

Abstract
Oxalate decarboxylase (OxDC) catalyzes the conversion of oxalate into formate and carbon dioxide in a remarkable reaction that requires manganese and dioxygen. Previous studies have shown that replacing an active-site loop segment Ser161-Glu162-Asn163-Ser164 in the N-terminal domain of OxDC with the cognate residues Asp161-Ala162-Ser-163-Asn164 of an evolutionarily related, Mn-dependent oxalate oxidase gives a chimeric variant (DASN) that exhibits significantly increased oxidase activity. The mechanistic basis for this change in activity has now been investigated using membrane inlet mass spectrometry (MIMS) and isotope effect (IE) measurements. Quantitative analysis of the reaction stoichiometry as a function of oxalate concentration, as determined by MIMS, suggests that the increased oxidase activity of the DASN OxDC variant is associated with only a small fraction of the enzyme molecules in solution. In addition, IE measurements show that C–C bond cleavage in the DASN OxDC variant proceeds via the same mechanism as in the wild-type enzyme, even though the Glu162 side chain is absent. Thus, replacement of the loop residues does not modulate the chemistry of the enzyme-bound Mn(II) ion. Taken together, these results raise the possibility that the observed oxidase activity of the DASN OxDC variant arises from an increased level of access of the solvent to the active site during catalysis, implying that the functional role of Glu162 is to control loop conformation. A 2.6 Å resolution X-ray crystal structure of a complex between oxalate and the Co(II)-substituted ΔE162 OxDC variant, in which Glu162 has been deleted from the active site loop, reveals the likely mode by which the substrate coordinates the catalytically active Mn ion prior to C–C bond cleavage. The “end-on” conformation of oxalate observed in the structure is consistent with the previously published V/K IE data and provides an empty coordination site for the dioxygen ligand that is thought to mediate the formation of Mn(III) for catalysis upon substrate binding.
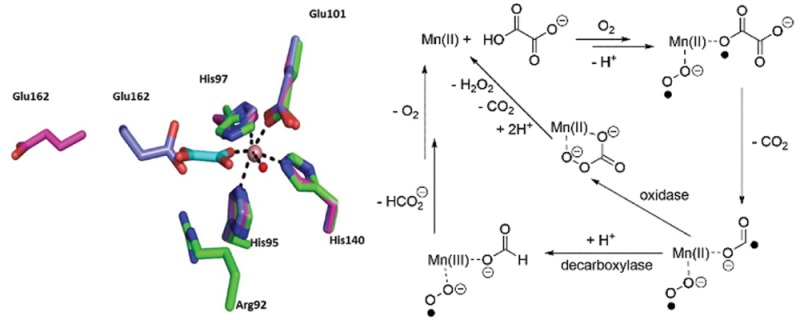
The enzymes oxalate decarboxylase (OxDC) and oxalate oxidase (OxOx) mediate the breakdown of the toxic metabolite oxalate (Scheme 1)1 and are thought to share a common evolutionary precursor on the basis of the structural similarity and sequence identity of their “cupin” domains (Figure 1).2,3 Even though OxDC and OxOx both require a bound manganese ion for activity,4−6 which is coordinated by identical ligands in the same octahedral geometry (Figure 1), these enzymes convert oxalate into different products.1 Elucidating the details of the catalytic and chemical mechanisms employed by these enzymes therefore provides an opportunity to understand how changes in protein environment might modulate metal reactivity.7−9 Thus, it was recently reported that replacing the conformationally mobile active-site loop (Ser161-Glu162-Asn163-Ser164) in the N-terminal, Mn-binding domain of Bacillus subtilis OxDC with residues from a cognate loop (Asp161-Ala162-Ser-163-Asn164) in Ceriporiopsis subvermispora OxOx (the only characterized oxalate oxidase that possesses two cupin domains)12,13 gave a variant of B. subtilis OxDC (DASN OxDC)a that exhibited considerably elevated oxidase and decreased decarboxylase activities.14 This remarkable finding was rationalized by assuming that the first two steps in the OxDC- and OxOx-catalyzed reactions are identical and result in the formation of a Mn-bound formyl radical anion (Scheme 2), which then partitions to give formate or CO2 depending on whether Glu162 is present to protonate the carbon atom.14 Thus, the absence of a residue (equivalent to Glu162) that is capable of mediating general acid/base catalysis in the OxOx active site11,15 results in the production of CO2 and hydrogen peroxide, perhaps via a peroxycarbonate intermediate (Scheme 2). Alternate explanations can be advanced for the switch in reaction specificity observed for the DASN OxDC chimeric enzyme, including altered oxalate coordination to Mn(II) and oxidation of the formyl radical anion in solution after release from the active site due to an altered loop conformation. Furthermore, if Glu162 mediates the initial proton-coupled electron transfer (PCET) step leading to C–C bond cleavage in Mn-bound oxalate,16−18 then it is hard to understand how its absence in OxOx might permit that enzyme to employ the same initial chemical steps as those leading to radical anion in the OxDC-catalyzed reaction.
Scheme 1. Reactions Catalyzed by Oxalate Decarboxylase (OxDC) and Oxalate Oxidase (OxOx).

Figure 1.

Ribbon representation of OxDC (left, PDB entry 1UW8)10 and OxOx (right, PDB entry 1FI2)11 showing the structural similarity of the cupin domains and Mn-binding sites in these enzymes. OxDC is constructed from two domains (N-terminal, blue; C-terminal, green) in both of which Mn(II) (orange spheres) is coordinated by conserved histidine and glutamate residues. OxOx has a single cupin domain (yellow) but also contains a tightly bound Mn(II) (orange sphere) coordinated by conserved histidine and glutamate residues. Oxygen atoms of metal-bound waters are rendered as red spheres.
Scheme 2. Hypothetical Scheme Showing How Partitioning of a Common Formyl Radical Anion Intermediate Might Lead to Different Enzyme Activities in OxDC and OxOx14,

We note that the metal oxidation states and dioxygen-binding site remain to be experimentally defined.
We now report investigations on a series of OxDC active-site loop variants using X-ray crystallography, membrane inlet mass spectrometry (MIMS),19,20 and 13(V/K) isotope effect measurements.17,21 These studies provide the first structural insights into the mode by which oxalate coordinates the N-terminal Mn(II) ion in OxDC and suggest that the functional role of Glu162 may be to “orient” the active-site loop into a conformation that precludes solvent access during turnover rather than to mediate the generation of a substrate-based radical intermediate as proposed previously.16
Materials and Methods
Materials
All chemicals and reagents were purchased from Fisher Scientific (Pittsburgh, PA) or Sigma-Aldrich (St. Louis, MO), unless otherwise stated. [13C2]Oxalic acid (99% 13C) was purchased from Cambridge Isotope Laboratories (Tewksbury, MA), and an oxalate assay kit was obtained from Trinity Biotech USA (Jamestown, NY). Nickel-nitrilotriacetic acid agarose (Ni-NTA) was supplied by Qiagen (Germantown, MD). DNA primers were synthesized by Integrated DNA Technologies, Inc. (Coralville, IA), and DNA sequencing was performed in the DNA Sequence Core at the University of Michigan (Ann Arbor, MI). Protein concentrations were determined using the CoomassiePlus Protein Assay reagent obtained from ThermoFisher Scientific (Waltham, MA), and BT Chelex 100 resin was purchased from Bio-Rad (Hercules, CA). ICP-MS measurements of metal content were taken at the Center for Applied Isotope Studies at the University of Georgia (Athens, GA).
Expression and Purification of C-Terminally Tagged, Mn-Containing OxDC Variants
Plasmids containing genes encoding C-terminally tagged WT OxDC and the DASN OxDC chimeric variant were generously provided by S. Bornemann (John Innes Centre, Norwich, U.K.). The QuickChange Site-Directed Mutagenesis System (Stratagene, La Jolla, CA) was used to prepare all other OxDC loop variants. All recombinant, Mn-containing C-terminally His-tagged proteins were expressed in Escherichia coli strain BL21(DE3) following published procedures,10,16 except that expression was induced with 0.8 mM isopropyl β-d-1-thiogalactopyranoside in the presence of 5 mM MnCl2 after the bacteria had been heat shocked for 15 min at 42 °C. OxDC loop variants were then purified by metal affinity chromatography on a Ni-NTA column. The fractions containing the desired protein were pooled and subjected to multiple buffer exchange steps at 4 °C to remove imidazole, and free metal was removed by passage over BT Chelex 100 resin. Recombinant enzymes were stored in 50 mM Tris buffer (pH 8.5) containing 500 mM NaCl and were concentrated, if necessary, using an Amicon Centriprep YM-30 filter unit from Millipore (Billerica, MA), with the enzyme concentration being measured by a Bradford assay using bovine serum albumin as the standard.22 Expression and purification of the His6-tagged Co(II)-containing OxDC variant, which lacked the Glu162 residue (ΔE162 OxDC), were accomplished by similar protocols,23 except 2 mM CoCl2 was added to the growth medium after the cells had been heat shocked and the soluble, recombinant enzyme was purified using a Talon cobalt column. The eluate was dialyzed overnight in the storage buffer [50 mM Tris-HCl (pH 8.5) containing 500 M NaCl] followed by a further purification step on a 320 mL Sephacryl S-100 gel filtration column.
Steady-State Kinetic Assays
The steady-state rate of enzyme-catalyzed oxalate breakdown could be determined in a continuous assay by measuring 13CO2 production and O2 consumption using MIMS. These assays were performed using an Extrel EXM-200 quadruple mass spectrometer with an inlet probe, as described in detail elsewhere.19,20,24 Thus, solutions containing [13C2]oxalate (0–100 mM) dissolved in 50 mM succinate buffer (pH 4.0) were equilibrated with air, dioxygen, or helium, and reaction was initiated by the addition of enzyme. Standard curves for the conversion of ion current into concentration were determined for CO2 and O2 using published methods.19,20 The addition of various known concentrations of solid K2CO3 to aqueous AcOH (pH 2.0) was used to calibrate the CO2 measurements, while buffer solutions containing known amounts of O2 were obtained by mixing known volumes of O2-saturated buffer and buffer from which all dioxygen had been removed by purging with helium. In the MIMS assay, initial reaction rates were determined from the slope of the linear portion of the curve after initiating reaction by the addition of enzyme. The reaction was quenched by addition of NaOH, and an aliquot was then taken out of the MIMS chamber to determine formate production using an end-point assay as described elsewhere.17 In a similar manner, oxidase activity was measured using a standard horseradish 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonate) (ABTS)–H2O2–peroxidase system coupled assay,12 for which control samples in which either oxalate or peroxidase was absent were required to correct for the ability of the OxDC loop variants to oxidize ABTS directly.12,21 The pH profile of the DASN OxDC variant was determined using the standard peroxidase–ABTS coupled assay except that reactions were performed in phosphate-succinate buffer from pH 3.5 to 7.0. Measurements were taken at specific substrate and enzyme concentrations in duplicate, and the data were analyzed to obtain the values of V and V/K by standard computer-based methods.25 The “specificity switch” value for all OxDC loop variants (Table 1) was calculated using the following expression:
| 1 |
where MUT(kcat/KM/Mn)OX and MUT(kcat/KM/Mn)DEC are computed for the oxidase and decarboxylase activities of the OxDC loop variant, respectively. Similarly, WT(kcat/KM/Mn)OX and WT(kcat/KM/Mn)DEC are calculated for the cognate activities of WT OxDC. The denominator denotes that the concentration of enzyme was corrected for Mn incorporation in the calculation of kcat.
Table 1. Steady-State Kinetic Parameters for the Decarboxylase and Oxidase Activities of C-Terminally His6-Tagged, WT OxDC and Selected OxDC Loop Variants.
| decarboxylase
activityb |
oxidase
activityc |
|||||||
|---|---|---|---|---|---|---|---|---|
| enzyme | Mn/monomera | KM (mM) | kcat (s–1) | kcat/KM/Mn (M–1 s–1) | KM (mM) | kcat (s–1) | kcat/KM/Mn (M–1 s–1) | specificity switchd |
| WT OxDC | 1.4 | 8 ± 1 | 60 ± 2 | 5700 ± 840 | 5.0 ± 0.2 | 0.13 ± 0.02 | 19 ± 3 | 1 |
| DASNe | 1.6 | 16 ± 7 | 0.4 ± 0.1 | 17 ± 8 | 3.0 ± 0.3 | 4.1 ± 0.4 | 800 ± 190 | 14100 (225000)f |
| DESNe | 1.5 | 6 ± 1 | 40 ± 1 | 4400 ± 660 | 11 ± 2 | 0.10 ± 0.01 | 11 ± 1 | 0.75 (9) |
| DENS | 1.6 | 10 ± 1 | 63 ± 3 | 3900 ± 840 | 4.0 ± 0.4 | 0.05 ± 0.01 | 9 ± 2 | 1 |
| DDSN | 1.5 | 3 ± 1 | 9 ± 2 | 2400 ± 910 | 17 ± 2 | 0.79 ± 0.03 | 32 ± 4 | 2 |
| DDNS | 1.5 | 10 ± 2 | 45 ± 3 | 2900 ± 680 | 16 ± 2 | 0.49 ± 0.04 | 20 ± 3 | 32 |
| SDNSg | 1.1 | 3.1 ± 0.7 | 29 ± 2 | 9000 ± 200 | ndi | ndi | ndi | ndi |
| SQNSg | 0.5 | 10.0 ± 0.2 | 0.3 ± 0.1 | 60 ± 20 | ndi | ndi | ndi | ndi |
| SANSh | 1.4 | ndi | inactive | ndi | ndi | ndi | ndi | ndi |
| ΔE162 | 1.3 | 1.6 ± 0.7 | 0.01 ± 0.005 | 5 ± 2 | 3 ± 1 | 0.05 ± 0.03 | 14 ± 9 | 840 |
The metal content value has been reported previously for WT OxDC and is included here for ease of comparison.17
Reaction mixtures consisted of WT OxDC or the loop variant (5 μM) and potassium oxalate (0–80 mM) dissolved in 100 mM citrate buffer containing 300 μM o-phenylenediamine (pH 4.2) at 25 °C (total volume of 1 mL). Turnover was initiated by the addition of enzyme. After incubation at 25 °C for 1 min, the reaction was quenched by the addition of 1.1 M aqueous NaOH (10 μL) and the formate quantified using the NADH absorption at 340 nm in an assay mixture containing formate dehydrogenase (pH 7.8).17
Reaction mixtures consisted of WT OxDC or the loop variant (0.5 μM) and potassium oxalate (0–50 mM) dissolved in 50 mM succinate buffer (pH 4.0) at 25 °C (total volume of 1 mL). Turnover was initiated by the addition of enzyme, and hydrogen peroxide was quantified using the horseradish peroxidase–ABTS assay. Initial rates were determined from the linear portion of the continuous assay.20
The specificity switch value is calculated using eq 1 (see Materials and Methods).
Steady-state kinetic parameters have been reported previously for this OxDC loop variant.14 The values reported herein, however, have been redetermined for the recombinant enzyme expressed in our laboratory using reaction conditions identical to those reported in the earlier study.
The specificity switch value in parentheses is that reported previously and is included in the table for ease of comparison. We note, however, that this earlier values was calculated using the following expression,14 which differs from the equation (eq 1) used in our study: {[MUT(Vmax)OX][WT(Vmax)DEC]}/{[MUT(Vmax)DEC][WT(Vmax)OX]} (eq 2), where MUT(Vmax)OX and MUT(Vmax)DEC are the Vmax values for the oxidase and decarboxylase activities of the OxDC loop variant, respectively. Similarly, WT(Vmax)OX and WT(Vmax)DEC are the cognate values of WT OxDC.
Values taken from ref (21) are included for ease of comparison. The specific activities reported for the oxidase activity of the SDNS and SQNS OxDC variants were 0.05 and 0.56 unit/mg, respectively. Note that both of these variants lacked a C-terminal His tag, and the assay conditions were different from those employed in this study.
Values taken from ref (27) are included for ease of comparison. The specific activity reported for the oxidase activity of the SANS OxDC variant was 0.02 unit/mg. This variant was reported to lack detectable decarboxylase activity.
Not determined.
Cocrystallization of the Co(II)-Substituted ΔE162 OxDC Variant with Oxalate
Purified Co(II)-containing ΔE162 OxDC was concentrated to 5 mg/mL in 100 mM Tris-HCl (pH 8.5) containing 500 mM NaCl and 10 mM potassium oxalate. Crystals were obtained by the vapor diffusion method with hanging drop geometry by mixing protein/oxalate (2 μL) and well solution (2 μL), using conditions similar to those used to crystallize the E162A OxDC variant (PDB entry 2UY9).27 Thus, the well solution contained 100 mM Ches (pH 9.5) and 10% PEG 6000. Crystals, which were rectangular prisms (0.05 mm × 0.025 mm × 0.01 mm), appeared after 1 week at 17 °C. These crystals were then cryoprotected in a well solution containing 25% glycerol and flash-frozen in liquid nitrogen.
Data Collection, Structure Determination, and Crystallographic Refinement of the Co(II)-Substituted ΔE162 OxDC Variant/Oxalate Complex
The Co(II)-substituted ΔE162 OxDC variant complex with oxalate crystallized in space group R32 with the following unit cell dimensions: a = b = 154.619 Å, and c = 121.954 Å. Data were collected to 2.6 Å resolution at the National Synchrotron Light Source (NSLS) of the Brookhaven National Laboratory (Upton, NY) at beamline X25 equipped with a Pilatus 6M detector (Table 2). Phasing was achieved by molecular replacement28 (Phaser-MR29) using WT OxDC (PDB entry 1UW8),12 after removing all ligands, metals, and water molecules, as the search model. The structure was refined using alternating rounds of manual rebuilding in COOT30 followed by simulated annealing in PHENIX31 using individual B factor refinement (occupancy was not refined). The oxalate ligand was placed in the corresponding density when the R factor reached 22%. The resulting model contained residues 6–382 of the enzyme with one molecule per asymmetric unit, one oxalate, two Co(II) ions, and 233 waters. The model was refined to 2.6 Å resolution with Rwork and Rfree values of 17.5 and 22.4%, respectively (Table 2). The Ramachandran plot shows that 94.9% of residues fall in the most favored regions, with 4.8% (17 residues) in the allowed regions and only 0.3% in the disallowed regions.
Table 2. Crystallographic Data Collection and Refinement Statisticsa.
| Data Collection | |
| resolution (highest-resolution shell) (Å) | 24.78–2.60 (2.68–2.60)b |
| X-ray source | X25, NSLS |
| wavelength (Å) | 0.979 |
| space group | R32 |
| cell dimensions (Å) | a = b = 156.97, c = 121.95 |
| no. of reflections observed (unique) | 896022 (17137) |
| completeness (%) | 98.9 (99.1) |
| Rmerge (%)b | 18.8 (47.1) |
| I/σ(I) | 13.6 (2.4) |
| redundancy | 9.8 (8.2) |
| Refinement | |
| no. of protein residues/water atoms per asymmetric unit | 375/233 |
| no. of other ligands per asymmetric unit | 2 Co(II), 1 oxalate |
| no. of reflections (work/free) | 17122/1712 |
| Rwork, Rfree (%) | 17.5, 22.4 |
| resolution (Å) | 24.78–2.60 |
| average B factor (Å2) | 37.9 |
| protein (Å2) | 35.2 |
| Co(II) (Å2) | 37.2 |
| oxalate (Å2) | 40.9 |
| water (Å2) | 39.1 |
| rmsd for bond lengths (Å) | 0.008 |
| rmsd for bond angles (deg) | 1.105 |
Data for the highest-resolution shell are given in parentheses.
Rmerge = ∑hkl∑i|Ihkl,i – ⟨Ihkl⟩I|/∑hkl∑i|Ihkl,i|, where ⟨Ihkl⟩ is the mean intensity of the multiple Ihkl,i observations for symmetry-related reflections.
Kinetic Isotope Effect Nomenclature
In this work, 13(V/K) represents the ratio of Vmax/KM for the 12C-containing substrate relative to the 13C-containing substrate.32
Kinetic Isotope Effect Measurements
The internal competition method17,33,34 was used to measure the primary 13C kinetic isotope effect (IE) for the conversion catalyzed by the DASN OxDC variant, with the 12C/13C ratio in CO2(g) produced in the reaction or indirectly from formate oxidation being analyzed by isotope ratio mass spectrometry (IRMS).1713C was present in the carbon atoms of oxalate at natural abundance in these experiments, and the observed IEs are therefore associated with steps up to, and including, the first irreversible step in the mechanism (cleavage of the C–C bond in the substrate).17 Analyses to determine the isotopic ratios (R values) were performed on CO2(g) isolated directly from the DASN-catalyzed partial conversion of oxalate. Enzymatic reactions were performed by incubation of the DASN OxDC variant with oxalate (1 mM) at 25 °C in either 50 mM 1,4-bis(2-hydroxyethyl)piperazine (pH 4.2) or 50 mM piperazine (pH 5.7). All gases were passed over Ascarite to remove CO2(g) prior to use, and buffer solutions were sparged with N2(g) before use. The presence of oxygen is required for oxidase activity, so equal volumes of O2-saturated water (10 mL) and buffer were mixed to give the desired O2 concentration in the experiment (total volume of 20 mL). Reactions were initiated by the addition of enzyme and quenched by increasing the solution pH to 7.5 using either 1 N Tris-H2SO4 buffer (pH 7.8) (reaction at pH 4.2) or 1 N Tris-H2SO4 buffer (pH 7.5) (reaction at pH 5.7), after 2 and 4 h, respectively. After quenching, the solution was passed through an Amicon ultrafiltration system and an aliquot (50 μL) taken to determine the fraction of conversion, f, using an oxalate assay kit. H2O2 was removed from the sample aliquot by the addition of catalase (10 μL of 10000 units/mg) because it interferes with the oxalate detection kit. After 10 min, CCl4 (50 μL) was added and the sample centrifuged to remove the precipitated catalase. The resulting aqueous phase was taken and assayed for residual oxalate to determine the fraction of reaction. Formate in the sample was quantitated from NADH production upon the addition of formate dehydrogenase (FDH).17
Observed IEs were analyzed using standard procedures and assumptions17 using the following equation (eq 3):
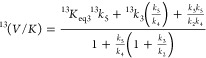 |
3 |
where 13(V/K) is the 13C IE, 13k3 and 13k5 correspond to the intrinsic isotope effects on the microscopic rate constants k3 and k5, respectively, in the minimal kinetic mechanism (Scheme 3), and 13Keq3 is the equilibrium isotope effect for the reversible conversion of the Michaelis complex to the first intermediate.
Scheme 3. Minimal Kinetic Mechanism for the OxDC-Catalyzed Reaction Up to, and Including, the First Irreversible Step.

Results
Steady-State Kinetics of OxDC Loop Variants
A series of C-terminally tagged, Mn-containing OxDC variants (DASN,a DESN, DDSN, DDNS, DENS, and ΔE162) were prepared and their steady-state kinetic parameters determined using standard assays that measured formate or hydrogen peroxide production for the decarboxylase or oxidase activities, respectively (Table 1). These experiments employed assay conditions similar to those used by Burrell and co-workers14 and confirmed that the DASN OxDC variant exhibits significantly increased oxidase activity. As reported previously, reintroduction of the glutamate side chain into the loop (DESN variant) restored decarboxylase activity when compared with that of DASN, even if the remaining loop residues were altered (DENS variant) (Table 1). We also investigated the effect of introducing an aspartate residue at that position. The DDNS and DDSN OxDC variants both exhibited significant decarboxylase activity. Finally, we investigated the effect of deleting Glu162 from the active-site loop. The decarboxylase activity of the resulting Mn-containing ΔE162 OxDC variant, as judged on the basis of kcat/KM/Mn,b was decreased approximately 1000-fold relative to that of the wild-type enzyme. On the other hand, this variant retained oxidase activity comparable to that of WT OxDC (Table 1).
X-ray Crystal Structure of the Co(II)-Substituted ΔE162 OxDC/Oxalate Complex
Removal of Glu162, for which the equivalent residue is alanine in the OxOx active-site loop, diminished both catalytic activities. To examine the extent to which removal of this residue had perturbed the active site, we obtained diffraction quality crystals of the Co(II)-substituted ΔE162 OxDC variant; it has been shown previously that the presence of Co(II) permits oxalate binding but does not support catalysis.4,23 Phases were calculated using WT OxDC (PDB entry 1UW8)10 as the model for molecular replacement to give a 2.6 Å resolution structure (Figure 2A) composed of residues 6–382 (one protein chain) containing two Co(II) ions and one molecule of oxalate (Table 2). This structure was then superimposed with those of WT OxDC in which the N-terminal active-site loop adopts an “open” (PDB entry 1J58)36 and “closed” (PDB entry 1UW8)10 conformation (Figure 2B). Deleting Glu162 had no significant impact on the overall fold of the enzyme (rmsd of 0.22 Å) and resulted in a loop that adopted a shape similar to that of the “closed” conformation that has been reported previously,10 although we acknowledge the possibility of some bias introduced by the choice of model for molecular replacement.
Figure 2.

(A) Ribbon representation of the X-ray crystal structure of the Co(II)-substituted ΔE162 OxDC variant color ramped from the N-terminus to the C-terminus (blue to red, respectively) and showing the location of the Co(II) ion (magenta sphere). (B) Overlay of the active-site loop (boxed, S-NS) in the ΔE162 OxDC variant (green) with the cognate loops (SENS) of WT OxDC in the “open” (magenta, PDB entry 1J58) and “closed” (blue, PDB entry 1UW8) conformation. The Co(II) ion is rendered as a magenta sphere.
In addition to demonstrating the relatively small structural impact of deleting Glu162, we observed an oxalate molecule bound within the N-terminal domain of the Co(II)-substituted ΔE162 OxDC variant (Figure 3) but not in the C-terminal domain. This is further evidence of the view that catalysis is associated with the N-terminal Mn-binding site in WT OxDC10,14,27,37 and provides a firm structural basis for understanding the catalytic mechanism used by the enzyme. As expected from the similarity of metal–ligand bond lengths for Co(II) and Mn(II) in the Scripps Metalloprotein Database,38 the Co(II) ion in the deletion variant is coordinated in the same manner as Mn(II) in WT OxDC; both metal ions bind three histidines (residues His95, His97, and His104 with coordinate bond lengths of 2.3, 2.2, and 2.1 Å, respectively) and one glutamate (Glu101 with a coordinate bond length of 2.1 Å) (Figure 3). In addition to these four protein ligands, the two remaining sites on Co(II) are occupied by a water and an oxalate O, giving an overall octahedral coordination geometry. As expected on the basis of heavy-atom IE measurements,17 the oxalate ligand is bound in an “end on” conformation with one oxygen atom of the carboxylate moiety coordinated to the metal ion with a coordination bond that is 1.7 Å in length. The second carboxylate group on oxalate lies sufficiently close to form a hydrogen bond with Arg92 and Thr165 (3.5 and 3.4 Å, respectively). While the positioning of Arg92 remains unchanged in the oxalate-bound structure compared to that in the WT structure, Thr165 adopts a different rotamer, allowing it to form the hydrogen bond with the substrate (Figure 3). We note that the mode of coordination observed in the ΔE162 OxDC variant contrasts with a bidentate interaction between oxalate and the Mn(II) ion present in a putative OxDC (a monocupin with function assigned on the basis of 23% sequence identity) from Thermotoga maritima (PDB entry 1O4T), which was crystallized at pH 8.39 Oxalate exists as a dianion at that solution pH; therefore, it is not surprising that binding of ligand to the Mn(II) center in this putative OxDC is bidentate, and we note that the decarboxylase activity assigned to this Thermotoga enzyme remains to be demonstrated. Indeed, if the Thermotoga enzyme is an oxalate decarboxylase, it is likely to function via a mechanism different from that of the B. subtilis ortholog given that it lacks both the SENS loop motif and other features of the oxalate-binding site. Perhaps of greater importance for understanding the catalytic mechanism used by WT OxDC is the fact that the Glu162 side chain in the WT OxDC holoenzyme (PDB entry 1UW8)10 occupies a position that would clash sterically with substrate if oxalate binds in the position seen in the Co(II)-substituted ΔE162 OxDC/oxalate complex (Figure 3B). To explore this further, we also made a model of WT OxDC with the oxalate in a “side on” conformation by placing the carboxylate oxygens in the two positions occupied by water in the wild-type structure (Figure 3C). This binding mode of oxalate also forms a steric clash with the Glu162 side chain (distance of 2.1 Å).
Figure 3.

(A) N-Terminal active site in the X-ray crystal structure of the ΔE162 OxDC/oxalate complex. Carbon atoms in the protein and bound oxalate are colored green and cyan, respectively. The Co(II) ion is rendered as a pink sphere. (B) Overlay of the active sites of the Co(II)-substituted ΔE162 OxDC variant (C, green), WT OxDC (C, purple) with the Glu162 side chain in the “closed” orientation (PDB entry 1UW8), and WT OxDC (C, magenta) with the Glu162 side chain in the “open” orientation (PDB entry 1J58). Only the metal from the Co(II)-substituted ΔE162 OxDC variant is shown for the sake of clarity. (C) N-Terminal active site of WT OxDC (C and Mn, purple) with oxalate (C, orange) modeled in a “side on” conformation by placing the carboxylate oxygens in the two positions occupied by waters (blue spheres) in the wild-type structure (PDB entry 1UW8). The steric clash with Glu162 is denoted by an orange line. The oxygen atoms of metal-bound water molecules are rendered as red spheres, and metal–ligand coordinate bonds are shown as black dashed lines in all three images.
Membrane Inlet Mass Spectrometric Characterization of the DASN OxDC Variant
Although the DASN OxDC variant exhibits significantly increased oxidase and decreased decarboxylase activity based on hydrogen peroxide and formate production, respectively, direct measurements of CO2 release under steady-state conditions have not yet been reported. We therefore determined 13CO2 production from uniformly labeled [13C2]oxalate under acidic conditions (pH 4.0) using a continuous MIMS-based assay.19,20 Using the isotopically labeled substrate ensured that dissolved 12CO2 could not affect the rate of increase in the ion current at the m/z 45 (13CO2) peak associated with the decarboxylase and/or oxidase activity of the DASN OxDC variant. Another advantage of using the MIMS was that real-time monitoring of the ion current for the m/z 32 peak allowed concomitant measurement of O2 consumption, thereby permitting a direct determination of reaction stoichiometry as a function of substrate concentration (Figure 4A). The DASN OxDC variant did indeed exhibit significant oxidase activity, but these direct mass spectrometric measurements indicated that the ratio of CO2 production and O2 consumption was not constant across the range of oxalate concentrations used in the experiment (Figure 4B). Thus, the ratio was approximately 2 at low substrate concentrations, as expected for oxalate oxidase activity. At oxalate concentrations of >20 mM, however, the ratio of CO2 production and O2 consumption increased to 3! To verify these MIMS measurements, we independently determined the rates of H2O2 and formate production using standard peroxidase–ABTS26 and FDH17 assays, respectively, under these reaction conditions (Figure 4A). The MIMS-derived Vmax for O2 consumption (4.6 ± 0.6 μmol min–1 mg–1) was comparable with that determined for hydrogen peroxide formation (5.6 ± 0.5 μmol min–1 mg–1). In addition, the Vmax for formate production (1.3 ± 0.1 μmol min–1 mg–1) gave a calculated rate of 12.4 μmol min–1 mg–1 for CO2 formation, based on the stoichiometries of the two activities and assuming that the Vmax for O2 consumption was 5.6 ± 0.5 μmol min–1 mg–1. This value derived from the independent rate measurements compares favorably with the observed MIMS-based rate of 12.5 μmol min–1 mg–1 for CO2 formation (Figure 4A).
Figure 4.

(A) Rates of 13CO2 production (●) and O2 consumption (■) when the DASN OxDC variant is incubated with varying concentrations of [1,2-13C2]oxalate in 50 mM succinate buffer (pH 4.0), as determined by the MIMS-based assay. Independent measurements of the rates of formate (▼) and H2O2 (▲) production under identical reaction conditions were performed using standard assays. (B) Ratio of 13CO2 production and O2 consumption as a function of oxalate concentration at pH 4.0, as calculated from the MIMS-based rate measurements (●). The solid line represents the theoretical stoichiometry calculated from eq 4 (Discussion) assuming that the fraction of enzyme exhibiting oxidase activity is 0.09.
Heavy-Atom Isotope Effect Determinations for the DASN OxDC Variant
We next determined 13(V/K) values for CO2 produced from oxalate in the DASN-catalyzed oxidase reaction. In these experiments, the substrate concentration was maintained at 1 mM to ensure that residual decarboxylase activity exhibited by the DASN OxDC variant would be negligible, based on our MIMS measurements (Figure 4B). The pH dependence of V/K for the DASN variant was obtained on the basis of the initial production rate of H2O2 that was measured by the peroxidase–ABTS assay over a range of 3.5–7.0 (Figure 5) and was similar to that seen for the decarboxylase activity of WT OxDC.17 We therefore performed reactions at pH 4.2 and 5.7, and isotope ratio mass spectrometry (IRMS) was used to determine the 12C/13C ratio in the CO2 produced from oxalate. Under these conditions, the vast majority of CO2 is formed by the oxidase activity of the DASN OxDC variant. Independent assays confirmed this assumption by showing that formate was produced from <3% of initial oxalate. The observed 13(V/K) values for the CO2 produced by the DASN OxDC variant were very similar in magnitude to, albeit slightly smaller than, those reported for the decarboxylase activity of WT OxDC (Table 3). Quantitative analysis using a minimal kinetic mechanism (Scheme 3) up to, and including, decarboxylation to give the putative Mn-bound formyl radical anion (Scheme 2) gave estimates of k5/k4 (5.7) and k3/k2 (0.78) at a solution pH of 4.0 (see the Supporting Information) even though the Glu162 side chain was replaced with alanine in this active-site loop variant. These values are almost identical to those computed from 13(V/K) IE measurements on WT OxDC for which the oxidase activity is only 0.3% of the decarboxylase activity (Table 1). Moreover, the bond orders of the carboxylate undergoing conversion to CO2 in the transition state were very similar to the corresponding values reported for the WT enzyme (see the Supporting Information).
Figure 5.

pH dependence of V/K for the DASN OxDC variant. The experimental data (●) were fitted to log(V/K) = log[C/(1 + K/[H+])]25 (—), which gave a pKa value of 4.6 ± 0.1.
Table 3. 13C Isotope Effects on the Oxidase Activity of the DASN OxDC Varianta.
|
13(V/K) (CO2) |
||
|---|---|---|
| pH (buffer) | WT OxDC | DASN loop variantb |
| 4.2 [1,4-bis(2-hydroxyethyl)piperazine] | 1.005 ± 0.001 | 1.0036 ± 0.0009 |
| 5.7 (piperazine) | 1.008 ± 0.001 | 1.006 ± 0.001 |
Values for WT OxDC have been reported previously and are included here for ease of comparison.17
Discussion
Many details of the catalytic mechanism by which OxDC catalyzes cleavage of the chemically unreactive C–C bond in oxalate remain the subject of speculation. Recent experimental40,41 and theoretical18 work has shown that the reaction proceeds via substrate-based radical intermediates, which are most likely generated by enzyme-bound Mn(III) formed during catalytic turnover.42 Strong evidence, derived primarily from X-ray crystallography,10,27 exists to support the idea that the active site is located in the N-terminal domain of the enzyme, although the C-terminal Mn(II) ion also appears to be required for optimal activity.4 Our observation of oxalate bound solely in the N-terminal domain of the Co(II)-substituted ΔE162 OxDC variant provides additional evidence of the presence of only one active site in the enzyme. Superimposing the Co(II)-substituted ΔE162 OxDC/oxalate complex onto that of WT OxDC also shows that the Glu162 side chain occupies a position in the holoenzyme that would sterically clash with substrate if oxalate binds in the same position in WT OxDC (Figure 3B). This finding therefore raises questions about the “true” orientation of the Glu162 carboxylate in the catalytically competent form of OxDC (the Michaelis complex). Examination of the WT OxDC structure (PDB entry 1UW8) in which the 161SENS164 loop adopts a “closed” conformation reveals that the Glu162 side chain forms a hydrogen bond to a Mn(II)-bound water,10 so the positioning of Glu162 results from this favorable interaction that stabilizes the structure of the active site in the absence of substrate. Support for this idea is provided by the flexibility of the 161SENS164 loop, which for the WT OxDC protein exhibits broken density and high B factors for residues Glu162 (64.24 Å2) and Asn163 (67.18 Å2) compared to the average B factor of 37.95 Å2. The 161SENS164 loop could therefore easily move outward, thereby allowing oxalate to bind at the location seen in the Co(II)-substituted ΔE162 OxDC/oxalate complex, and positioning Glu162 for catalysis. A second possibility is that the absence of Glu162 results in altered oxalate binding compared to that in the catalytically active conformation of the Michaelis complex. We therefore built a model of the active site in which both waters bound to the N-terminal Mn(II) ion in WT OxDC were replaced so that oxalate was coordinated to the metal in a side-on orientation (Figure 3C). Even when the enzyme is bound in this alternate orientation, a steric clash remains between oxalate and the Glu162 side chain in its observed orientation in the X-ray crystal structure of WT OxDC with a distance between O (oxalate) and the carboxylate oxygens of Glu162 of 2.1 Å (Figure 3C). Finally, it is also possible that when oxalate binds to the Mn(II) ion, with either monodentate or bidentate coordination, the Glu162 side chain adopts the conformation seen in the WT OxDC/formate complex (PDB entry 1J58)36 and does not occupy the active site simultaneously with oxalate. Evidence against this possibility, however, comes from V/K IE measurements and kinetic data showing the functional importance of Glu162 for decarboxylase activity.14,16,21 In the absence of additional evidence, we posit that the “end on” conformation of oxalate seen in the Co(II)-substituted ΔE162 OxDC active site does indeed represent the catalytically active orientation. Such a model is consistent with the quantitative interpretation of V/K IE data17,21,22 and also provides an empty coordination site for the dioxygen ligand that is thought to mediate the formation of Mn(III) for catalysis upon substrate binding.41,42 We therefore undertook identification of structural determinants that might enforce this monodentate binding mode within the enzyme active site. Accordingly, cavities within the X-ray crystal structure of WT OxDC in which the 161SENS164 adopts the “closed” conformation (PDB entry 1UW8)10 as well as the Co(II)-substituted ΔE162 OxDC variant were identified with the VOIDOO software package.43 No cavity in the N-terminal domain active site was found in the closed WT OxDC structure, whereas one cavity corresponding exactly in size and shape to the oxalate ligand was observed for the ΔE162 OxDC variant (Figure 6). As a result, we suggest that the location of the Glu162 side chain that is observed when the SENS loop is in a “closed” conformation does not represent the catalytically competent form of the enzyme. This also suggests that the oxalate position observed in the ΔE162 OxDC variant is the catalytically competent position.
Figure 6.
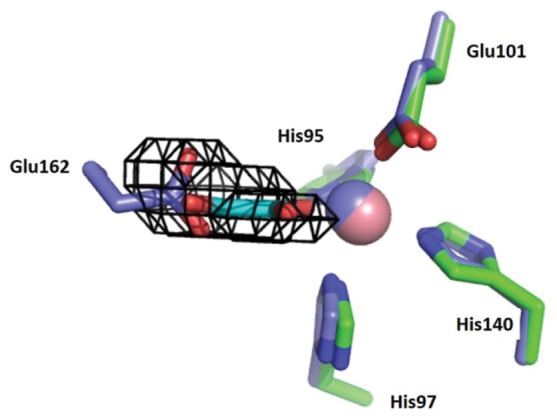
Overlay of the active-site loop “closed” conformer of WT OxDC (PDB entry 1UW8; C, blue; Mn, blue) and the Co(II)-substituted ΔE162 OxDC/oxalate complex (C, green; Co, pink). The VOIDOO cavity (black cages) was calculated for the ΔE162 OxDC/oxalate complex and predicts a cavity that overlays with both oxalate in the ΔE162 OxDC/oxalate complex and the side chain of Glu162 when the active-site loop in WT OxDC adopts a “closed” conformation. The oxalate present in the Co(II)-substituted ΔE162 OxDC variant is colored cyan.
As reported previously,14 the DASN OxDC variant showed substantially increased oxidase activity (Table 1). MIMS-based characterization of the DASN OxDC variant, coupled with a quantitative analysis of IE measurements (Supporting Information), yields new insights into the molecular details underlying the “specificity switch” originally observed by Bornemann and co-workers.14 We were initially surprised by the dependence of CO2/O2 stoichiometry on oxalate concentration (Figure 4B). Assuming that both the decarboxylase and oxidase activities are taking place at the same active site of the enzyme, however, one can write the following expression (eq 4):
| 4 |
where x is the fraction of active sites exhibiting oxidase activity, [S] is the substrate concentration, and {Vmax}OX and {Vmax}DEC are the steady-state Vmax values for the oxidase and decarboxylase activities, respectively. Similarly, {KM}OX and {KM}DEC are the steady-state KM values for the oxidase and decarboxylase activities, respectively. The MIMS-derived stoichiometry at 100 mM was used to determine a value of 0.09 for x, thereby permitting prediction of the CO2/O2 stoichiometry at all oxalate concentrations using eq 4. Indeed, the agreement between the calculated and experimental MIMS-derived values (Figure 4B) is striking. We therefore conclude that oxidase activity is associated with only 9% of the total number of active sites under these reaction conditions, so the overwhelming majority of the DASN OxDC molecules therefore give rise to decarboxylase activity. Moreover, the similarity of the 13(V/K) values for CO2 produced by WT OxDC and the DASN OxDC variant (Table 3) is consistent with the notion that the catalytic steps leading to C–C bond cleavage are identical in the two enzymes, even though the Glu162 side chain is absent. If the DASN OxDC variant were employing a different chemical mechanism for the initial decarboxylation of oxalate, it seems unlikely that there would be an absence of a contribution to the observed IE, so the oxidase activity is likely associated with altered partitioning of the formyl radical anion intermediate as originally proposed by Bornemann and co-workers (Scheme 2).14 Of course, this model also predicts that the Glu162 side chain is not required for the PCET step, leading to an oxalate-based radical intermediate (Scheme 2), as proposed on the basis of heavy-atom IE studies employing the T165V OxDC variant.16
It has also been shown that considerably more of the formyl radical anion intermediate reacts with spin-trapping reagents when oxalate is incubated with the DASN OxDC variant compared with WT OxDC.14 The substantially higher oxidase activity exhibited by the DASN OxDC variant might therefore be a consequence of the loss of formyl radical anion into bulk solvent where it could then undergo reaction with superoxide to form peroxycarbonic acid, which then spontaneously breaks down to give CO2 and hydrogen peroxide at pH 4.2. Of course, it is also possible that spin-trapping reagents have improved access to the active site of the DASN OxDC variant. In this case, the coupling of superoxide [formed by dioxygen-mediated production of Mn(III)42] and formate radical anion would take place on the enzyme with subsequent release of peroxycarbonic acid.
Given that both of these models are consistent with an increased level of access of solvent to the active site, the observed switch in reaction specificity exhibited by the DASN OxDC variant may merely reflect an absence of the negatively charged Glu162 side chain. There is no doubt that replacing Glu162 with aspartate restores decarboxylase activity (Table 1, DDSN and DDNS), showing that a negative charge at this position in the active-site loop is important for how the formyl radical anion intermediate partitions to products. Taken overall, our MIMS and IE data raise the possibility that the function of the Glu162 side chain is to “lock” the mobile 161SENS164 loop into a catalytically competent conformation that precludes solvent access during catalytic turnover.
Acknowledgments
Dr. Stephen Bornemann (John Innes Centre) is thanked for generously providing plasmids containing genes encoding His-tagged, WT B. subtilis OxDC and the DASN OxDC variant. We thank Dr. Alex Angerhofer (Florida) for useful discussions concerning spin-trapping studies of the WT enzyme and selected site-specific variants. We thank Michael E. Pique (The Scripps Research Institute, La Jolla, CA) for providing us with access to the data from the Scripps Metalloprotein Database. Data presented in this publication were collected at beamline X25. Use of the NSLS is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract DE-AC02-98CH10886.
Glossary
Abbreviations
- ABTS
2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonate)
- DASN OxDC
oxalate decarboxylase chimeric variant
- ΔE162 OxDC
oxalate decarboxylase deletion variant
- OxDC
oxalate decarboxylase
- OxOx
oxalate oxidase
- PCET
proton-coupled electron transfer
- WT
wild type
- rmsd
root-mean-square deviation
- MIMS
membrane inlet mass spectrometry
- IE
isotope effect
- FDH
formate dehydrogenase
- PDB
Protein Data Bank.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.6b00043.
An analysis of the IE determinations for the DASN OxDC variant (PDF)
Accession Codes
Coordinates and structure factors for the Co(II)-containing ΔE162 OxDC variant/oxalate complex have been deposited in the PDB as entry 5HI0.
Author Present Address
⊥ N.G.J.R.: School of Chemistry, Cardiff University, Park Place, Cardiff CF10 3AT, U.K.
Author Present Address
@ L.A.R.: U.S. Dairy Forage Research Center, 1925 Linden Dr., Madison, WI 53706.
Author Present Address
# S.E.C.: Department of Chemistry, Massachusetts Institute of Technology, 77 Massachusetts Ave., Cambridge, MA 02139.
Author Contributions
W.Z. and L.M.E. contributed equally to this work.
This work was supported by National Institutes of Health Grants DK061666 (to N.G.J.R.) and GM025154 (to D.N.S.). Partial support was provided through an Undergraduate Research Opportunities Program Grant from Boston University (to S.E.C.).
The authors declare no competing financial interest.
Footnotes
The nomenclature used here refers to the amino acid replacements for the active-site loop residues Ser161-Glu162-Asn163-Ser164 and is based on that previously employed by Burrell et al. (shown here in parentheses).14 Thus, the loop residues are Asp161-Ala162-Ser163-Asn164 in the DASN (SENS161-4DASN) variant, Asp161-Glu162-Ser163-Asn164 in the DESN (S161D/163-4SN) variant, Asp161-Asp162-Ser163-Asn164 in the DDSN (SENS161-4DDSN) variant, Asp161-Asp162-Asn163-Ser164 in the DDNS (SE161-2DD) variant, and Asp161-Glu162-Asn163-Ser164 in the DENS (S161D) variant. Residue numbering throughout this work refers to the oxalate decarboxylase encoded by the YvrK gene in B. subtilis.35
We have demonstrated elsewhere that activity is Mn-dependent.4 Given that WT OxDC and site-specific variants exhibit different levels of incorporation of Mn, the catalytic efficiencies are normalized for the amount of Mn in a given preparation.
Supplementary Material
References
- Svedruzic D.; Jonsson S.; Toyota C. G.; Reinhardt L. A.; Ricagno S.; Lindqvist Y.; Richards N. G. J. (2005) The enzymes of oxalate metabolism: unexpected structures and mechanisms. Arch. Biochem. Biophys. 433, 176–192. 10.1016/j.abb.2004.08.032. [DOI] [PubMed] [Google Scholar]
- Dunwell J. M.; Khuri S.; Gane J. P. (2000) Microbial relatives of the seed storage proteins of higher plants: Conservation of structure and diversification of function during evolution of the cupin superfamily. Microbiol. Mol. Biol. Rev. 64, 153–179. 10.1128/MMBR.64.1.153-179.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uberto R.; Moomaw E. W. (2013) Protein similarity networks reveal relationships among sequence, structure, and function with the cupin superfamily. PLoS One 8, e74477. 10.1371/journal.pone.0074477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moomaw E. W.; Angerhofer A.; Moussatche P.; Ozarowski A.; Garcia-Rubio I.; Richards N. G. J. (2009) Metal dependence of oxalate decarboxylase activity. Biochemistry 48, 6116–6125. 10.1021/bi801856k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner A.; Bowater L.; Fairhurst S. A.; Bornemann S. (2001) Oxalate decarboxylase requires manganese and dioxygen for activity. J. Biol. Chem. 276, 14627–14634. [DOI] [PubMed] [Google Scholar]
- Whittaker M. M.; Pan H.-Y.; Yukl E. T.; Whittaker J. W. (2007) Burst kinetics and redox transformations of the active site manganese ion in oxalate oxidase. J. Biol. Chem. 282, 7011–7023. 10.1074/jbc.M609374200. [DOI] [PubMed] [Google Scholar]
- Furnham N.; Sillitoe I.; Holliday G. L.; Cuff A. L.; Laskowski R. A.; Orengo C. A.; Thornton J. M. (2012) Exploring the evolution of novel enzyme functions within structurally defined protein superfamilies. PLoS Comput. Biol. 8, e1002403. 10.1371/journal.pcbi.1002403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khersonsky O.; Tawfik D. (2010) Enzyme promiscuity: a mechanistic and evolutionary perspective. Annu. Rev. Biochem. 79, 471–505. 10.1146/annurev-biochem-030409-143718. [DOI] [PubMed] [Google Scholar]
- Glasner M. E.; Gerlt J. A.; Babbitt P. C. (2006) Evolution of enzyme superfamilies. Curr. Opin. Chem. Biol. 10, 492–497. 10.1016/j.cbpa.2006.08.012. [DOI] [PubMed] [Google Scholar]
- Just V. J.; Stevenson C. E.; Bowater L.; Tanner A.; Lawson D. M.; Bornemann S. (2004) A closed conformation of Bacillus subtilis oxalate decarboxylase OxDc provides evidence for the true identity of the active site. J. Biol. Chem. 279, 19867–19874. 10.1074/jbc.M313820200. [DOI] [PubMed] [Google Scholar]
- Woo E. J.; Dunwell J. M.; Goodenough P. W.; Marvier A. C.; Pickersgill R. W. (2000) Germin is a manganese containing homohexamer with oxalate oxidase and superoxide dismutase activities. Nat. Struct. Biol. 7, 1036–1040. 10.1038/80954. [DOI] [PubMed] [Google Scholar]
- Escutia M. R.; Bowater L.; Edwards A.; Bottrill A. R.; Burrell M. R.; Polanco R.; Vicuña R.; Bornemann S. (2005) Cloning and sequencing of two Ceriporiopsis subvermispora bicupin oxalate oxidase allelic isoforms: Implications for the reaction specificity of oxalate oxidases and decarboxylases. Appl. Environ. Microbiol. 71, 3608–3616. 10.1128/AEM.71.7.3608-3616.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moussatche P.; Angerhofer A.; Imaram W.; Hoffer E.; Uberto K.; Brooks C.; Bruce C.; Sledge D.; Richards N. G. J.; Moomaw E. W. (2011) Characterization of Ceriporiopsis subvermispora oxalate oxidase expressed in Pichia pastoris. Arch. Biochem. Biophys. 509, 100–107. 10.1016/j.abb.2011.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrell M. R.; Just V. J.; Bowater L.; Fairhurst S. A.; Requena L.; Lawson D. M.; Bornemann S. (2007) Oxalate decarboxylase and oxalate oxidase activities can be interchanged with a specificity switch of up to 282,000 by mutating an active site lid. Biochemistry 46, 12327–12336. 10.1021/bi700947s. [DOI] [PubMed] [Google Scholar]
- Opaleye O.; Rose R. S.; Whittaker M. M.; Woo E. J.; Whittaker J. W.; Pickersgill R. W. (2006) Structural and spectroscopic studies shed light on the mechanism of oxalate oxidase. J. Biol. Chem. 281, 6428–6433. 10.1074/jbc.M510256200. [DOI] [PubMed] [Google Scholar]
- Saylor B. T.; Reinhardt L. A.; Lu Z.; Shukla M. S.; Nguyen L.; Cleland W. W.; Angerhofer A.; Allen K. N.; Richards N. G. J. (2012) A structural element that facilitates proton-coupled electron transfer in oxalate decarboxylase. Biochemistry 51, 2911–2920. 10.1021/bi300001q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhardt L. A.; Svedruzic D.; Chang C. H.; Cleland W. W.; Richards N. G. J. (2003) Heavy atom isotope effects on the reaction catalyzed by the oxalate decarboxylase from Bacillus subtilis. J. Am. Chem. Soc. 125, 1244–1252. 10.1021/ja0286977. [DOI] [PubMed] [Google Scholar]
- Molt R. W. Jr.; Lecher A. M.; Clark T.; Bartlett R. J.; Richards N. G. J. (2015) Facile Csp2-Csp2 bond cleavage in oxalic acid-derived radicals. J. Am. Chem. Soc. 137, 3248–3252. 10.1021/ja510666r. [DOI] [PubMed] [Google Scholar]
- Tu C. K.; Swenson E. R.; Silverman D. N. (2007) Membrane inlet for mass spectrometric measurements of nitric oxide. Free Radical Biol. Med. 43, 1453–1457. 10.1016/j.freeradbiomed.2007.07.026. [DOI] [PubMed] [Google Scholar]
- Moral M. E. G.; Tu C. K.; Richards N. G. J.; Silverman D. N. (2011) Membrane inlet for mass spectrometric measurement of catalysis by enzymatic decarboxylases. Anal. Biochem. 418, 73–77. 10.1016/j.ab.2011.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svedruzic D.; Liu Y.; Reinhardt L. A.; Wroclawska E.; Cleland W. W.; Richards N. G. J. (2007) Investigating the roles of putative active site residues in the oxalate decarboxylase from Bacillus subtilis. Arch. Biochem. Biophys. 464, 36–47. 10.1016/j.abb.2007.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254. 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Campomanes P.; Kellett W. F.; Easthon L. M.; Ozarowski A.; Allen K. N.; Angerhofer A.; Rothlisberger U.; Richards N. G. J. (2014) Assigning the EPR fine structure parameters of the Mn(II) centers in Bacillus subtilis oxalate decarboxylase by site-directed mutagenesis and DFT/MM calculations. J. Am. Chem. Soc. 136, 2313–2323. 10.1021/ja408138f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moral M. E. G.; Tu C. K.; Imaram W.; Angerhofer A.; Silverman D. N.; Richards N. G. J. (2011) Nitric oxide reversibly inhibits Bacillus subtilis oxalate decarboxylase. Chem. Commun. 47, 3111–3113. 10.1039/c0cc04946h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleland W. W. (1979) Statistical analysis of enzyme kinetic data. Methods Enzymol. 63, 103–138. 10.1016/0076-6879(79)63008-2. [DOI] [PubMed] [Google Scholar]
- Requena L.; Bornemann S. (1999) Barley (Hordeum vulgare) oxalate oxidase is a manganese-containing enzyme. Biochem. J. 343, 185–190. 10.1042/bj3430185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Just V. J.; Burrell M. R.; Bowater L.; McRobbie I.; Stevenson C. E.; Lawson D. M.; Bornemann S. (2007) The identity of the active site of oxalate decarboxylase and the importance of the stability of active-site lid conformations. Biochem. J. 407, 397–406. 10.1042/BJ20070708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin A.; Teplyakov A. (2010) Molecular replacement with MOLREP. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 22–25. 10.1107/S0907444909042589. [DOI] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674. 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkoczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L. W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 213–221. 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 60, 2126–2132. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Northrop D. B. (1977) Determining the absolute magnitude of hydrogen isotope effects. In Isotope Effects on Enzyme-Catalyzed Reactions (Cleland W. W., O’Leary W. H., Northrop D. B., Eds.) pp 122–152, University Park Press, Baltimore. [Google Scholar]
- O’Leary M. H. (1980) Determination of heavy atom isotope effects on enzyme-catalyzed reactions. Methods Enzymol. 64, 83–104. 10.1016/S0076-6879(80)64006-3. [DOI] [PubMed] [Google Scholar]
- Weiss P. M. (1991) Heavy-atom isotope effects using the isotope-ratio mass spectrometer. In Enzyme Mechanism from Isotope Effects (Cook P. F., Ed.) pp 292–311, CRC Press, Boca Raton, FL. [Google Scholar]
- Tanner A.; Bornemann S. (2000) Bacillus subtilis YvrK is an acid-induced oxalate decarboxylase. J. Bacteriol. 182, 5271–5273. 10.1128/JB.182.18.5271-5273.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand R.; Dorrestein P. C.; Kinsland C.; Begley T. P.; Ealick S. E. (2002) Structure of oxalate decarboxylase from Bacillus subtilis at 1.75 Å resolution. Biochemistry 41, 7659–7669. 10.1021/bi0200965. [DOI] [PubMed] [Google Scholar]
- Karmakar T.; Periyasamy G.; Balasubramanian S. (2013) CO2 migration pathways in oxalate decarboxylase and clues about its active site. J. Phys. Chem. B 117, 12451–12460. 10.1021/jp4074834. [DOI] [PubMed] [Google Scholar]
- Castagnetto J. M.; Hennesey S. W.; Roberts V. A.; Getzoff E. D.; Tainer J. A.; Pique M. E. (2002) MDB: The metalloprotein database and browser at the Scripps Research Institute. Nucleic Acids Res. 30, 379–382. 10.1093/nar/30.1.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzenbacher R.; von Delft F.; Jaroszewski L.; Abdubek P.; Ambing E.; Biorac T.; Brinen L. S.; Canaves J. M.; Cambell J.; Chiu H.-J.; Dai X.; Deacon A. M.; DiDonato M.; Elsliger M.-A.; Eshagi S.; Floyd R.; Godzik A.; Grittini C.; Grzechnik S. K.; Hampton E.; Karlak C.; Klock H. E.; Koesema E.; Kovarik J. S.; Kreusch A.; Kuhn P.; Lesley S. A.; Levin I.; McMullan D.; McPhillips T. M.; Miller M. D.; Morse A.; Moy K.; Ouyang J.; Page R.; Quijano K.; Robb A.; Spraggon G.; Stevens R. C.; van den Bedem H.; Velasquez J.; Vincent J.; Wang X.; West B.; Wolf G.; Xu Q.; Hodgson K. O.; Wooley J.; Wilson I. A. (2004) Crystal structure of a putative oxalate decarboxylase (TM1287) from Thermotoga maritima at 1.95 Å resolution. Proteins: Struct., Funct., Genet. 56, 392–395. 10.1002/prot.20016. [DOI] [PubMed] [Google Scholar]
- Imaram W.; Saylor B. T.; Centonze C.; Richards N. G. J.; Angerhofer A. (2011) EPR spin trapping of an oxalate-derived free radical in the oxalate decarboxylase reaction. Free Radical Biol. Med. 50, 1009–1015. 10.1016/j.freeradbiomed.2011.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twahir U. T.; Stedwell C. N.; Lee C. T.; Richards N. G. J.; Polfer N. C.; Angerhofer A. (2015) Observation of superoxide production during catalysis of Bacillus subtilis oxalate decarboxylase at pH 4. Free Radical Biol. Med. 80, 59–66. 10.1016/j.freeradbiomed.2014.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W.; Wilcoxen J.; Britt R. D.; Richards N. G. J. (2016) Formation of hexacoordinate Mn(III) in Bacillus subtilis oxalate decarboxylase requires catalytic turnover. Biochemistry 55, 429–434. 10.1021/acs.biochem.5b01340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleywegt G. J.; Jones T. A. (1994) Detection, delineation, measurement and display of cavities in macromolecular structures. Acta Crystallogr., Sect. D: Biol. Crystallogr. 50, 178–185. 10.1107/S0907444993011333. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.