessary to achieve cross-coupling; without any of these components, only trace cross-coupled product was observed (see Supporting Information). This rules out a strictly photoredox-based coupling via a radical addition–elimination mechanism, as reported by Fensterbank with vinyl sulfones and halides.10
Using these conditions, we set out to examine the scope of this reaction within the context of various alkenyl halides (Scheme 3). Using the alkylsilicate 1a and alkenyl iodide 2a, the resulting cross-coupled product 3a was obtained in 58% isolated yield after 24 h. However, the reaction is not limited to alkenyl iodides, as alkenyl bromides could also be employed. Electron-rich, electron-neutral, and electron-deficient β-bromostyrenes as well as a 2-bromoindene (2b–e) were competent alkenyl cross-coupling partners. In addition to giving good yields of their respective cross-coupled products and being amendable to scale-up (e.g., both 2d and 2e could be performed on a gram scale), the geometry of the double bond was virtually unaffected throughout the reaction. This latter fact again discriminates this process from an addition–elimination mechanism.10 On a small scale, 2d produced the cross-coupled product in a 15:1 ratio of cis/trans isomers. Upon scale-up, the pure cis isomer was obtained in 76% yield. This is important given the typical degradation of stereoselectivity that occurs for styryl halides irradiated in the presence of Ir-based photocatalysts.12,17 Additionally, nonstyryl or styryl-like bromides were similarly well-tolerated toward cross-coupling. An acryl bromide 2f as well as more substituted cyclic 2g, exo-cyclic 2h, and acyclic 2i alkenyl bromides underwent facile cross-coupling. Additionally, 2g′ served equally well under the reaction conditions and gave a comparable yield of the cross-coupled product, indicating the interchangeability of iodides and bromides in this reaction manifold. Although the free hydroxyl group in 2j was not tolerated, its TBS-protected analogue 2k was cross-coupled with ease and in excellent yield. As with the styryl systems (2b–d), the geometry of the double bond was conserved during cross-coupling.
Scheme 3. Scope of Alkenyl Halides in Ni/Photoredox Cross-Coupling.

Reaction run on gram scale using white or blue LEDs; all yields are isolated yields after purification.
We next explored the scope of the reaction within the context of various alkylsilicates (Scheme 4).18 We found that moderate to excellent yields were obtained for the cross-coupling of a range of primary and secondary alkylsilicates with 2a. Secondary alkylsilicates (1b–d) performed well, with the exception of a bicyclic species, 1d. Primary systems faired equally well. Benzyl (1e) and allylic (1f) silicates, which produce stabilized radicals, underwent cross-coupling successfully. Additionally, isobutyl, alkene-containing, and hexylsilicates were also amendable to cross-coupling (1g–i). Various functional groups that were tolerated include ether, amide, pyridyl, and amine substituents (1j–m).
Scheme 4. Scope of Alkyl Bis(catecholato)Silicates in Ni/Photoredox Cross-Coupling.
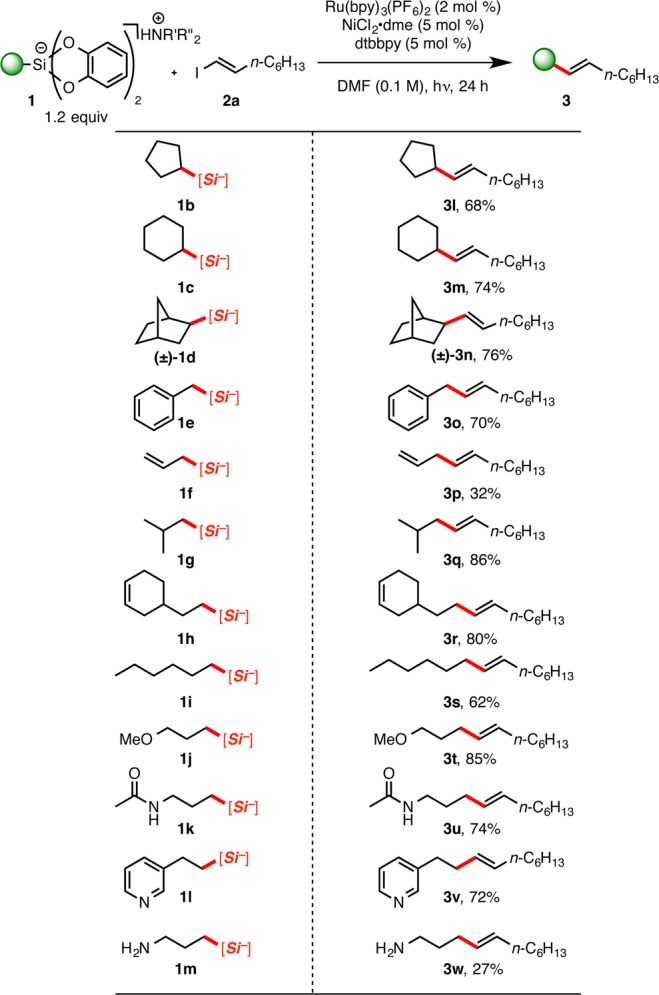
R′ = R″ = Et or R′ = H, R″ = i-Pr; all yields are isolated yields after purification.
The substrate-diversity of the reaction was next assessed via the coupling of various alkenyl bromides with a range of alkylsilicates (Scheme 5). Both activated and nonactivated alkylsilicates performed well with several sterically and electronically disparate alkenyl bromides. Again, the geometry of the double bond was unaffected during cross-coupling (e.g., compounds 3ac–3ag). Interestingly, although primary amine-containing silicates were previously shown to couple well with aryl bromides,9 they proved to be rather recalcitrant when coupling with alkenyl bromides (3ab, 3ag, and 3al). Although the failure to generate 3ag is somewhat expected given the instability of such systems, the low yield or failure of other systems cannot be readily explained.
Scheme 5. Alkenyl Bromide Scope with Primary and Secondary Alkyl Bis(catecholato)alkylsilicates.
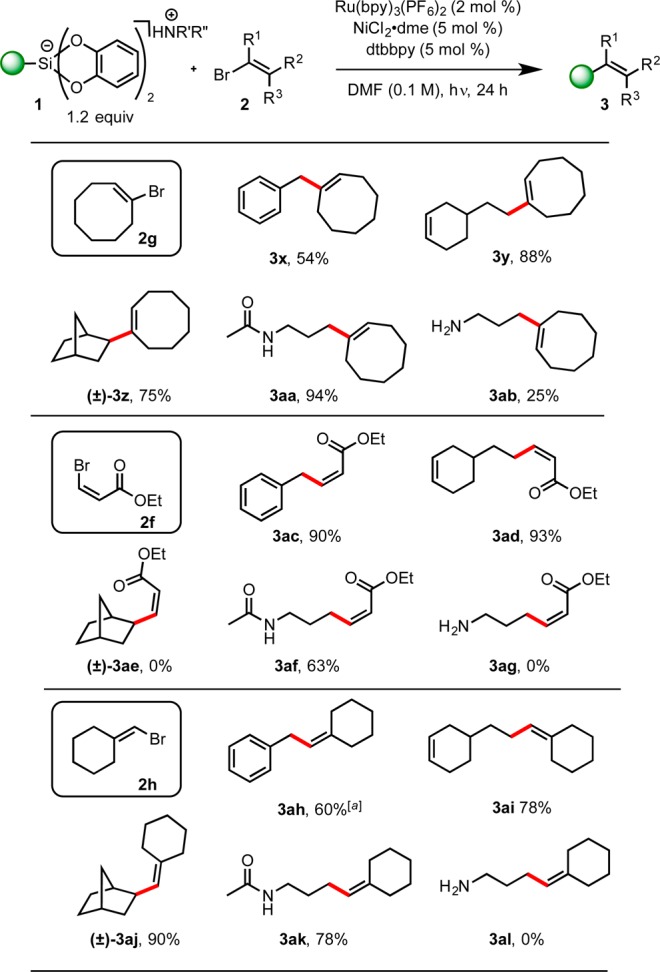
Inseparable bibenzyl impurity in 1H NMR; R′ = R″ = Et or R′ = H, R″ = i–Pr; all yields are isolated yields after purification
Finally, to demonstrate the robust nature of the developed cross-coupling protocol, the viability of alkenyl chlorides as cross-coupling partners was assessed. Remarkably, under the same conditions utilized for alkenyl bromides and iodides, near-identical reactivity was observed (Scheme 6). The cross-coupled product of 2l with 1a was obtained in 50% yield. In addition, a representative β-chlorostyryl derivative 2m reacted with ease with 1j. Similarly, cross-coupling of 2n with benzylsilicate 1e was accomplished successfully and in comparable yield to its bromide congener (90% vs 80%, respectively).
Scheme 6. Ni/Photoredox-Catalyzed Cross-Coupling Alkenyl Chlorides with Alkyl Bis(catecholato)silicates.
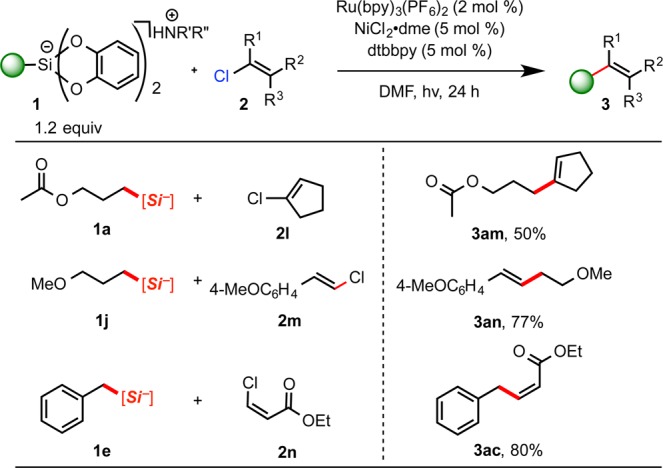
R′ = R″ = Et or R′ = H, R″ = i-Pr; all yields are isolated yields after purification.
In summary, ammonium organobis(catecholato)silicates, which serve as powerful radical precursors, and alkenyl halides can be readily cross-coupled via dual photoredox/Ni catalysis. By exploiting the relatively low oxidation potential of primary and secondary (bis-catecholato)alkylsilicates, a variety of primary and secondary radicals can be accessed using a practical ruthenium photocatalyst. Subsequently, these radicals are amenable to nickel-mediated cross-coupling with a diverse array of alkenyl halides, including yet unexplored alkenyl chlorides. The developed method complements existing protocols for alkenyl cross-coupling via dual catalysis and enables access to several previously inaccessible fragments. Additionally, the mild reaction conditions allow functional group tolerance, and complete stereochemical fidelity is exhibited.
Acknowledgments
We thank Kingson Lin (University of Pennsylvania) for the preparation of alkylsilicates. We thank NIGMS (RO1 GM113878) for financial support of this research.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.orglett.6b00024.
Experimental details and spectral data (PDF)
Author Contributions
† N.R.P. and C.B.K. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Metal-Catalyzed Cross-Coupling Reactions; de Meijere A., Diederich F., Eds.; Wiley-VCH: Weinheim, Germany, 2004. [Google Scholar]
- Hartwig J. F.Organotransition Metal Chemistry: From Bonding to Catalysis, 3rd ed.; University Science: Sausalito, CA, 2010. [Google Scholar]
- a Tellis J. C.; Primer D. N.; Molander G. A. Science 2014, 345, 433. 10.1126/science.1253647. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zuo Z.; Ahneman D. T.; Chu L.; Terrett J. A.; Doyle A. G.; MacMillan D. W. C. Science 2014, 345, 437. 10.1126/science.1255525. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Osawa M.; Nagai H.; Akita M. Dalton Trans. 2007, 827. 10.1039/b618007h. [DOI] [PubMed] [Google Scholar]; d Kalyani D.; McMurtrey K. B.; Neufeldt S. R.; Sanford M. S. J. Am. Chem. Soc. 2011, 133, 18566. 10.1021/ja208068w. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Ye Y.; Sanford M. S. J. Am. Chem. Soc. 2012, 134, 9034. 10.1021/ja301553c. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Rueping M.; Koenigs R. M.; Poscharny K.; Fabry D. C.; Leonori D.; Vila C. Chem. - Eur. J. 2012, 18, 5170. 10.1002/chem.201200050. [DOI] [PubMed] [Google Scholar]; g Sahoo B.; Hopkinson M. N.; Glorius F. J. Am. Chem. Soc. 2013, 135, 5505. 10.1021/ja400311h. [DOI] [PubMed] [Google Scholar]; h Shu X. Z.; Zhang M.; He Y.; Frei H.; Toste F. D. J. Am. Chem. Soc. 2014, 136, 5844. 10.1021/ja500716j. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Tasker S. Z.; Jamison T. F. J. Am. Chem. Soc. 2015, 137, 9531. 10.1021/jacs.5b05597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez O.; Tellis J. C.; Primer D. N.; Molander G. A.; Kozlowski M. C. J. Am. Chem. Soc. 2015, 137, 4896. 10.1021/ja513079r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakaya I.; Primer D. N.; Molander G. A. Org. Lett. 2015, 17, 3294. 10.1021/acs.orglett.5b01463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Khatib M.; Serafim R. A. M.; Molander G. A. Angew. Chem., Int. Ed. 2016, 55, 254. 10.1002/anie.201506147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Primer D. N.; Karakaya I.; Tellis J. C.; Molander G. A. J. Am. Chem. Soc. 2015, 137, 2195. 10.1021/ja512946e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molander G. A. J. Org. Chem. 2015, 80, 7837. 10.1021/acs.joc.5b00981. [DOI] [PubMed] [Google Scholar]
- Jouffroy M.; Primer D. N.; Molander G. A. J. Am. Chem. Soc. 2016, 138, 475. 10.1021/jacs.5b10963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corce V.; Chamoreau L.-M.; Derat E.; Goddard J.-P.; Ollivier C.; Fensterbank L. Angew. Chem., Int. Ed. 2015, 54, 11414. 10.1002/anie.201504963. [DOI] [PubMed] [Google Scholar]
- a Matsuoka D.; Nishigaichi Y. Chem. Lett. 2014, 43, 559. 10.1246/cl.131132. [DOI] [Google Scholar]; b Matsuoka D.; Nishigaichi Y. Chem. Lett. 2015, 44, 163. 10.1246/cl.140940. [DOI] [Google Scholar]
- Noble A.; McCarver S. J.; MacMillan D. W. C. J. Am. Chem. Soc. 2015, 137, 624. 10.1021/ja511913h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishigaichi Y.; Suzuki A.; Takuwa A. Tetrahedron Lett. 2007, 48, 211. 10.1016/j.tetlet.2006.11.057. [DOI] [Google Scholar]
- Prier C. K.; Rankic D. A.; MacMillan D. W. C. Chem. Rev. 2013, 113, 5322. 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Jutand A.; Negri S. Organometallics 2003, 22, 4229. 10.1021/om030298c. [DOI] [Google Scholar]; b Fahey D. R.; Mahan J. E. J. Am. Chem. Soc. 1977, 99, 2501. 10.1021/ja00450a017. [DOI] [Google Scholar]
- Note that the source of the irradiation (CFL, white or blue LEDs) had no effect on the overall outcome of the reaction.
- For examples, see:; a Noble A.; MacMillan D. W. C. J. Am. Chem. Soc. 2014, 136, 11602. 10.1021/ja506094d. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Singh K.; Staig S. J.; Weaver J. D. J. Am. Chem. Soc. 2014, 136, 5275. 10.1021/ja5019749. [DOI] [PubMed] [Google Scholar]
- Note that both diisopropylammonium and triethylammonium counterions show no discernible difference in reactivity or yield.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.