

Abstract
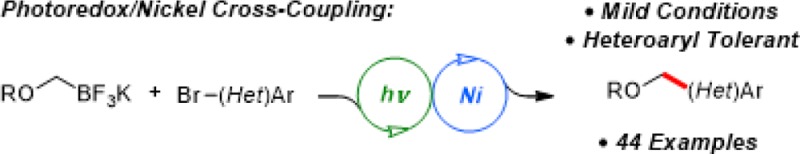
Single-electron transmetalation has emerged as an enabling paradigm for the cross-coupling of Csp3 hybridized organotrifluoroborates. Cross-coupling of α-alkoxymethyltrifluoroborates with aryl and heteroaryl bromides has been demonstrated by employing dual catalysis with a combination of an iridium photoredox catalyst and a Ni cross-coupling catalyst. The resulting method enables the alkoxymethylation of diverse (hetero)arenes under mild, room-temperature conditions.
Visible-light mediated photoredox catalysis has become recognized as an empowering technology for radical-mediated chemistry in recent years.1−3 By transiently generating catalytic quantities of both a single-electron oxidant and reductant within the confines of the same flask, typically inert substrates can be activated for reaction. Furthermore, the controlled radical generation afforded by these catalysts opens the door to a host of net redox neutral transformations that would have been previously impossible using stoichiometric reagents, where unproductive reductant/oxidant quenching and radical side products are an inevitable shortcoming.4 To date, many new and innovative reactions have been designed around the use of photoredox single-electron transfer (SET) catalysts, leading to a resurgence in radical-mediated methods for C–C bond formation.
Of particular note, dual catalytic photoredox/Ni cross-coupling has recently emerged as a powerful tool for the construction of Csp2–Csp3 bonds.5,6 Here, a single-electron activation mode has served as an ideal platform to circumvent the problems associated with traditional alkyl cross-coupling.7 By exploiting the greater stability of Csp3 derived radicals and their propensity to combine with Ni complexes,8 a new paradigm for the transmetalation of traditionally obstinate alkyl nucleophiles has been established. Cross-couplings that historically defaulted to sensitive reagents9−11 or harsh conditions12,13 to overcome the high activation energy for transmetalation can now be achieved at ambient temperatures with mildly basic additives. Consequently, since the initial report, several methods for the cross-coupling of Csp3 radicals (α-alkoxy,14 α-amino,15 benzylic,5 secondary alkyl6) with various electrophiles (aryl,5,6,15 alkenyl14) have been demonstrated.
Moving forward, we sought to apply this chemistry broadly to primary α-alkoxymethyl systems. Stabilized by donation from the α-oxygen, these radicals are well-behaved and facile to generate.16 Furthermore, their generation through chemical and photochemical oxidation of potassium organotrifluoroborate salts has already been well established.17−19
Knowing the chemical characteristics of these radicals, we recognized an opportunity to take commercially available and readily accessible primary α-alkoxymethyltrifluoroborates and show more broadly the advantages of a radical mediated cross-coupling. Through this approach to late stage ether synthesis, there was an effort to highlight the broad functional group tolerance and mild conditions employed. Moreover, benzylic ethers are an important motif in pharmaceutical20−22 and supramolecular chemical architectures,23,24 and dissonant approaches to their synthesis are useful in their own right. Although the use of α-alkoxy anions25,26 as a disconnection in cross-coupling has been reported previously for alkoxymethylzincs,27,28 alkoxymethylstannanes,29−32 and alkoxymethylboron derivatives,33,34 the current methods are severely limited by the use of toxic and/or air- and moisture-sensitive reagents under often harsh conditions that inhibit the incorporation of reactive functional groups.
Consequently, successful application of a photoredox cross-coupling with these reagents serves the dual purpose of (1) providing a platform to expand and improve a currently under-utilized disconnection and (2) validating photoredox cross-coupling as a general manifold for engaging alkyl-based radicals in cross-coupling, where similar reduction potentials are a single unifying feature among structurally dissimilar reagents (Figure 1).
Figure 1.
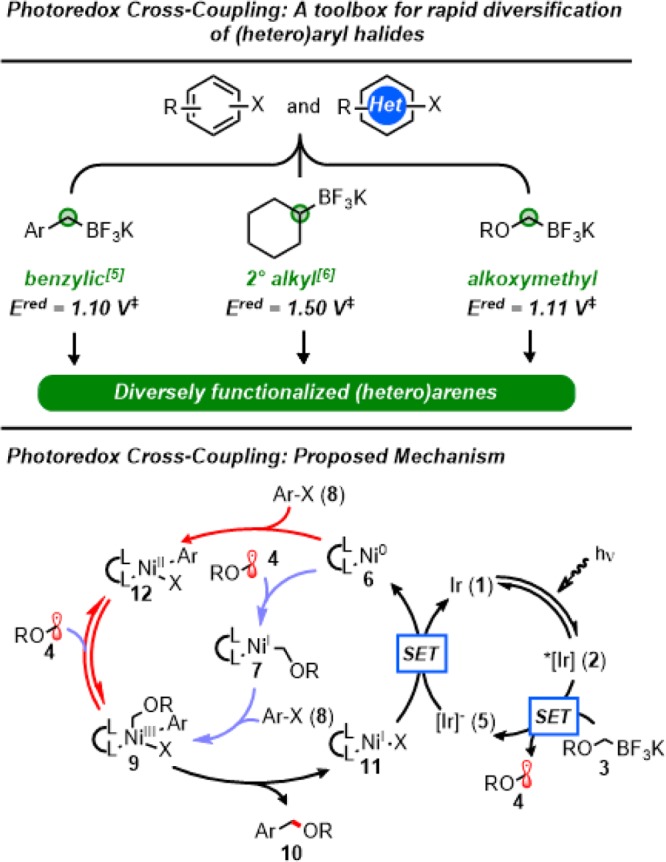
Photoredox cross-coupling as a general manifold for cross-coupling of diverse Csp3 derived radicals; proposed catalytic cycle for photoredox cross-coupling. ‡Reduction potentials are reported against the saturated calomel electrode (SCE).
The proposed catalytic cycle for this transformation is outlined in Figure 1. First, visible light excitation of photoredox catalyst {Ir[dF(CF3)ppy]2(bpy)}·PF6 (1) produces the long-lived photoexcited state, {*IrIII[dF(CF3)ppy]2(bpy)}·PF6 (2). This excited state complex is sufficiently oxidizing [Ered1/2 (*IrIII/IrII) = +1.32 V vs SCE35] to perform a single-electron oxidation of the C–B bond of the alkoxymethyltrifluoroborate 3 [Ered1/2 = +1.11 V vs SCE19] to the corresponding α-alkoxy radical 4, returning the reduced ground state photocatalyst 5. Addition of 4 to ligated Ni0 complex 6 affords alkyl-NiI species 7, which can engage aryl halide 8 in oxidative addition. The generated NiIII complex 9 then reductively eliminates rapidly to provide the desired benzylic ether 10, returning ligated NiI–X species 11. Reduction of 11 by the ground state of the reduced photocatalyst 5 concurrently returns both catalytic cycles to their start.
As noted previously,36 calculations suggest that oxidative addition may occur either from Ni0 or NiI-alkyl at room temperature. In addition, the generated NiIII intermediate is predicted to exist in equilibrium with free radical 4 and ArNiIIX complex 12 through reversible homolysis of the Ni–C bond. Based on the low barrier for radical addition at Ni0, the currently favored pathway is outlined in blue (Figure 1).
Initial evaluation of the proposed α-alkoxymethyl cross-coupling commenced with an extensive solvent screen. It should be noted for these reactions that solubility of the reagents is a crucial component for productive reactivity because insoluble material disrupts light penetration and can serve to decouple the integrated catalytic cycles. In our experience, maintaining the homogeneity of the reaction mixture has led to significant improvements. Using methyl 4-bromobenzoate, potassium [(benzyloxy)methyl]trifluoroborate (1.2 equiv), NiCl2·dme (5.0 mol %), dtbbpy (5.0 mol %), and Ir catalyst 1 (2.0 mol %), a cosolvent system consisting of dioxane with added DMA was determined to provide the right balance of solubility and reactivity for the dual catalytic cross-coupling.
An investigation of additives, proposed to be key for sequestering generated BF3 during trifluoroborate oxidation, also improved the yields significantly. Without added base, the reactions tend to stall out around 40–60% conversion (∼10 h), leading us to suspect that byproduct inhibition is principally responsible. Among those bases examined, K2HPO4 led to full consumption of the starting material after 16 h.
With reasonably effective conditions in hand, alternate ligands and Ni sources were examined to identify scaffolds that might be useful in extending this chemistry. Although a number of these Ni sources and ligands served as competent catalysts (see Supporting Information for full details), our model ligand remained a clear front-runner in both conversion and yield. Considering the commercial availability and low cost of this established catalytic system (NiCl2·dme, dtbbpy), we moved forward with this combination to evaluate the full scope of this reaction.
Beginning with the aryl bromide partner (Figure 2), it was important to emphasize the ability of this catalyst system to engage a range of electronically differentiated aryl bromides. Reaction with electron-withdrawing 4-bromoacetophenone and electron-neutral 4-bromotoluene led to isolation of 15 and 24 in 95 and 62% yields, respectively. Even electron-rich bromides were competent electrophiles, as reaction with 4-bromoanisole gave rise to product 23, albeit in a modest 52% yield.
Figure 2.
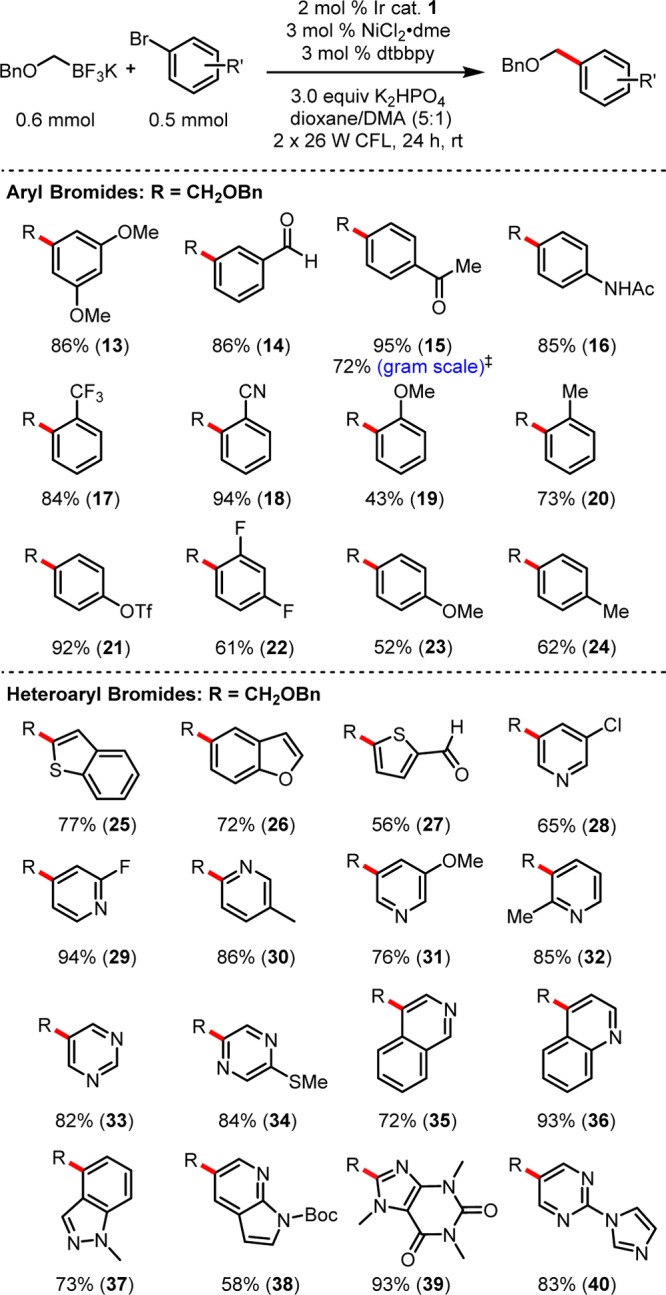
α-Alkoxymethyltrifluoroborate photoredox cross-coupling: halide scope. ‡Using 1.0 mol % Ir catalyst 1, 1.5 mol % Ni/ligand, 48 h.
Ortho-substitution was tolerated in trifluoromethyl-, nitrile-, methyl-, and methoxy-substituted aryl bromides leading to ethers 17–20. Selective cross-coupling was observed exclusively at the bromide for both a bifunctional aryl triflate and a heteroaryl chloride, affording products 21 and 28. Aldehydes contained in products 14 and 27 survived the reaction untouched. Considering the sensitivity of aldehydes to reduction under Pd-catalysis37 as well as their electrophilicity toward harsher organometallic nucleophiles in cross-coupling, this method provides the opportunity to circumvent protection and/or oxidation–reduction sequences that may be required to access the same structural architecture. To ensure the scalability of this transformation, a gram-scale reaction was performed with 4-bromoacetophenone using reduced catalyst loadings (1.0 mol % Ir catalyst 1 and 1.5 mol % NiCl2·dme). Although extended reaction times were required for full conversion (48 h), the desired ether 15 was obtained in good yield (72%).
The generality for the cross-coupling with heteroaryl substructures makes this method particularly attractive for late-stage diversification. In addition to the products derived from well-behaved benzofuran- and benzothiophene-containing bromides (25, 26), more challenging pyrazine-, pyrimidine-, quinoline-, and isoquinoline-containing bromides gave the desired products in excellent yields (33–36). Bromopyridines substituted at all regioisomeric positions could also be used to access products 28–32 in good yields. Even 8-bromocaffeine could be used to access product 39 in excellent yield. Unfortunately, protection was required for indazole- and azaindole-containing bromides, affording products 37 and 38, respectively. Although satisfactory methods for cross-coupling of these protic heteroaryl bromides remain elusive, it should be noted that previous Pd complex-catalyzed conditions [3 mol % Pd(OAc)2, 6 mol % RuPhos, 3 equiv of Cs2CO3, dioxane/H2O, 100 °C] have proved wholly ineffective or low yielding for a number of the heteroaryl bromides shown.33 Therefore, this method represents a marked improvement over the current state of the art.
To highlight further the heteroaryl tolerance of this coupling, 5-bromopyrimidine was used as an electrophile to evaluate the alkoxymethyltrifluoroborate scope (Figure 3). To this end, a pyridyl-containing organotrifluoroborate was successfully employed to yield compound 41 in reasonable yield, showing tolerance of a heteroaryl functional unit in both reaction partners. Aryl chlorides were also tolerated on the trifluoroborate partner to give the dichlorinated arene 42 with no competitive reaction at the chlorides.
Figure 3.
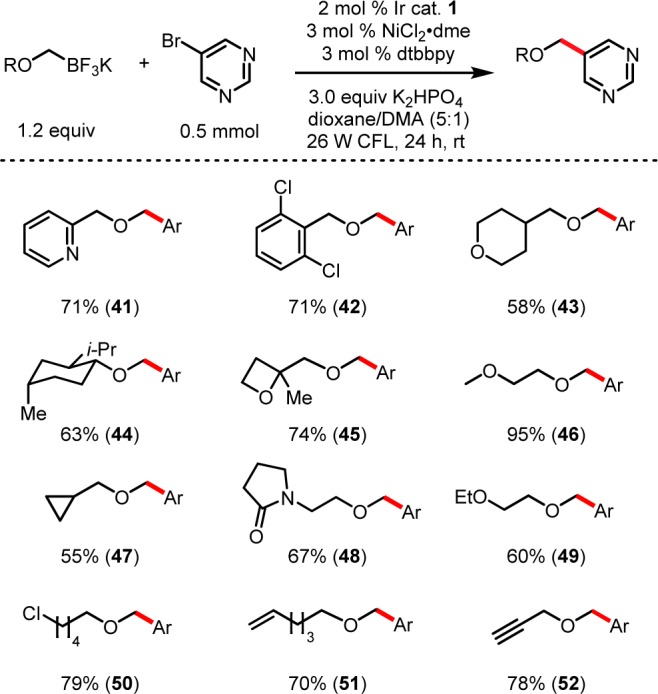
α-Alkoxymethyltrifluoroborate photoredox cross-coupling: organotrifluoroborate scope.
As expected, ethers were tolerated throughout the functionalized organotrifluoroborate core to give products such as 43, 45, and 46. For 46, the ability to incorporate PEG linkers on target molecules at a late stage could serve as a means to modulate in vivo drug solubility.38 Steric bulk, although distal from the reacting radical, led to isolation of menthol derivative 44 and tert-butyl ether product 55 (Figure 4) in acceptable yields.
Figure 4.
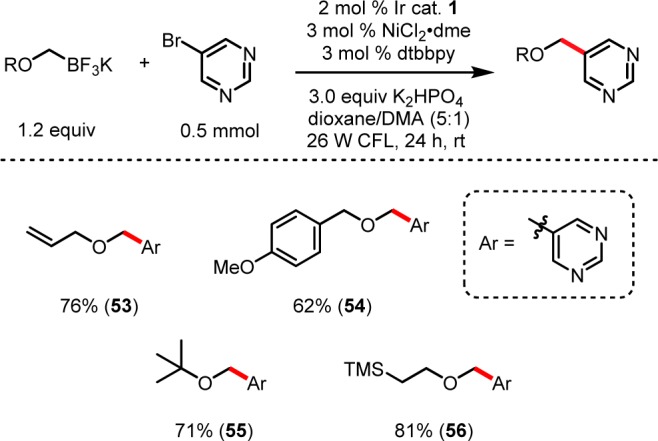
α-Alkoxy photoredox cross-coupling: formation of protected alcohols.
Beyond the largely aliphatic functional groups shown, amides and alkyl chlorides were untouched under the reaction conditions, leaving a site for further manipulation in products 48 and 50. Also to be noted is that products containing alkenes and alkynes were isolated in excellent yields (51–53). Considering the intermediacy of radical intermediates during this process, the tolerance of these functional handles for later derivatization is particularly intriguing.
As a final testament to the utility of this transformation, a number of α-alkoxymethyltrifluoroborates substituted with simple protecting groups were prepared (Figure 4). Currently, the facile preparation of many benzylic alcohols is predicated on commercial access to the corresponding esters and aldehydes. Even in cases where these precursors are readily available, the generated alcohols can be sensitive to ambient oxidation to afford a mixture of alcohol, aldehyde, and other oxidation byproducts.39,40 Consequently, a method for the preparation of protected benzylic alcohol variants from the far more widely available aryl and heteroaryl halides serves as an attractive method for reliable synthesis and long-term storage. To that end, allyl-, para-methoxybenzyl-, tert-butyl-, and trimethylsilylethyl-protected α-alkoxymethyltrifluoroborates were synthesized and employed as cross-coupling partners to give products 53–56. Subsequent deprotection of these protected alcohols allows the formation of benzylic alcohols in two steps from readily accessible bromoarenes.
In summary, room-temperature conditions for the cross-coupling of α-alkoxymethyltrifluoroborates with an array of aryl bromides has been communicated. Previous reports for the cross-coupling of these substrates with aryl chlorides have taken place at 100 °C in the presence of a strong aqueous base and have suffered from significant limitations in scope when applied to both heteroaryl chlorides and -bromides. Consequently, the developed couplings address key substrate limitations of the former methods. Furthermore, the delineated procedures provide a method for the synthesis of benzylic ethers that can be readily deprotected to afford the corresponding alcohols, many of which are challenging to access in high purity.
Finally, although the products derived from these reactions can often be made by other means, the use of these reagents serves to validate further the photoredox cross-coupling manifold as a means of engaging Csp3 nucleophiles in alkyl transfer under mild conditions. Most importantly, the ability to cross-couple late stage bromides with an ever-expanding library of Csp3 hybridized nucleophiles using a single robust catalyst system is anticipated to enable practitioners to diversify their synthetic molecular architecture quickly and expand three-dimensional chemical space.41
Acknowledgments
This research was generously supported by the National Institute of General Medical Sciences (R01-GM113878) and TÜBİTAK. We thank Frontier Scientific for providing several organoboron reagents used in this research and Aldrich for iridium salts used in the preparation of the photoredox catalysts.
Supporting Information Available
Experimental procedures and spectra data. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.orglett.5b01463.
The authors declare no competing financial interest.
Supplementary Material
References
- Tucker J. W.; Stephenson C. R. J. J. Org. Chem. 2012, 77, 1617. [DOI] [PubMed] [Google Scholar]
- Prier C. K.; Rankic D. A.; MacMillan D. W. C. Chem. Rev. 2013, 113, 5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkinson M. N.; Sahoo B.; Li J.-L.; Glorius F. Chem.—Eur. J. 2014, 20, 3874. [DOI] [PubMed] [Google Scholar]
- Lloyd-Jones G. C.; Ball L. T. Science 2014, 345, 381. [DOI] [PubMed] [Google Scholar]
- Tellis J. C.; Primer D. N.; Molander G. A. Science 2014, 345, 433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Primer D. N.; Karakaya I.; Tellis J. C.; Molander G. A. J. Am. Chem. Soc. 2015, 137, 2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jana R.; Pathak T. P.; Sigman M. S. Chem. Rev. 2011, 111, 1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitenfeld J.; Ruiz J.; Wodrich M. D.; Hu X. J. Am. Chem. Soc. 2013, 135, 12004. [DOI] [PubMed] [Google Scholar]
- Han C.; Buchwald S. L. J. Am. Chem. Soc. 2009, 7532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pompeo M.; Froese R. D. J.; Hadei N.; Organ M. G. Angew. Chem., Int. Ed. 2012, 51, 11354. [DOI] [PubMed] [Google Scholar]
- Vila C.; Giannerini M.; Hornillos V.; Fañanás-Mastral M.; Feringa B. L. Chem. Sci. 2014, 5, 1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreher S. D.; Dormer P. G.; Sandrock D. L.; Molander G. A. J. Am. Chem. Soc. 2008, 130, 9257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L.; Zhao S.; Joshi-Pangu A.; Diane M.; Biscoe M. R. J. Am. Chem. Soc. 2014, 136, 14027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble A.; McCarver S. J.; MacMillan D. W. C. J. Am. Chem. Soc. 2015, 137, 624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo Z.; Ahneman D.; Chu L.; Terrett J.; Doyle A. G.; MacMillan D. W. C. Science 2014, 345, 437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S.-Y.; Zhang F.-M.; Tu Y.-Q. Chem. Soc. Rev. 2011, 40, 1937.21286642 [Google Scholar]
- Molander G. A.; Colombel V.; Braz V. A. Org. Lett. 2011, 13, 1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasu Y.; Koike T.; Akita M. Adv. Synth. Catal. 2012, 354, 3414. [Google Scholar]
- Miyazawa K.; Yasu Y.; Koike T.; Akita M. Chem. Commun. 2013, 49, 7249. [DOI] [PubMed] [Google Scholar]
- Lawrence N. J.; Rennison D.; Woo M.; McGown A. T.; Hadfield J. A. Bioorg. Med. Chem. Lett. 2001, 11, 51. [DOI] [PubMed] [Google Scholar]
- Thompson A. M.; Blaser A.; Anderson R. F.; Shinde S. S.; Franzblau S. G.; Ma Z.; Denny W. A.; Palmer B. D. J. Med. Chem. 2009, 52, 637. [DOI] [PubMed] [Google Scholar]
- Güven O. O.; Erdoğan T.; Göker H.; Yildiz S. Bioorg. Med. Chem. Lett. 2007, 17, 2233. [DOI] [PubMed] [Google Scholar]
- Helms B.; Mynar J. L.; Hawker C. J.; Fréchet J. M. J. J. Am. Chem. Soc. 2004, 126, 15020. [DOI] [PubMed] [Google Scholar]
- Percec V.; Heck J.; Lee M.; Ungar G.; Alvarez-Castillo A. J. Mater. Chem. 1992, 2, 1033. [Google Scholar]
- Hutchinson D. K.; Fuchs P. L. J. Am. Chem. Soc. 1987, 109, 4930. [Google Scholar]
- Tamao K.; Ishida N.; Ito Y.; Makoto K. Org. Synth. 1990, 69, 96. [Google Scholar]
- Blanc R.; Groll K.; Bernhardt S.; Stockmann P.; Knochel P. Synthesis 2014, 46, 1052. [Google Scholar]
- Sakata K.; Urabe D.; Inoue M. Tetrahedron Lett. 2013, 54, 4189. [Google Scholar]
- Masanori K.; Takashi S.; Toshimi O.; Hiroshi S.; Toshihiko M. Chem. Lett. 1984, 1225. [Google Scholar]
- Majeed A. J.; Antonsen O.; Benneche T.; Undheim K. Tetrahedron 1989, 45, 993. [Google Scholar]
- Falck J. R.; Patel P. K.; Bandyopadhyay A. J. Am. Chem. Soc. 2007, 129, 790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goli M.; He A.; Falck J. R. Org. Lett. 2011, 13, 344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molander G. A.; Canturk B. Org. Lett. 2008, 10, 2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai N.; Yonaga M.; Tanaka K. Org. Lett. 2012, 14, 1278. [DOI] [PubMed] [Google Scholar]
- Koike T.; Akita M. Inorg. Chem. Front. 2014, 1, 562. [Google Scholar]
- Gutierrez O.; Tellis J. C.; Primer D. N.; Molander G. A.; Kozlowski M. C. J. Am. Chem. Soc. 2015, 137, 4896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang A.; Yang Z.; Liu J.; Gui Q.; Chen X.; Tan Z.; Shi J.-C. Synth. Commun. 2014, 44, 280. [Google Scholar]
- Harris J. M.; Chess R. B. Nat. Rev. Drug Discovery 2003, 2, 214. [DOI] [PubMed] [Google Scholar]
- Urakami K.; Kobayashi C.; Miyazaki Y.; Nishijima K.; Yoshimura Y.; Hashimoto K. Chem. Pharm. Bull. 2000, 48, 1299. [DOI] [PubMed] [Google Scholar]
- Abend A. M.; Chung L.; Bibart R. T.; Brooks M.; McCollum D. G. J. Pharm. Biomed. Anal. 2004, 34, 957. [DOI] [PubMed] [Google Scholar]
- Lovering F.; Bikker J.; Humblet C. J. Med. Chem. 2009, 52, 6752. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.