

Abstract
The incretin hormones, glucose‐dependent insulinotropic peptide and glucagon‐like peptide‐1, are secreted from intestinal K‐ and L cells, respectively, with the former being most abundant in the proximal small intestine, whereas the latter increase in number towards the distal gut. Although an overlap between K‐ and L cells can be observed immunohistochemically or in murine models expressing fluorescent markers under the control of the two hormone promoters, the majority (>80%) of labeled cells seems to produce only one of these hormones. Transcriptomic analysis showed a close relationship between small intestinal K‐ and L cells, and glucose sensing mechanisms appear similar in both cell types with a predominant role of electrogenic glucose uptake through sodium‐coupled glucose transporter 1. Similarly, both cell types produce the long‐chain fatty acid sensing G‐protein‐coupled receptors, FFAR1 (GPR40) and FFAR4 (GPR120), but differ in the expression/functionality of other lipid sensing receptors. GPR119 and FFAR2/3, for example, have clearly documented roles in glucagon‐like peptide‐1 secretion, whereas agonists for the endocannabinoid receptor type 1 have been found to show largely selective inhibition of glucose‐dependent insulinotropic peptide secretion. In conclusion, although K‐ and L cell populations overlap and share key molecular nutrient‐sensing mechanisms, subtle differences between the responsiveness of the different cell types might be exploited to differentially modulate glucose‐dependent insulinotropic peptide or glucagon‐like peptide‐1 secretion.
Keywords: Gastric inhibitory polypeptide/glucose‐dependent insulinotropic peptide, Glucagon‐like peptide‐1, Secretion
Introduction
Glucose‐dependent insulinotropic polypeptide (GIP) and glucagon‐like peptide‐1 (GLP‐1) are gut hormones secreted from specialized enteroendocrine cells within the intestinal epithelium. They are released postprandially and act as circulating markers of food consumption, enabling the body to respond appropriately to food‐derived elevations of blood nutrient concentrations. This is crucial for the control of blood glucose concentrations, as costimulation of pancreatic β‐cells by GIP and GLP‐1 approximately doubles the amount of insulin released in response to an elevation in ambient (blood) glucose concentrations. GLP‐1 and GIP are hence often termed ‘incretins,’ and underlie the ‘incretin effect’ – a well‐documented observation that intravenous glucose infusion at a rate that simulates postprandial blood glucose levels triggers only about half as much insulin release as a matched oral glucose challenge. After the discovery that the insulinotropic effect of GLP‐1 is preserved in most people with type 2 diabetes1, GLP‐1 mimetics and inhibitors of GLP‐1 degradation by dipeptidyl peptidase 4 have been developed and licensed for the treatment of type 2 diabetes2.
Although a wealth of evidence supports the idea that both GLP‐1 and GIP underlie the incretin effect, there are important differences in the activity and plasma profiles of the two hormones. GIP, for example, stimulates glucagon secretion from pancreatic α‐cells, whereas GLP‐1 inhibits α‐cell activity. GLP‐1, in contrast, has anorexigenic properties, whereas GIP seems to have no effects on food intake. GIP is instead considered pro‐adipogenic, based on several observations, including that knockout of the GIP‐receptor3 or immunoneutralization of circulating GIP4 are protective against diet‐induced obesity in rodents. As the incretin action of GIP seems to be impaired in patients with type 2 diabetes1, it might be postulated that a stimulation of GLP‐1 and an inhibition of GIP secretion could be a therapeutic objective in overweight patients with type 2 diabetes. Further understanding the differences between these two hormones should enable a more targeted approach to their exploitation for the treatment of diabetes and obesity. The present review will focus on the physiology of the enteroendocrine cells secreting these two hormones, the GIP‐expressing K cells and the proglucagon/GLP‐1‐expressing L cells.
Location: overlapping but distinctive populations of K‐ and L cells
GIP‐containing cells are found at highest density in the duodenum in a number of species5, 6, 7. GLP‐1‐containing cells can also be found in the proximal small intestine, but increase in number towards the distal small intestine, and in contrast to K cells are also numerous in the colon and rectum (Figure 1). Based on immunohistochemical staining, so‐called LK cells have been described, which were immune‐positive for both GIP and GLP‐18, 9. In transgenic mice expressing fluorescent markers under the control of either the GIP or the proglucagon promoter, cells producing the ‘other’ incretin have also been observed; that is, GIP in cells labeled by the proglucagon promoter, and GLP‐1 in cells labeled by the GIP promoter10; however, these double‐positive cells only amounted to ~10–20% of the total K‐ and L cell population. Overall secretory responses from cell populations in vivo or in vitro are thus likely to be dominated by the remaining ~80% of single‐positive cells that produced only GLP‐1 or GIP6, and as we describe below, selective stimulation or inhibition of either GIP or GLP‐1 secretion is therefore possible. Nevertheless, the observation that K‐ and L cells additionally produce other hormones, such as CCK10, challenges the traditional classification of enteroendocrine cells according to their expression of one (or sometimes two) specific hormones, and suggests a more plastic expression profile that could be affected by external factors, such as the recent exposure of the intestine to nutrients and other luminal stimulants. Given the relatively rapid turnover of enteroendocrine cells in the small intestine every ~5 days11, it seems plausible that recent nutritional availability could result in changes to the overall enteroendocrine cell population within days or weeks. In a recent study to identify the effects of a high‐fat diet on mouse L cells, however, we observed a general downregulation of many enteroendocrine cell‐specific genes rather than a switch to the preferential production of an alternative hormone12.
Figure 1.
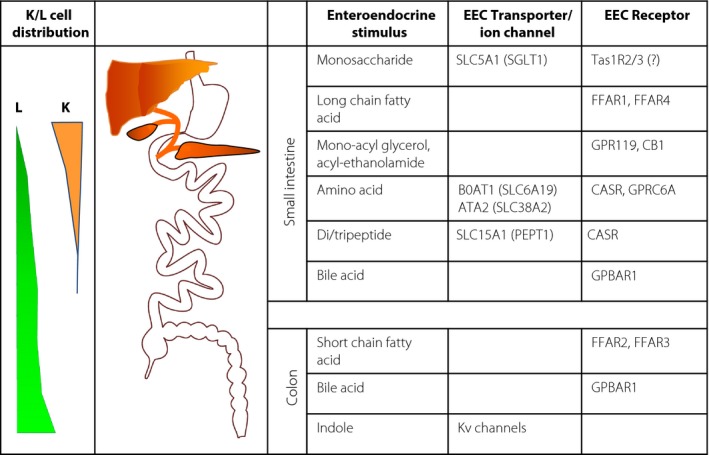
L‐ and K cell distribution and stimulus detection machinery. The majority of K cells are more proximally located than L cells. Fasting and postprandial glucose‐dependent insulinotropic polypeptide (K cells) and glucagon‐like peptide‐1 (L cells) secretion likely reflect the dynamic gradient of different intestinal stimuli along the gut. Physiological activation of the L‐ and K cell detection pathways is shown, involving transporters/ion channels (linked to altered cellular electrical activity) and G‐protein‐coupled receptors, differing between the small intestine and colon. ATA2, amino acid transporter A2 (solute carrier [Slc] Slc38A2); BOAT1, system B(0) neutral amino acid transporter AT1 (Slc6A19); CASR, calcium‐sensing receptor; CB1, cannabinoid receptor 1; EEC, enteroendocrine cell; FFAR, free fatty‐acid receptor; GPBAR1, G‐protein coupled bile‐acid receptor 1; GPR119, G‐protein coupled receptor 119; GPRC6A, G‐protein coupled receptor classC 6A; Kv‐channels, voltage gated potassium channels; SGLT1, sodium‐coupled glucose transporter 1 (Slc5A1); Tas1R, taste receptor type 1.
Glucose sensing: similar mechanisms operate in both K‐ and L cells
Given the importance of both GIP and GLP‐1 for the incretin effect, one of the most investigated secretory stimuli of gut hormone secretion is glucose. Both K‐ and L cells in mixed primary intestinal epithelial cultures failed to respond to glucose when the sodium‐coupled glucose transporter 1 (SGLT1) was inhibited either pharmacologically or genetically13, 14, 15, 16. Indeed, a wealth of in vitro and in vivo data have suggested that the rapid elevations in plasma GIP and GLP‐1 concentrations after glucose ingestion are directly linked to the electrogenic uptake of glucose by K‐ and L cells, resulting in membrane depolarization, voltage‐gated calcium entry and enhanced rates of vesicular exocytosis17. More extensive phenotyping of global SGLT1 knockout mice, however, showed differences between the release patterns of GIP and GLP‐1, which are likely related to the different locations of K‐ and L cells along the gastrointestinal tract axis18. In this mouse model, the GIP response to an oral glucose tolerance test was abolished, consistent with the proposed SGLT1‐dependent coupling of glucose absorption to GIP secretion in K cells. By contrast, whereas the early GLP‐1 response ~5 min after a glucose gavage was abrogated in SLGT1 knockout mice or in mice treated with an SGLT1 inhibitor13, 18, elevated plasma GLP‐1 concentrations were observed at later time‐points18. The findings support the idea that the rapid secretion of GLP‐1 and GIP from the proximal small intestine after a glucose load is linked to SGLT1‐dependent glucose absorption, but suggest that alternative sensory mechanisms operate in the distal gut. Inhibition of glucose absorption in the upper gastrointestinal tract in SGLT1 knockout mice results in a dramatic increase in glucose delivery to the distal gut with its higher density of L cells18, likely underpinning the delayed elevation of GLP‐1 levels in these mice. The mechanism by which this glucose load is sensed by the distal ileum and/or colon remains unclear. Candidate pathways include the bacterial fermentation of distally‐delivered carbohydrate to metabolites, such as short chain fatty acids that are then sensed by G‐protein‐coupled receptors, such as GPR4319, or metabolism of the sugar within L cells resulting in the activation of an alternative downstream signaling pathway. Neither of these hypotheses has yet been validated experimentally.
Whereas the SGLT1‐dependent pathway is common to K‐ and L cells, we observed some differences between the responsiveness of the glucose‐sensing machinery underlying GIP and GLP‐1 secretion from small intestinal primary murine epithelial cultures. Whereas α‐methyl‐glucopyranoside, a non‐metabolizable SGLT1 substrate, enhanced GLP‐1 secretion in the absence of other stimuli16, it only became an effective GIP secretagogue at elevated cyclic adenosine monophosphate (cAMP) levels14. Interestingly, the responsiveness to tolbutamide, an inhibitor of adenosine triphosphate‐sensitive potassium (KATP) channels, showed the reverse dependence, triggering GIP responses in the absence, but not presence, of the cAMP‐raising agents, forskolin and 3‐isobutyl‐1‐methylxanthine (IBMX)14. KATP channel closure has itself been postulated as a mechanism underlying K‐ and L cell glucose sensing, similar to its well‐established role in mediating glucose‐dependent insulin secretion from pancreatic β‐cells. In support of this idea, KATP channel inhibition enhanced GIP and GLP‐1 secretion from intestinal cultures, triggered GLP‐1 secretion from the enteroendocrine cell line, GLUTag20, and increased GLP‐1 release from perfused rat intestine21. However, there is little data supporting the idea that KATP channel closure triggers gut hormone secretion in an intact animal. Mice lacking the KATP channel subunit Kir6.2 did not show impaired glucose‐triggered gut hormone secretion22, 23; and in humans, the KATP channel inhibitor, glibenclamide, had no effects on GIP and GLP‐1 concentrations before or during an oral glucose tolerance test24. KATP‐channel closure has, however, been suggested to underlie fructose‐stimulated GLP‐1 secretion25. Fructose is not a substrate for sodium‐coupled monosaccharide transport, and instead enters cells through the facilitative transporter GLUT5. Interestingly, fructose does not stimulate GIP secretion in healthy rodents and humans25, but GIP release in response to fructose has been reported in diabetic mouse models22, 26. However, opening of KATP‐channels with diazoxide did not abolish fructose‐stimulated GIP secretion in diabetic mice, and even the GLP‐1 response to oral fructose remained intact in mice lacking the KATP channel subunit, Kir6.223. In view of the widespread agreement that KATP channels are expressed and functional in K‐ and L cells, the results unanimously support the idea that the resting KATP conductance is already very small in native K‐ and L cells in vivo, and that any differences observed between in vivo and in vitro experiments might reflect differences in the metabolic status of the enteroendocrine cells under the different conditions.
Another physiological glucose‐sensing mechanism involves the G‐protein‐coupled receptor heterodimer of TAS1R2 and TAS1R3, that underlies sweet taste sensation in the tongue. Impaired postprandial GLP‐1 levels have been observed in mice lacking α‐gustducin, a component of the downstream taste receptor signaling pathway27, and both GIP and GLP‐1 secretion have been reported from the GLUTag cell line in response to high concentrations of artificial sweeteners28. We, however, were unable to show a role for TAS1R2/3 in hormone secretion from murine K‐ or L cells in primary culture14, 16, and artificial sweeteners failed to elevate plasma incretin hormone levels in human volunteers29. Other studies also failed to show artificial sweetener‐stimulated GIP secretion in mice in vivo 22 or GLP‐1 secretion in a perfused rat intestinal preparation21, questioning the importance of TASR1R2/3 in incretin secreting cells.
G‐protein‐coupled receptors: candidates for selectively targeting K‐ and L cells
The development of transgenic mice with fluorescently tagged K‐ or L cells has enabled the transcriptomic analysis of these different cell types. Similarities and differences between K‐ and L cell populations were observed for the expression of a number of G‐protein‐coupled receptors. Both K‐ and L cell populations, for example, were found to express messenger ribonucleic acids (mRNAs) encoding the free‐fatty acid receptors, FFAR1 (GPR40) and FFAR4 (GPR120)14, 16. GPR40 activation has, for example, been linked to the stimulation of GLP‐1 secretion in experiments using Gpr40 knockout mice30, as well as by the application of GPR40 agonists to the perfused rat intestine31. Recent data, by contrast, convincingly showed a reduction in lard‐triggered GIP‐responses in mice lacking Gpr120 32. In primary intestinal cultures from mice lacking either Gpr40 or Gpr120, however, we observed diminished secretion of both GIP and GLP‐1 in response to oleate (Reimann and Gribble unpublished observation). Thus, although the data linking GPR120 activation to GIP secretion and GPR40 activation to GLP‐1 release are robust, it is currently unclear whether either of these receptors plays a relatively greater role in any particular enteroendocrine cell type.
Other lipid‐derived stimulants, such as mono‐acyl‐glycerol, activate GPR119, mRNA for which is expressed in both K‐ and L cells14, 16. Whereas a small molecule GPR119 agonist elevated GLP‐1 and GIP concentrations in mice33, however, the natural GPR119 ligand oleoylethanolamide (OEA) was not a good stimulus of GIP‐secretion from primary epithelial cultures14. GLP‐1 secretion, by contrast, was stimulated by OEA from both small and large intestinal‐derived murine cultures34. In the colon, this was mediated by GPR119, as shown by the loss of OEA‐triggered GLP‐1 release in colonic cultures from mice lacking GPR119 specifically in proglucagon expressing cells. Similar experiments carried out in small intestinal cultures from these mice, however, showed that the OEA‐triggered GLP‐1 secretory response in the upper gastrointestinal tract was not GPR119‐dependent34, suggesting that OEA recruits alternative mechanisms in duodenal/jejunal L cells, and that this pathway might not be sufficiently active in K cells to trigger GIP secretion.
The related compounds, mono‐arachidonoyl‐glycerol and arachidonoylethanolamine (anandamide), are agonists for the endocannabinoid receptor, CB1 (encoded by the gene Cnr1), which is predominantly Gi‐coupled. Cnr1 is highly expressed in small intestinal K‐ and L cells, with a tendency for higher expression in K‐ than L cells, but was not detected in L cells from the colon35. Consistent with the known Gi coupling of CB1, anandamide was shown to inhibit IBMX‐triggered GIP secretion in vivo, an effect that was blocked by the CB1‐antagonist AM251. Interestingly, GLP‐1 secretion from the same cultures was not affected by anandamide, despite the relatively high expression of Cnr1 in small intestinal L cells. Pretreatment of mice with anandamide delayed GIP, but not GLP‐1 responses to an oral glucose tolerance test35. These findings raise questions about why the same receptor is apparently more effectively coupled to inhibition of secretion in K‐ than L cells, despite relatively high expression levels in both.
Ligands for other predominantly Gi‐coupled receptors expressed in both K‐ and L cells, by contrast, tend to have similar effects on both GIP and GLP‐1 secretion. Somatostatin strongly suppressed the elevation of cAMP triggered by forskolin and/or IBMX in both cell types, and the effect was at least partly mediated through SSTR535. The most likely sources of somatostatin are nearby intestinal D cells, this being an example of how paracrine signals can integrate responses to luminal signals within the epithelium. Galanin, most likely secreted from enteric neurons, similarly recruits GALR1 in both K‐ and L cells, resulting in a Gi‐dependent suppression of cAMP levels36, and exemplifying that enteroendocrine cells likely integrate responses to luminal nutrients with signals arriving through the enteric nervous system. Gi‐coupled receptors, such as GALR1, have been reported to inhibit electrically excitable cells through activation of G‐protein activated inwardly rectifying potassium (GIRK) channels, mediated by the G‐protein βγ‐subunit. Interestingly, whereas both K‐ and L cells showed enriched expression of mRNAs encoding GIRK channels, and although GIRK‐inhibition had no effect on the ability of galanin to inhibit GLP‐1 secretion, only GLP‐1, but not GIP, secretion could be inhibited by co‐application of the GIRK‐activator, ML297, with IBMX or glucose36. This might point to a difference in the role of potassium conductances in the stimulus secretion coupling of K‐ and L cells, but further work assessing possible differences in the electrical activity of these enteroendocrine cell types and its relationship to hormone secretion is required.
Given the calcium dependence of a number of proteins involved in the exocytotic pathway, predominantly Gq‐coupled receptors should be good targets to stimulate incretin secretion. The similar expression levels in K‐ and L cells of mRNAs encoding the Gq‐coupled receptor, FFAR1, have been mentioned above. The related short‐chain fatty acid receptor FFAR2 (GPR43) by contrast seems more abundant in L‐ than K cells (Affymetrix chip array probe 1425216 RMA‐values are 2,143 for fluorescently tagged L‐ and 154 for fluorescently tagged K cells isolated from the small intestine, with the latter value being similar to values observed for the non‐fluorescent control cells; Reimann and Gribble unpubl.), supporting the reported importance of GPR43 in short‐chain fatty acid stimulated GLP‐1 secretion19. However, whereas fluorescent reporter mice for FFAR2 had only a few labeled enteroendocrine cells, reporter mice for the other short chain fatty acid receptor FFAR3 (GPR41) showed strong labeling of a number of enteroendocrine cells in the small intestine, including K‐ and L cells37. A recent publication reported a blunting of GLP‐1, but not GIP, secretion in response to orally‐administered butyrate in Ffar3 knockout mice38, and further work will be required to clarify the relative roles of these receptors in incretin‐secreting cells. Another Gq‐coupled receptor, presumably underlying modulation of enteroendocrine secretion by the enteric nervous system, as it is activated by neuromedin C and gastrin‐releasing peptide, is the bombesin receptor 2. Bombesin receptor 2 mRNA was found to be selectively enriched in L cells, but not K cells, and, consistent with this finding, bombesin increased calcium concentrations in L cells, but not K cells, and triggered GLP‐1, but not GIP, secretion from primary small epithelial cultures as well as in a perfused intestinal preparation39.
Whether any of the predominantly Gq‐coupled receptors will be good targets for selective stimulation of GLP‐1 secretion in vivo will have to await further research. It should be noted, however, that a recent report of a so‐called FFAR1 ‘superagonist,’ capable of triggering robust GLP‐1 secretory responses, stresses the importance of the dual action of such compounds in recruiting both Gq‐ and Gs‐coupled pathways40. This latter notion is supported by our observation that raising intracellular cAMP levels with forskolin and IBMX boosts GIP and GLP‐1 secretory responses to a number of agents that elevate enteroendocrine cell cytosolic Ca2+ concentrations, in primary epithelial cultures17. A predominantly Gs‐coupled receptor expressed in L cells, but not K cells, is the melanocortin receptor 4, and increased GLP‐1 and PYY secretion in response to agonists has been shown41, although currently the nature or source of the physiological ligand for this receptor on L cells is uncertain. Other predominantly Gs‐coupled receptors, expression of which is enriched in L cells, include GPR119 (see above) and the bile acid‐sensitive receptor, GPBAR1 (TGR5). Although there seems to be some expression of Gpbar1 in K cells, it is not enriched compared with the surrounding cells (Reimann and Gribble unpublished observation), making GPBAR1 one of the more promising receptors to ‘selectively’ stimulate GLP‐1 secretion. However, we recently showed that bile acids need to access the basolateral rather than the apical/luminal surface of L cells to stimulate GLP‐1 secretion through GPBAR142, and a similar observation was reported for agonists of GPR4031. As melanocortin receptor 4 was also located to the basolateral side41, the interesting question arises if any GPCRs directly sample the luminal contents. If all L cell GPCRs are instead located on the basolateral membrane, the potential hope to develop agents with limited systemic bioavailability to avoid off‐target side‐effects as a result of action on other cells expressing the receptors in question would be unfounded.
Conclusion
Although the success of GLP‐1 analogs/mimetics in the treatment of type 2 diabetes and the correlation of strongly elevated postprandial GLP‐1 levels after Roux‐Y gastric bypass surgery43 strongly suggests benefits of recruiting endogenous GLP‐1 reserves as a not yet exploited treatment alternative, the situation for GIP is less clear. Arguments have been put forward for developing both GIP‐receptor agonists and antagonists44. The differences observed in murine K‐ and L cells suggest that it is possible to elevate one incretin preferentially by external stimuli, but further work will be required before translation into a clinical therapy.
Disclosure
The authors declare no conflict of interest.
Acknowledgments
Research in the Reimann and Gribble laboratories is currently funded by the Wellcome Trust (grants 106262/Z/14/Z and 106263/Z/14/Z), Full4Health (grants FP7/2011‐2015 no :266408) and the Medical Research Council (MRC; grant MRC_ MC_UU_12012/3).
J Diabetes Investig 2016; 7: 13–19
This article is based on the presentations given by the authors at a symposium, Incretin 2015, July 29‐31, 2015, Vancouver, BC Canada.
References
- 1. Nauck MA, Heimesaat MM, Orskov C, et al Preserved incretin activity of glucagon‐like peptide 1 [7‐36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type‐2 diabetes mellitus. J Clin Invest 1993; 91: 301–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Drucker DJ, Nauck MA. The incretin system: glucagon‐like peptide‐1 receptor agonists and dipeptidyl peptidase‐4 inhibitors in type 2 diabetes. Lancet 2006; 368: 1696–1705. [DOI] [PubMed] [Google Scholar]
- 3. Miyawaki K, Yamada Y, Ban N, et al Inhibition of gastric inhibitory polypeptide signaling prevents obesity. Nat Med 2002; 8: 738–742. [DOI] [PubMed] [Google Scholar]
- 4. Fulurija A, Lutz TA, Sladko K, et al Vaccination against GIP for the treatment of obesity. PLoS ONE 2008; 3: e3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cho HJ, Kosari S, Hunne B, et al Differences in hormone localisation patterns of K and L type enteroendocrine cells in the mouse and pig small intestine and colon. Cell Tissue Res 2015; 359: 693–698. [DOI] [PubMed] [Google Scholar]
- 6. Svendsen B, Pedersen J, Albrechtsen NJ, et al An analysis of cosecretion and coexpression of gut hormones from male rat proximal and distal small intestine. Endocrinology 2015; 156: 847–857. [DOI] [PubMed] [Google Scholar]
- 7. Sjölund K, Sandén G, Håkanson R, et al Endocrine cells in human intestine: an immunocytochemical study. Gastroenterology 1983; 85: 1120–1130. [PubMed] [Google Scholar]
- 8. Mortensen K, Christensen LL, Holst JJ, et al GLP‐1 and GIP are colocalized in a subset of endocrine cells in the small intestine. Regul Pept 2003; 114: 189–196. [DOI] [PubMed] [Google Scholar]
- 9. Theodorakis MJ, Carlson O, Michopoulos S, et al Human duodenal enteroendocrine cells: source of both incretin peptides, GLP‐1 and GIP. Am J Physiol Endocrinol Metab 2006; 290: E550–E559. [DOI] [PubMed] [Google Scholar]
- 10. Habib AM, Richards P, Cairns LS, et al Overlap of endocrine hormone expression in the mouse intestine revealed by transcriptional profiling and flow cytometry. Endocrinology 2012; 153: 3054–3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cheng H, Leblond CP. Origin, differentiation and renewal of the four main epithelial cell types in the mouse small intestine. III. Entero‐endocrine cells. Am J Anat 1974; 141: 503–519. [DOI] [PubMed] [Google Scholar]
- 12. Richards P, Pais R, Habib AM, et al High fat diet impairs the function of glucagon‐like peptide‐1 producing L‐cells. Peptides 2015; 3. pii: S0196‐9781(15)00189‐8. doi: 10.1016/j.peptides.2015.06.006. [Epub ahead of print]. PMID:26145551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gorboulev V, Schürmann A, Vallon V, et al Na(+)‐D‐glucose cotransporter SGLT1 is pivotal for intestinal glucose absorption and glucose‐dependent incretin secretion. Diabetes 2012; 61: 187–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Parker HE, Habib AM, Rogers GJ, et al Nutrient‐dependent secretion of glucose‐dependent insulinotropic polypeptide from primary murine K cells. Diabetologia 2009; 52: 289–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Parker HE, Adriaenssens A, Rogers G, et al Predominant role of active versus facilitative glucose transport for glucagon‐like peptide‐1 secretion. Diabetologia 2012; 55: 2445–2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Reimann F, Habib AM, Tolhurst G, et al Glucose sensing in L cells: a primary cell study. Cell Metab 2008; 8: 532–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Psichas A, Reimann F, Gribble FM. Gut chemosensing mechanisms. J Clin Invest 2015; 125: 908–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Powell DR, Smith M, Greer J, et al LX4211 increases serum glucagon‐like peptide 1 and peptide YY levels by reducing sodium/glucose cotransporter 1 (SGLT1)‐mediated absorption of intestinal glucose. J Pharmacol Exp Ther 2013; 345: 250–259. [DOI] [PubMed] [Google Scholar]
- 19. Tolhurst G, Heffron H, Lam YS, et al Short‐Chain Fatty Acids Stimulate Glucagon‐Like Peptide‐1 Secretion via the G‐Protein‐Coupled Receptor FFAR2. Diabetes 2012; 61: 364–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Reimann F, Gribble FM. Glucose‐sensing in glucagon‐like peptide‐1‐secreting cells. Diabetes 2002; 51: 2757–2763. [DOI] [PubMed] [Google Scholar]
- 21. Kuhre RE, Frost CR, Svendsen B, et al Molecular mechanisms of glucose‐stimulated GLP‐1 secretion from perfused rat small intestine. Diabetes 2015; 64: 370–382. [DOI] [PubMed] [Google Scholar]
- 22. Ogata H, Seino Y, Harada N, et al KATP channel as well as SGLT1 participates in GIP secretion in the diabetic state. J Endocrinol 2014; 222: 191–200. [DOI] [PubMed] [Google Scholar]
- 23. Seino Y, Ogata H, Maekawa R, et al Fructose induces glucose‐dependent insulinotropic polypeptide, glucagon‐like peptide‐1 and insulin secretion: role of adenosine triphosphate‐sensitive K(+) channels. J Diabetes Investig 2015; 6: 522–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. El‐Ouaghlidi A, Rehring E, Holst JJ, et al The dipeptidyl peptidase 4 inhibitor vildagliptin does not accentuate glibenclamide‐induced hypoglycemia but reduces glucose‐induced glucagon‐like peptide 1 and gastric inhibitory polypeptide secretion. J Clin Endocrinol Metab 2007; 92: 4165–4171. [DOI] [PubMed] [Google Scholar]
- 25. Kuhre RE, Gribble FM, Hartmann B, et al Fructose stimulates GLP‐1 but not GIP secretion in mice, rats, and humans. Am J Physiol Gastrointest Liver Physiol 2014; 306: G622–G630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Flatt PR, Kwasowski P, Bailey CJ. Stimulation of gastric inhibitory polypeptide release in ob/ob mice by oral administration of sugars and their analogues. J Nutr 1989; 119: 1300–1303. [DOI] [PubMed] [Google Scholar]
- 27. Jang HJ, Kokrashvili Z, Theodorakis MJ, et al Gut‐expressed gustducin and taste receptors regulate secretion of glucagon‐like peptide‐1. Proc Natl Acad Sci U S A 2007; 104: 15069–15074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Margolskee RF, Dyer J, Kokrashvili Z, et al T1R3 and gustducin in gut sense sugars to regulate expression of Na+‐glucose cotransporter 1. Proc Natl Acad Sci U S A. 2007; 104: 15075–15080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ma J, Bellon M, Wishart JM, et al Effect of the artificial sweetener, sucralose, on gastric emptying and incretin hormone release in healthy subjects. Am J Physiol Gastrointest Liver Physiol 2009; 296: G735–G739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Edfalk S, Steneberg P, Edlund H. Gpr40 is expressed in enteroendocrine cells and mediates free fatty acid stimulation of incretin secretion. Diabetes 2008; 57: 2280–2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Christensen LW, Kuhre RE, Janus C, et al Vascular, but not luminal, activation of FFAR1 (GPR40) stimulates GLP‐1 secretion from isolated perfused rat small intestine. Physiol Rep 2015; 3(9): pii: e12551. doi: 10.14814/phy2.12551. PMID:26381015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Iwasaki K, Harada N, Sasaki K, et al Free fatty acid receptor GPR120 is highly expressed in enteroendocrine K cells of the upper small intestine and has a critical role in GIP secretion after fat ingestion. Endocrinology 2015; 156: 837–846. [DOI] [PubMed] [Google Scholar]
- 33. Chu ZL, Carroll C, Alfonso J, et al A role for intestinal endocrine cell‐expressed g protein‐coupled receptor 119 in glycemic control by enhancing glucagon‐like Peptide‐1 and glucose‐dependent insulinotropic Peptide release. Endocrinology 2008; 149: 2038–2047. [DOI] [PubMed] [Google Scholar]
- 34. Moss CE, Glass LL, Diakogiannaki E, et al Lipid derivatives activate GPR119 and trigger GLP‐1 secretion in primary murine L‐cells. Peptides 2015; 2. pii: S0196‐9781(15)00195‐3. doi: 10.1016/j.peptides.2015.06.012. [Epub ahead of print]. PMID:26144594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Moss CE, Marsh WJ, Parker HE, et al Somatostatin receptor 5 and cannabinoid receptor 1 activation inhibit secretion of glucose‐dependent insulinotropic polypeptide from intestinal K cells in rodents. Diabetologia 2012; 55: 3094–3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Psichas P, Glass LL, Reimann F, et al Galanin inhibits GLP‐1 and GIP secretion via the GAL1 receptor in enteroendocrine L and K cells. Br J Pharmacol 2015; 12. doi: 10.1111/bph.13407. [Epub ahead of print]. PMID: 26661062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nøhr MK, Pedersen MH, Gille A, et al GPR41/FFAR3 and GPR43/FFAR2 as cosensors for short‐chain fatty acids in enteroendocrine cells vs FFAR3 in enteric neurons and FFAR2 in enteric leukocytes. Endocrinology 2013; 154: 3552–3564. [DOI] [PubMed] [Google Scholar]
- 38. Lin HV, Frassetto A, Kowalik EJ, et al Butyrate and propionate protect against diet‐induced obesity and regulate gut hormones via free fatty acid receptor 3‐independent mechanisms. PLoS ONE 2012; 7: e35240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Svendsen B, Pais R, Engelstoft MS, et al GLP1 and GIP‐producing cells rarely overlap and differ by bobesin receptor‐2 expression and responsiveness. J Endocrinol 2016; 228(1): 39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hauge M, Vestmar MA, Husted AS, et al GPR40 (FFAR1) ‐ Combined Gs and Gq signaling in vitro is associated with robust incretin secretagogue action ex vivo and in vivo. Mol Metab. 2015; 4: 3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Panaro BL, Tough IR, Engelstoft MS, et al The melanocortin‐4 receptor is expressed in enteroendocrine L cells and regulates the release of peptide YY and glucagon‐like peptide 1 in vivo. Cell Metab 2014; 20: 1018–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Brighton CA, Rievaj J, Kuhre RE, et al Bile acids trigger GLP‐1 release predominantly by accessing basolaterally‐located G‐protein coupled bile acid receptors. Endocrinology 2015; 156: 3961–3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jørgensen NB, Jacobsen SH, Dirksen C, et al Acute and long‐term effects of Roux‐en‐Y gastric bypass on glucose metabolism in subjects with Type 2 diabetes and normal glucose tolerance. Am J Physiol Endocrinol Metab 2012; 303: E122–E131. [DOI] [PubMed] [Google Scholar]
- 44. Irwin N, Flatt PR. Therapeutic potential for GIP receptor agonists and antagonists. Best Pract Res Clin Endocrinol Metab 2009; 23: 499–512. [DOI] [PubMed] [Google Scholar]