

Abstract
The Dsb protein family is responsible for introducing disulfide bonds into nascent proteins in prokaryotes, stabilizing the structure of many proteins. Helicobacter pylori HP0231 is a Dsb-like protein, shown to catalyze disulfide bond formation and to participate in redox homeostasis. Notably, many H. pylori virulence factors are stabilized by the formation of disulfide bonds. By employing H. pylori HP0231 deficient strains we analyzed the effect of lack of this bacterial protein on the functionality of virulence factors containing putative disulfide bonds. The lack of H. pylori HP0231 impaired CagA translocation into gastric epithelial cells and reduced VacA-induced cellular vacuolation. Moreover, H. pylori HP0231 deficient bacteria were not able to colonize the gastric mucosa of mice, probably due to compromised motility. Together, our data demonstrate an essential function for H. pylori HP0231 in gastric colonization and proper function of bacterial virulence factors related to gastric pathology.
Introduction
Helicobacter pylori infection is one of the most common bacterial infections worldwide. Chronic gastritis induced by H. pylori represents the first step of a pathological cascade that can eventually progress to cancer [1,2]. In order to cope with the acidic conditions H. pylori encounters in the stomach, the bacterium produces urease, thereby hydrolyzing urea and increasing the pH locally [3]. In addition, motility mediated by flagella and cell morphology is essential for successful colonization of the gastric epithelium [4].
The immune response towards the bacterium as well as the degree of gastric pathology have been related to the presence of certain bacterial virulence factors. Of those, the Cag Pathogenicity Island (CagPAI) and VacA are considered the major pathogenic factors, and their presence defines populations at higher risk for gastric cancer development [5]. The CagPAI encodes CagA and several genes composing a type IV secretion system (T4SS), capable of translocating CagA into host epithelial cells. Once translocated, CagA interacts with a number of host proteins and activates signaling cascades that contribute to pathology [6]. Conversely, VacA is a secreted cytotoxin which is internalized, forming vacuoles in the host cells, and causing changes in mitochondrial potential [7] or disrupting autophagy [8].
Disulfide bonds, which in prokaryotes are introduced by proteins of the Dsb family, stabilize the structure of many proteins. Dsb proteins are crucial for correct folding or assembly of different pathogenic factors such as toxins, adhesins or components of type III secretion systems. Hence, loss of disulfide bond formation due to mutations in dsb genes were found to reduce pathogenesis of bacteria and viruses [9–12]. Dsb proteins have been largely studied in bacteria such as E. coli. In contrast, the disulfide bond formation machinery of H. pylori is still less characterized. H. pylori possesses different putative Dsb homologs: HP0595 (DsbI) was identified as a DsbB homolog, HP0377 as DsbC, and HP0265 as CcdA homolog [13,14]. However, it has been recently shown that HP0377 is a cytochrome c maturation (CcmG) protein playing also a role in Dsb isomerization pathway [15]. Initial structural studies proposed HP0231 as a DsbG homolog [16], however, more recent data showed HP0231 to have DsbA-activity [17,18]. Notably, disulfide formation mediated by HP0231 is important for folding, solubilization, production and secretion of H. pylori cysteine rich protein (HcpE), which is suggested to contribute to bacterial virulence [18]. Interestingly, presence of disulfide bonds has been reported in bacterial virulence factors such as VacA [19] or components of the T4SS [20], suggesting these are potential targets of the H. pylori Dsb system and specifically of HP0231.
In order to elucidate whether HP0231 is required for proper functionality of virulence factors containing disulfide bonds such as VacA and the T4SS, we generated HP0231-deficient H. pylori strains. In addition, we studied the impact of loss of HP0231 in gastric colonization in mice. Our results suggest an important role for HP0231 in the establishment of the infection and in determining bacterial virulence.
Material and Methods
Bacterial strains and growth conditions
H. pylori wild type P12 [21], G27 [22], PMSS1 and SS1 [23] or mutant strains PMSS1ΔcagA, PMSS1ΔcagE [24], P12ΔvacA [25], P12ΔggT [26], G27ΔcagA (generated by natural transformation of J99 ΔcagA DNA into H. pylori strain G27) and G27ΔcagE [27] were cultured on WC dent agar plates under microaerophilic conditions. All the bacterial strains used for experiments were sub-cultured no more than 3 times to avoid possible genotypic and phenotypic changes.
Generation of H. pylori strains by natural transformation
Plasmids to generate HP0231 knockout strains and the corresponding HP0231 complemented strains were previously described [17]. To knock out hp0231, a PCR fragment, which was composed with the upstream region of hp0231 gene followed by a chloramphenicol cassette and the downstream region of hp0231 gene, was cloned into pGEM-T (Promega, USA) easy vector to generate the knockout plasmid pGMTeasy-Δ231. To complement the knockout strains with HP0231, the hp0231 coding region was amplified from H. pylori 26695 genome DNA and was inserted into the shuttle vector pHeL3 to construct the vector pHeL3-231+. The plasmid was then introduced into knockout strains by natural transformation.
To naturally transform H. pylori, 10μg of plasmid DNA were introduced into around 4x108 H. pylori bacterial cells during exponential growth phase. After one-day incubation on WC dent plates, bacteria were transferred onto WC dent plates containing antibiotics for selection. Single colonies were picked, grown and analyzed by PCR and western blot (S1A and S1B Fig). No changes in growth were detected between wild type and HP0231 knockout strains (S1C Fig).
H. pylori growth curve
H. pylori cells growing at exponential phase on WC dent plates were resuspended in PBS and used to inoculate Brucella Broth dent liquid medium containing 10% FCS at an optical density (OD) of 0.5. Cultures were kept shaking under microaerophilic conditions. Samples were taken at different time points and cell density was determined by measuring OD600.
Cell culture and infections
AGS (ATCC CRL-1739) and MKN45 [28] were cultured in DMEM (Gibco, USA) containing 10% FCS, at 37°C and 5%CO2. Cells were routinely tested for Mycoplasma contamination.
For infection experiments, 1x105 cells were seeded on a well of a 24-well plate. After 24h, cells were infected with different H. pylori strains at MOI 20. After 6h infection, cells were photographed, supernatants collected and cell lysates obtained for western blot.
Western blot
Cells were lysed using SDS sample buffer (62.5 mM Tris-HCl (pH 6.8), 2% w/v SDS, 10% glycerol, 50 mM DTT, 0.01% w/v bromophenol blue), while proteins from supernatants of OD 1 bacterial cultures were extracted after 24 hours incubation by TCA precipitation and resuspended in Laemmli buffer and 5xSDS-buffer as previously reported [29]. Equal amounts of lysate were loaded on SDS-PAGE gels, separated proteins were transferred to nitrocellulose membrane (Protran, Germany) and membranes blocked before applying primary antibodies. CagA phosphorylation was detected using a rabbit anti-CagA antiserum (kindly provided by Dr. R. Vogelmann, University Hospital Mannheim II, Mannheim, Germany), and a p-Tyr antibody (Millipore, USA). VacA antibody AK197, CagI antibody AK293 and CagL antibody AK271 have been previously described [30,31]. HP0231 was detected by using an anti-HP0231 serum (1:500) obtained after immunization of C57BL/6 mice with HP0231 recombinantly produced in our lab. β-actin was detected with anti-β-actin antibody (Sigma-Aldrich, USA).
CagA translocation (β-lactamase activity)
TEM-1 β-lactamase activity was determined as described elsewhere [32]. Briefly, AGS cells were infected with H. pylori P12 [TEM-1-CagA] for 2.5 hours, and cells were loaded subsequently with the fluorescent substrate CCF4-AM in the respective loading solution (LiveBLAzer-FRET B/G loading kit; Invitrogen) supplemented with 1 mM probenecid (Sigma). Cells were incubated at room temperature in the dark for 90 min. For quantification of translocation, infected cells were measured with a Clariostar plate reader (BMG Labtech) using an excitation wavelength of 405 nm (10 nm bandwidth), and emission at 460 nm (20 nm bandwidth, blue fluorescence) and 530 nm (15 nm bandwidth, green fluorescence). CagA translocation was defined as the emission ratio at 460 nm(sample-blank) divided by 530 nm(sample-blank) (Blue-to-Green ratio).
ELISA
1x105 AGS cells were infected by different H. pylori strains and the supernatant was collected after 6h infection. The secretion of Interleukin-8 (IL8) was detected using IL-8 Elisa kit, according to manufacturer’s instructions (eBioscience, USA).
Vacuolation Assay
Cell vacuolation was monitored by counting vacuole formation in H. pylori-infected AGS cells. Briefly, 5x104 AGS cells were seeded into 6-well plates and infected with different H. pylori strains at MOI 100 for 24h. High magnification pictures (400x) were taken at five random positions for each sample. Cells that showed vacuoles were counted and percentage of cells showing vacuoles in total cell number was calculated.
H. pylori gamma-glutamyltranspeptidase activity assay
H. pylori gamma-glutamyltranspeptidase activity was monitored by following the cleavage of g-glutamyl-p-nitroanilide (gGpNA). 108 cells of different H. pylori strains were resuspended in 200 μl of Tris-buffer pH8.0, which contained 20 mmol/L glycyl-glycine, 2.5 mmol/L L-γ-glutamyl-p-nitroanilide (Sigma, USA) and incubated at 37°C for 10min. The release of p-nitroanilide was then detected by spectrophotometry at 405nm. All assays were performed in triplicates.
Bacterial Binding Assay
Human gastric cancer cells were seeded in 96-well plates (1.5x105 cells per well). Different H. pylori strains were labeled by cell tracer CFDA-SE (Life Technologies, USA) in PBS at 37°C for 30min. After washing three times with PBS, labeled H. pylori were added at MOI 10 to the cells and incubated for 1h at 37°C. Samples were washed with PBS three times and fixed with 100μL 4% paraformaldehyde (PFA) for 5min. Finally, the samples were resuspended with 200μL FACS buffer and analyzed by flow cytometry. Analysis was performed with a FACS CyAn (Beckman Coulter) and the FlowJo software.
Immunofluorescence
5x104 AGS cells were seeded on a well of 4-well chamber slide (Thermo Fisher Scientific, USA). After 24h, AGS cells were infected by CFDA-SE labeled H. pylori PMSS1 or PMSS1Δhp0231 at MOI 5 for 1h at 37°C. Samples were washed 5 times with PBS and fixed with 4% PFA for 10min. After washing twice with PBS, samples were stained with pre-warmed deep red/PBS (1:1000 dilution; Thermo Fisher Scientific, USA) for 30min at 37°C. After washing with PBS, samples were mounted with DAPI (Vector laboratories, USA) and fluorescence pictures were taken by confocal microscopy (Olympus Life Science, Japan) (600x magnification).
Motility Assays
H. pylori motility was assessed on soft agar plates as previously described [17,33] with slightly modifications. Briefly, the soft agar plates used in this study were composed of 0.35% (w/v) agar, 10% (v/v) FCS, 2.8%(w/v) Brucella Broth base, 5 μg/mL trimethoprim, 5 μg/mL amphotericin B, 10 μg/mL vancomycin and 5 μg/mL cefsulodin. H. pylori were inoculated with a sterile pipette tip into a soft agar plate. Migration of the bacteria on the plate was monitored and pictures were taken after 2 to 3 days of culture.
Mouse experiments
6–8 weeks old C57BL/6 mice were used for H. pylori infection. Mice were orally infected with 4x108 bacteria three times every two days and sacrificed after 7 days or 1 month. Stomach pieces were weighted and homogenized in Brain-Heart infusion medium. Gastric homogenate dilutions were plated on WC dent plates containing 200 μg/mL bacitracin, 10 μg/mL nalidixic acid and 3 μg/mL polymyxin B and antibiotics for selection. After 4 to 6 days incubation, H. pylori colonies were counted.
Ethics statement
All animal studies were conducted in compliance with European guidelines for the care and use of laboratory animals and were approved by the Government of Oberbayern (AZ- 55.2-1-54-2532-147-12).
Results
HP0231 is essential for CagA translocation
As proteins composing the H. pylori T4SS were suggested to form disulfide bonds to maintain structural integrity, we sought to investigate whether lack of HP0231 could alter T4SS functionality by analyzing CagA translocation into gastric epithelial cells. To this end, we deleted hp0231 in different H. pylori strains (S1A and S1B Fig) and used them for infection of AGS cells. H. pylori wild type bacteria induced a hummingbird phenotype (Fig 1A and S2A Fig). No such phenotypic changes were induced in cells infected with bacteria lacking hp0231 or when using an isogenic strain lacking CagA, whereas complementation of hp0231 reconstituted cell elongation. These data suggested impaired CagA translocation in the absence of HP0231. To further analyze CagA translocation after hp0231 deletion, lysates from infected cells were subjected to western blot to detect CagA phosphorylation. Infection of cells with wild type bacteria resulted in CagA tyrosine phosphorylation (Fig 1B and S2B Fig), while in the absence of hp0231, phosphorylation of CagA could not be detected. As expected, CagA was not phosphorylated after infecting the cells with strains lacking cagA or cagE or infected with H. pylori SS1 (Fig 1B).
Fig 1. Lack of H. pylori HP0231 impairs CagA translocation.

(A) Representative pictures of AGS cells infected with the H. pylori strain PMSS1 and the indicated isogenic mutant strains at MOI 20. Pictures of the cultures were taken after 6 hours of infection at 100x (upper panel) and 400x (lower panel) magnification. (B) and (C) CagA phosphorylation detected by western blot after infection of AGS cells with wild type H. pylori PMSS1 and isogenic mutant strains for 6 hours at MOI 20. β-actin was used as a protein loading control. One representative blot from three independent experiments is shown. (D) CagA translocation measured as β-lactamase activity in cells infected with H. pylori strains producing TEM-1-CagA. The numbers indicated are mean values ± S.D. of blue-to-green fluorescence ratios obtained from four independent experiments performed in duplicate. ***p≤0.001; ANOVA, Bonferroni’s multiple comparison test. (E) Levels of phosphorylated and total p65 detected by western blot in lysates from AGS cells infected for 6 hours with the indicated H. pylori strains at MOI 50. One representative blot from three independent experiments is shown. (F) IL-8 secretion after infection of AGS cells for 6 hours with the indicated H. pylori strains at MOI 50. Results of three independent experiments expressed as mean ± S.D. are shown. **p≤0.01, ***p≤0.001; ANOVA, Bonferroni’s multiple comparison test. Asterisks on top of the bars indicate significances relative to uninfected cells.
Since genetic manipulation of H. pylori often leads to secondary genetic changes resulting in impairment of CagA translocation, cells were infected with bacteria in which the hp0231 gene had been complemented. In contrast to hp0231 deficient bacteria, hp0231-complemented H. pylori strains translocated CagA, as detected by CagA phosphorylation (Fig 1C and S2C Fig), excluding off-target effects after deletion of hp0231.
Defects on CagA translocation were further confirmed by performing CagA translocation assays using a β-lactamase activity reporter assay. β-lactamase activity was observed when using CagA from wild type H. pylori, while no activity was detected when CagA from the hp0231 isogenic knockout strain was analyzed (Fig 1D and S2D Fig).
The presence of a functional T4SS has been related to the activation of different signaling cascades in gastric epithelial cells. Thus, we next analyzed whether lack of hp0231 affected T4SS-mediated signaling and focused on canonical NF-κB. Infection of AGS cells with wild type bacteria induced phosphorylation of p65 (Fig 1E), indicating activation of NF-κB upon infection. Notably, p65 phosphorylation was not detected in cells infected with bacteria lacking hp0231. Similar results were obtained when infecting the cells with bacteria lacking cagE, as expected, while the absence of cagA still led to p65 phosphorylation (Fig 1E). Complementation of the gene in the knockout strain led to activation of NF-κB at similar levels as observed in the wild type situation (Fig 1E), confirming that absence of hp0231 attenuates host signaling events induced by H. pylori.
As activation of NF-κB by H. pylori initiates a pro-inflammatory response, mainly translating into IL-8 expression by gastric epithelial cells, we assessed the effect of deleting hp0231 in IL-8 secretion. Cells infected with wild type H. pylori PMSS1 and the cagA deficient isogenic strain secreted IL-8 in response to the bacteria (Fig 1F). Upon deletion of hp0231 only slight levels of IL-8 were detected, similar to the levels observed after infection with bacteria lacking cagE. Complementation of hp0231 again restored IL-8 secretion (Fig 1F).
The T4SS is a protein complex spanning both bacterial membranes and containing several essential protein components. We have shown previously that two of these proteins, CagI and CagL, are produced at strongly reduced levels when genes encoding several other secretion apparatus proteins are missing, suggesting that production of normal CagI and CagL levels depends on correct assembly of these other components within the secretion apparatus [31]. Interestingly, we found that CagI and CagL are strongly reduced in levels after deletion of hp0231 (S2E and S2F Fig), indicating that hp0231 is also involved in correct assembly of the secretion apparatus.
Together, these results show that lack of hp0231 alters H. pylori T4SS functionality, thereby not only hampering CagA translocation into gastric epithelial cells but also affecting pro-inflammatory responses towards the bacterium.
Absence of HP0231 reduces H. pylori-induced vacuolation in host cells
VacA is another major virulence determinant of H. pylori potentially forming disulfide bonds. Therefore, we analyzed whether lack of hp0231 influenced VacA functionality in vitro by comparing cellular vacuolation upon infection with wild type and knockout bacteria. Infection with wild type bacteria resulted in vacuole formation in host cells (Fig 2A and 2B) that was reduced in the absence of hp0231 to a similar extent as when infecting cells with VacA deficient bacteria. Vacuolation was restored when hp0231 was complemented in the knockout strain (Fig 2A and 2B). Notably, the bacteria lacking hp0231 expressed VacA at similar levels as wild type bacteria (Fig 2C). However, lack of hp0231 highly impaired VacA secretion (Fig 2D), indicating that defects on HP0231 alter VacA secretion but not its expression.
Fig 2. Lack of HP0231 reduces H. pylori vacuolation capacity.

(A) Representative pictures of AGS cells infected with different H. pylori strains for 24 hours at MOI 100. Arrows indicate vacuoles. (400x magnification). (B) Percentage of cells showing vacuoles in response to H. pylori infection. NH4Cl was used as positive control. Five high power field (400x) were counted for each experiment. Results (mean ± S.D.) of three independent experiments are shown. ***p≤0.001; ANOVA, Bonferroni’s multiple comparison test. Asterisks on top of the bars indicate significances relative to uninfected cells. (C) VacA and HP0231 expression detected by western blot. (D) VacA levels in supernatants (SN) and pellets of 24 hours H. pylori cultures detected by western blot. (E) H. pylori gGT activity assay. G27Δggt was used as negative control, while recombinant gGT (HpgGT) was used as a positive control. Results from three independent experiments (mean ± S.D.) are shown. ***p≤0.001; ANOVA, Bonferroni’s multiple comparison test. Asterisks on top of the bars indicate significances relative to uninfected cells.
To exclude unspecific effects of deletion of hp0231, we analyzed whether lack of hp0231 also altered the function of other virulence factors. Specifically, we focused on gGT, since no disulfide bonds have been reported in its structure [34]. No changes in gGT activity were detected when hp0231 was depleted (Fig 2E), confirming that the effects induced by HP0231 are specific for bacterial factors forming disulfide bonds.
HP0231 is required for gastric colonization
Next, we examined the effects of deleting hp0231 in an infection model in vivo. C57BL/6 mice were infected for 7 and 30 days and gastric colonization was assessed by counting colony formation from stomach homogenates. Bacteria lacking hp0231 were not able to colonize the stomach of mice at either time point of analysis (Fig 3 and S3A Fig), while complementation of the gene reverted the phenotype observed. These results indicate that HP0231 is essential for gastric colonization.
Fig 3. H. pylori HP0231deficient bacteria cannot colonize the gastric mucosa of mice.
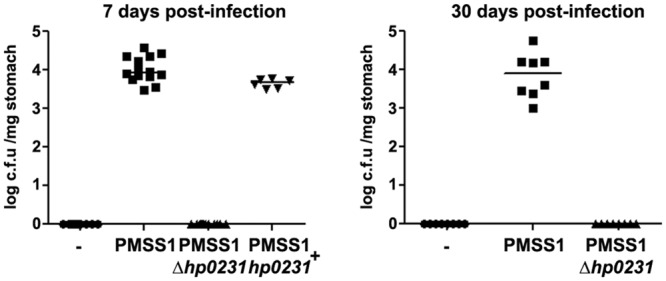
C57/BL6 mice were infected with H. pylori PMSS1 wild type or isogenic strains lacking or showing complementation of the hp0231 gene. Colony forming units (c.f.u) were examined after plating serial dilutions of stomach homogenates. Each dot represents one mouse. Horizontal bars indicate medians.
Lack of HP0231 does not affect bacterial binding but impairs motility
To colonize the gastric epithelium, H. pylori needs to bind to epithelial cells using different adhesins. As mice infected with hp0231 deficient bacteria were not colonized, we first analyzed whether lack of hp0231 induced defects in H. pylori binding to epithelial cells. Thus, human gastric epithelial cells were incubated with fluorescence-labeled bacteria and binding was determined by flow cytometry and immunofluorescence. Lack of hp0231 did not affect binding of H. pylori to gastric epithelial cells, since no differences in binding were observed between wild type and hp0231 knockout bacteria (Fig 4A and 4B), while lack of the adhesins BabA and SabA slightly reduced bacterial binding (S3B Fig).
Fig 4. HP0231 does not alter bacterial binding but it alters motility.

(A) Human gastric cancer epithelial cells were infected with CFDA-SE-labeled H. pylori PMSS1 or the PMSS1hp0231 mutant strain and binding was analyzed by FACS. Geometric Mean Fluorescence Intensity (GeoMFI) from three independent experiments (left) and a representative histogram (right) are shown. ***p≤0.001; ANOVA, Bonferroni’s multiple comparison test. Asterisks on top of the bars indicate significances relative to uninfected cells. (B) Immunofluorescence and corresponding bright field pictures of AGS cells co-incubated for 1 hour with the indicated H. pylori strains (green). Nuclei were stained with DAPI (blue) and cell membranes with deep red (red). Magnification 600x. (C) H. pylori motility assessed after 2 days incubation on soft agar plates.
As no differences in bacterial binding to host cells were detected, we then analyzed whether defects in motility could explain the lack of gastric colonization observed for the hp0231 knockout strains. Indeed, in the absence of hp0231 H. pylori showed reduced motility, as previously reported in other bacterial backgrounds [17] (Fig 4C and S3C Fig), which may explain the defects in colonization detected.
Discussion
Disulfide bond formation is an essential post-translational modification to maintain the proper conformational structure of many proteins. In bacteria, the Dsb system is mainly responsible for introducing disulfide bonds, however, not much data on Dsb proteins of H. pylori have been reported to date. Recently, H. pylori HP0231 was reported as a dimeric oxidoreductase that catalyzes disulfide bond formation in the periplasm [17]. In addition, it was found that HP0231 is important for folding and secretion of HcpE, a protein suggested to play a role in virulence [18]. As other virulence factors of H. pylori were suggested to form disulfide bonds, we analyzed whether HP0231 was important to maintain their functionality. Thus, we first focused on the T4SS. We observed that CagA translocation was impaired in the absence of hp0231, suggesting that lack of hp0231 may destabilize the structure of the T4SS and therefore, virulence factors as CagA are prevented from being injected into host cells.
Two cysteine residues (C771 and C782) located near the C terminus of the p55 subunit of VacA were shown to form a disulfide bond [19], while mutation of either of these cysteines to serine resulted in decreased secretion of VacA [35]. Upon deletion of hp0231, we observed impaired vacuolating activity, which may be attributed to the loss of the disulfide bond stabilizing both cysteines important for the secretion of the toxin. Indeed, impaired VacA secretion was detected in bacteria lacking hp0231.
Our observations not only would strengthen the crucial role previously suggested for HP0231 in bacterial virulence, but also indicate that the functionality of two of the most important H. pylori virulence determinants is highly dependent on their correct folding controlled by HP0231. Nevertheless, structural studies would be necessary in order to identify specific conformational changes in bacterial proteins upon deletion of hp0231. Although H. pylori might possess other proteins involved in disulfide bond formation, our data indicate that they cannot compensate for the lack of hp0231 concerning functionality of the T4SS and VacA.
Studies examining the role of another Dsb protein of H. pylori (DsbI) showed that impairment of disulfide bond formation highly reduced the ability of H. pylori to colonize the stomach of mice [36]. In line, we observed that bacteria lacking hp0231 were not able to colonize the gastric mucosa of mice. Impaired colonization was not due to reduced binding of H. pylori to gastric epithelial cells, but most likely to compromised motility of the hp0231 deficient bacteria. Defects in motility were already reported for the H. pylori strain N6 when hp0231 was deleted [17]. Our results indicate that this is not a strain specific effect, but that HP0231 is important to maintain bacterial motility in general. Notably, the defects on motility observed for the hp0231 deficient bacteria were not attributed to defects in the structure or distribution of the flagella but to changes in the morphology of the cells [17]. These morphological changes concomitant to a lack of motility would highly hamper colonization, and thus the bacteria would be rapidly cleared.
H. pylori infection is still a highly prevalent infection worldwide and current therapeutic strategies based on antibiotic treatment have failed mostly due to an increased rate of antibiotic resistance. Thus, the search of novel therapies based on specific inhibitors represents an interesting alternative to antibiotics. Our data suggest that HP0231 might potentially be a good target to develop inhibitors for the treatment of H. pylori infection. Blocking HP0231 in bacteria might induce morphological and structural changes, which, in the one hand, would reduce bacterial virulence once the bacterium has colonized the stomach and, on the other hand, would contribute to bacterial clearance.
Together, we provide evidence of a novel and important role for H. pylori HP0231 in bacterial virulence and colonization of the gastric mucosa. Moreover, our results should encourage the development of specific Dsb inhibitors to treat this widespread infection.
Supporting Information
(A) Expression of hp0231 assessed by PCR. bp, base pair. (B) Expression of HP0231 detected by western blot. CagA was used as a control. (C) H. pylori growth curve. Growth was determined by measuring optical density at the indicated times. Results from one representative experiment conducted in triplicate is shown.
(TIF)
(A) Representative pictures of AGS cells infected with the indicated H. pylori strains for 6 hours at MOI 20. (B) and (C) CagA phosphorylation detected by western blot in AGS cells infected for 6 hours with H. pylori at MOI 20. β-actin was used as protein loading control. (D) TEM-1-CagA expression levels detected by western blot. (E) CagI and CagL protein expression levels detected by western blot. Arrows denote specific bands.
(TIF)
(A) C57/BL6 mice were infected with H. pylori SS1 wild type or the isogenic strain lacking the hp0231. Colony forming units (c.f.u) were examined after plating serial dilutions of stomach homogenates. Each dot represents one mouse. Horizontal bars indicate medians. (B) Human gastric cancer epithelial cells were infected with CFDA-SE-labeled H. pylori G27 or isogenic mutant strains and binding was analyzed by FACS. Cells were gated on FSC/SSC followed by live/dead discrimination. Geometric Mean Fluorescence Intensity (GeoMFI) from three independent experiments are shown. **p≤0.01, ***p≤0.001; ANOVA, Bonferroni’s multiple comparison test. Asterisks on top of the bars indicate significances relative to uninfected cells. (C) H. pylori motility was assessed after 2 days incubation on soft agar plates.
(TIF)
Acknowledgments
Authors want to thank R. Semper, A. Javaheri and C. Bolz for valuable technical advice.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by the Polish National Science Centre, 2012/05/B/NZ1/00039, https://www.ncn.gov.pl (to EKJK), and the China Scholarship Council, 2011630085, http://en.csc.edu.cn/ (to YZ). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Correa P (1992) Human gastric carcinogenesis: a multistep and multifactorial process—First American Cancer Society Award Lecture on Cancer Epidemiology and Prevention. Cancer Res 52: 6735–6740. [PubMed] [Google Scholar]
- 2.Ohnishi N, Yuasa H, Tanaka S, Sawa H, Miura M, Matsui A, et al. (2008) Transgenic expression of Helicobacter pylori CagA induces gastrointestinal and hematopoietic neoplasms in mouse. Proc Natl Acad Sci U S A 105: 1003–1008. 10.1073/pnas.0711183105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schreiber S, Konradt M, Groll C, Scheid P, Hanauer G, Werling HO, et al. (2004) The spatial orientation of Helicobacter pylori in the gastric mucus. Proc Natl Acad Sci U S A 101: 5024–5029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martinez LE, Hardcastle JM, Wang J, Pincus Z, Tsang J, Hoover TR, et al. (2015) Helicobacter pylori strains vary cell shape and flagellum number to maintain robust motility in viscous environments. Mol Microbiol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sgouras DN, Trang TT, Yamaoka Y (2015) Pathogenesis of Helicobacter pylori Infection. Helicobacter 20 Suppl 1: 8–16. 10.1111/hel.12251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pachathundikandi SK, Tegtmeyer N, Backert S (2013) Signal transduction of Helicobacter pylori during interaction with host cell protein receptors of epithelial and immune cells. Gut Microbes 4: 454–474. 10.4161/gmic.27001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rassow J, Meinecke M (2012) Helicobacter pylori VacA: a new perspective on an invasive chloride channel. Microbes Infect 14: 1026–1033. 10.1016/j.micinf.2012.07.002 [DOI] [PubMed] [Google Scholar]
- 8.Raju D, Hussey S, Ang M, Terebiznik MR, Sibony M, Galindo-Mata E, et al. (2012) Vacuolating cytotoxin and variants in Atg16L1 that disrupt autophagy promote Helicobacter pylori infection in humans. Gastroenterology 142: 1160–1171. 10.1053/j.gastro.2012.01.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peek JA, Taylor RK (1992) Characterization of a periplasmic thiol:disulfide interchange protein required for the functional maturation of secreted virulence factors of Vibrio cholerae. Proc Natl Acad Sci U S A 89: 6210–6214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu J (1998) Inactivation of DsbA, but not DsbC and DsbD, affects the intracellular survival and virulence of Shigella flexneri. Infect Immun 66: 3909–3917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu J, Kroll JS (1999) DsbA: a protein-folding catalyst contributing to bacterial virulence. Microbes Infect 1: 1221–1228. [DOI] [PubMed] [Google Scholar]
- 12.Stenson TH, Weiss AA (2002) DsbA and DsbC are required for secretion of pertussis toxin by Bordetella pertussis. Infect Immun 70: 2297–2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Raczko AM, Bujnicki JM, Pawlowski M, Godlewska R, Lewandowska M, Jagusztyn-Krynicka EK (2005) Characterization of new DsbB-like thiol-oxidoreductases of Campylobacter jejuni and Helicobacter pylori and classification of the DsbB family based on phylogenomic, structural and functional criteria. Microbiology 151: 219–231. [DOI] [PubMed] [Google Scholar]
- 14.Kaakoush NO, Kovach Z, Mendz GL (2007) Potential role of thiol:disulfide oxidoreductases in the pathogenesis of Helicobacter pylori. FEMS Immunol Med Microbiol 50: 177–183. [DOI] [PubMed] [Google Scholar]
- 15.Roszczenko P, Grzeszczuk M, Kobierecka P, Wywial E, Urbanowicz P, Wincek P, et al. (2015) Helicobacter pylori HP0377, a member of the Dsb family, is an untypical multifunctional CcmG that cooperates with dimeric thioldisulfide oxidase HP0231. BMC Microbiol 15: 135 10.1186/s12866-015-0471-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yoon JY, Kim J, Lee SJ, Kim HS, Im HN, Yoon HJ, et al. (2011) Structural and functional characterization of Helicobacter pylori DsbG. FEBS Lett 585: 3862–3867. 10.1016/j.febslet.2011.10.042 [DOI] [PubMed] [Google Scholar]
- 17.Roszczenko P, Radomska KA, Wywial E, Collet JF, Jagusztyn-Krynicka EK (2012) A novel insight into the oxidoreductase activity of Helicobacter pylori HP0231 protein. PLoS One 7: e46563 10.1371/journal.pone.0046563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lester J, Kichler S, Oickle B, Fairweather S, Oberc A, Chahal J, et al. (2015) Characterization of Helicobacter pylori HP0231 (DsbK): role in disulfide bond formation, redox homeostasis and production of Helicobacter cystein-rich protein HcpE. Mol Microbiol 96: 110–133. 10.1111/mmi.12923 [DOI] [PubMed] [Google Scholar]
- 19.Gangwer KA, Mushrush DJ, Stauff DL, Spiller B, McClain MS, Cover TL, et al. (2007) Crystal structure of the Helicobacter pylori vacuolating toxin p55 domain. Proc Natl Acad Sci U S A 104: 16293–16298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barden S, Lange S, Tegtmeyer N, Conradi J, Sewald N, Backert S, et al. (2013) A helical RGD motif promoting cell adhesion: crystal structures of the Helicobacter pylori type IV secretion system pilus protein CagL. Structure 21: 1931–1941. 10.1016/j.str.2013.08.018 [DOI] [PubMed] [Google Scholar]
- 21.Backert S, Muller EC, Jungblut PR, Meyer TF (2001) Tyrosine phosphorylation patterns and size modification of the Helicobacter pylori CagA protein after translocation into gastric epithelial cells. Proteomics 1: 608–617. [DOI] [PubMed] [Google Scholar]
- 22.Xiang Z, Censini S, Bayeli PF, Telford JL, Figura N, Rappuoli R, et al. (1995) Analysis of expression of CagA and VacA virulence factors in 43 strains of Helicobacter pylori reveals that clinical isolates can be divided into two major types and that CagA is not necessary for expression of the vacuolating cytotoxin. Infect Immun 63: 94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee A, O'Rourke J, De Ungria MC, Robertson B, Daskalopoulos G, Dixon MF (1997) A standardized mouse model of Helicobacter pylori infection: introducing the Sydney strain. Gastroenterology 112: 1386–1397. [DOI] [PubMed] [Google Scholar]
- 24.Arnold IC, Lee JY, Amieva MR, Roers A, Flavell RA, Sparwasser T, et al. (2011) Tolerance rather than immunity protects from Helicobacter pylori-induced gastric preneoplasia. Gastroenterology 140: 199–209. 10.1053/j.gastro.2010.06.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gebert B, Fischer W, Weiss E, Hoffmann R, Haas R (2003) Helicobacter pylori vacuolating cytotoxin inhibits T lymphocyte activation. Science 301: 1099–1102. [DOI] [PubMed] [Google Scholar]
- 26.Oertli M, Noben M, Engler DB, Semper RP, Reuter S, Maxeiner J, et al. (2013) Helicobacter pylori gamma-glutamyl transpeptidase and vacuolating cytotoxin promote gastric persistence and immune tolerance. Proc Natl Acad Sci U S A 110: 3047–3052. 10.1073/pnas.1211248110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guillemin K, Salama NR, Tompkins LS, Falkow S (2002) Cag pathogenicity island-specific responses of gastric epithelial cells to Helicobacter pylori infection. Proc Natl Acad Sci U S A 99: 15136–15141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Motoyama T, Hojo H, Watanabe H (1986) Comparison of seven cell lines derived from human gastric carcinomas. Acta Pathol Jpn 36: 65–83. [DOI] [PubMed] [Google Scholar]
- 29.Semper RP, Mejias-Luque R, Gross C, Anderl F, Muller A, Vieth M, et al. (2014) Helicobacter pylori-Induced IL-1beta Secretion in Innate Immune Cells Is Regulated by the NLRP3 Inflammasome and Requires the Cag Pathogenicity Island. J Immunol 193: 3566–3576. 10.4049/jimmunol.1400362 [DOI] [PubMed] [Google Scholar]
- 30.Schmitt W, Haas R (1994) Genetic analysis of the Helicobacter pylori vacuolating cytotoxin: structural similarities with the IgA protease type of exported protein. Mol Microbiol 12: 307–319. [DOI] [PubMed] [Google Scholar]
- 31.Pham KT, Weiss E, Jimenez Soto LF, Breithaupt U, Haas R, Fischer W (2012) CagI is an essential component of the Helicobacter pylori Cag type IV secretion system and forms a complex with CagL. PLoS One 7: e35341 10.1371/journal.pone.0035341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schindele F, Weiss E, Haas R, Fischer W (2016) Quantitative analysis of CagA type IV secretion by Helicobacter pylori reveals substrate recognition and translocation requirements. Mol Microbiol 100: 188–203. 10.1111/mmi.13309 [DOI] [PubMed] [Google Scholar]
- 33.Sanders L, Andermann TM, Ottemann KM (2013) A supplemented soft agar chemotaxis assay demonstrates the Helicobacter pylori chemotactic response to zinc and nickel. Microbiology 159: 46–57. 10.1099/mic.0.062877-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morrow AL, Williams K, Sand A, Boanca G, Barycki JJ (2007) Characterization of Helicobacter pylori gamma-glutamyltranspeptidase reveals the molecular basis for substrate specificity and a critical role for the tyrosine 433-containing loop in catalysis. Biochemistry 46: 13407–13414. [DOI] [PubMed] [Google Scholar]
- 35.Letley DP, Rhead JL, Bishop K, Atherton JC (2006) Paired cysteine residues are required for high levels of the Helicobacter pylori autotransporter VacA. Microbiology 152: 1319–1325. [DOI] [PubMed] [Google Scholar]
- 36.Godlewska R, Dzwonek A, Mikula M, Ostrowski J, Pawlowski M, Bujnicki JM, et al. (2006) Helicobacter pylori protein oxidation influences the colonization process. Int J Med Microbiol 296: 321–324. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Expression of hp0231 assessed by PCR. bp, base pair. (B) Expression of HP0231 detected by western blot. CagA was used as a control. (C) H. pylori growth curve. Growth was determined by measuring optical density at the indicated times. Results from one representative experiment conducted in triplicate is shown.
(TIF)
(A) Representative pictures of AGS cells infected with the indicated H. pylori strains for 6 hours at MOI 20. (B) and (C) CagA phosphorylation detected by western blot in AGS cells infected for 6 hours with H. pylori at MOI 20. β-actin was used as protein loading control. (D) TEM-1-CagA expression levels detected by western blot. (E) CagI and CagL protein expression levels detected by western blot. Arrows denote specific bands.
(TIF)
(A) C57/BL6 mice were infected with H. pylori SS1 wild type or the isogenic strain lacking the hp0231. Colony forming units (c.f.u) were examined after plating serial dilutions of stomach homogenates. Each dot represents one mouse. Horizontal bars indicate medians. (B) Human gastric cancer epithelial cells were infected with CFDA-SE-labeled H. pylori G27 or isogenic mutant strains and binding was analyzed by FACS. Cells were gated on FSC/SSC followed by live/dead discrimination. Geometric Mean Fluorescence Intensity (GeoMFI) from three independent experiments are shown. **p≤0.01, ***p≤0.001; ANOVA, Bonferroni’s multiple comparison test. Asterisks on top of the bars indicate significances relative to uninfected cells. (C) H. pylori motility was assessed after 2 days incubation on soft agar plates.
(TIF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.