

Abstract
Insulin secretion from the pancreatic β‐cell (referred to as β‐cell hereafter) plays a central role in glucose homeostasis. Impaired insulin secretion is a major factor contributing to the development of diabetes and, therefore, is an important target for treatment of the disease. Cyclic adenosine monophosphate is a key second messenger in β‐cells that amplifies insulin secretion. Incretins released by the gut potentiate insulin secretion through cyclic adenosine monophosphate signaling in β‐cells, which is the basis for the incretin‐based diabetes therapies now being used worldwide. Despite its importance, the interaction between glucose metabolism and incretin/cyclic adenosine monophosphate signaling in β‐cells has long been unknown. A recent study showed that cytosolic glutamate produced by glucose metabolism in β‐cells is a key signal in incretin‐induced insulin secretion. Here we review the physiological and pathophysiological roles of β‐cell glutamate signaling in incretin‐induced insulin secretion.
Keywords: Cyclic adenosine monophosphate, Glutamate, Incretin
Introduction
Insulin secretion from pancreatic β‐cells is critical for the maintenance of glucose homeostasis, and its impairment is associated closely with the pathogenesis and pathophysiology of diabetes. Mechanisms of insulin secretion have been investigated extensively over several decades. In the 1980s, the major intracellular signals in insulin secretion were identified by physiological, pharmacological and biochemical methods. These signals include Ca2+, adenosine triphosphate (ATP), cyclic adenosine monophosphate (cAMP) and phospholipid‐derived molecules, such as diacylglycerol (DAG) and inositol 1,4,5‐trisphosphate (IP3)1, 2, 3. Glucose is physiologically the most important fuel of insulin secretion (glucose‐induced insulin secretion [GIIS]).
Metabolic coupling factors (MCFs) in fuel‐induced insulin secretion (FIIS) have been reviewed elegantly by a recent paper4. Regulatory MCFs, such as citrate, malonyl‐coenzyme A (CoA), glutamate and adenine nucleotides, modulate metabolic networks involved in FIIS, whereas the effectory MCFs, such as ATP, cAMP and nicotinamide adenine dinucleotide phosphate, are directly involved in the triggering and amplification of insulin secretion. There are also interactions between glucose and free fatty acid in β‐cells. GIIS is thought to be associated with inhibition of free fatty acid oxidation and increased lipid synthesis in β‐cells. Malonyl‐CoA is known to be a key metabolite that lies at the crossroad of glucose metabolism and lipid metabolism, and is proposed to be an MCF for GIIS.
Amplification of GIIS by neuronal and hormonal inputs is also important for normal regulation of insulin secretion5. For example, acetylcholine, a neurotransmitter, stimulates insulin secretion through activation of muscarinic acetylcholine receptor in β‐cells6. Incretins, such as glucagon‐like peptide‐1 (GLP‐1) and glucose‐dependent insulinotropic polypeptide (GIP), which are secreted from the enteroendocrine cells in response to meal ingestion, potentiate insulin secretion in a glucose‐dependent manner through cAMP signaling7. However, how glucose metabolism and cAMP signaling interact with each other, and why incretin‐induced insulin secretion is glucose‐dependent was not known. We recently found that cytosolic glutamate produced through the malate‐aspartate (MA) shuttle in glucose metabolism in β‐cells is a key cell signal linking glucose metabolism to incretin/cAMP action to amplify insulin secretion. In the present review, we discuss the physiological and pathophysiological roles of glutamate signaling in β‐cells.
Glucose‐induced insulin secretion and its modulation by hormonal and neuronal inputs
GIIS involves two pathways, the triggering pathway and the metabolic amplifying pathway, and is modulated by various hormonal and neuronal inputs.
Triggering pathway
ATP generated in glucose metabolism in pancreatic β‐cells and Ca2+ influx are the principal signals in this pathway. Glucose is transported into the β‐cells by glucose transporter (GLUT2 in rodents and mainly GLUT1 in humans), and is metabolized in glycolysis and the tricarboxylic acid cycle. Increased glucose metabolism causes an increase in ATP concentration, closing the ATP sensitive K+ (KATP) channels, depolarizing the β‐cell membrane, opening the voltage‐dependent Ca2+ channels, thereby causing Ca2+ influx into β‐cells. The resultant rise in intracellular Ca2+ concentration ([Ca2+]i) triggers exocytosis of insulin granules8.
Metabolic amplifying pathway
It is known that in addition to ATP, various metabolic signals generated by glucose metabolism amplify insulin secretion. This pathway is called the “metabolic amplifying pathway.” Although this pathway is not fully understood, augmentation of the effect of Ca2+ on insulin secretion is thought to be involved8. A recent study suggested that three pyruvate cycling pathways – the pyruvate‐malate shuttle, the pyruvate‐citrate shuttle and the pyruvate‐isocitrate shuttle – might be associated with the metabolic amplifying pathway9.
Modulation of insulin secretion by hormones and neurotransmitters
The actions of hormones and neurotransmitters in insulin secretion are mediated by their specific receptors, most of which are guanine‐nucleotide‐binding regulatory protein (G protein)‐coupled receptors (GPCRs)10. GPCRs include Gs‐, Gq/11‐ and Gi/o‐protein‐coupled receptors. They principally mediate two signaling pathways: the cAMP signaling pathway and the phosphatidylinositol signaling pathway. Ligand binding to GPCR causes its conformational change and activates G protein.
Both Gs protein signaling and Gq/11‐protein signaling mediate amplification of GIIS. The incretins GLP‐1 and GIP are known to potentiate insulin secretion through activation of Gs protein signaling followed by cAMP signaling pathways, which include the protein kinase A‐dependent pathway and the Epac2A (also called cAMP‐GEFII)‐dependent pathway11, 12, 13.
Acetylcholine amplifies insulin secretion through Gq/11‐protein signaling by activation of phospholipase C‐β, which produces IP3 and DAG. IP3 triggers Ca2+ release from the endoplasmic reticulum, whereas DAG activates protein kinase C14. Gi/o‐protein signaling mediates an inhibitory effect on GIIS, and is known to regulate K+ and Ca2+ channels directly and indirectly through suppression of cAMP production10. Somatostatin, noradrenaline and ghrelin inhibit insulin secretion through activation of Gi‐protein signaling.
Incretin effects on insulin secretion
It is well known that oral glucose load produces a much greater insulin secretion than intravenous injection of glucose, which is now recognized as the “incretin effect.” Incretins, such as GIP and GLP‐1, are gut hormones released from the gut in response to ingestion of glucose or nutrients7. GIP, a 42 amino acid hormone, is secreted from K cells of the upper part of the intestine; GLP‐1, a 31 amino acid hormone, is secreted from L cells of the lower part of the intestine. GLP‐1 and GIP potentiate insulin secretion through cAMP signaling in a glucose‐dependent manner. It was found by perfusion of mouse pancreas that when the glucose concentration is increased in a stepwise manner from 2.8 to 12.5 mmol/L in the absence of cAMP‐increasing agents (i.e., 8‐Br‐cAMP or GLP‐1), no insulin secretion is induced, but the presence of the agents resulted in dramatic induction of insulin secretion15. Similar results also were found in Kir6.2 knockout mice, in which almost no GIIS was detected. These findings show that incretin/cAMP signaling is critical for the induction of glucose responsiveness in insulin secretion, as well as the potentiation of GIIS5. We found by total internal reflection fluorescence microscopy analysis that the cAMP analog 8‐Br‐cAMP enhanced the frequency of fusion events of insulin granules to the plasma membrane in the presence of glucose (but not by itself) in both the first phase and the second phase of potentiation12. It is now established that incretin/cAMP signaling potentiates GIIS by both protein kinase A‐dependent and Epac2A‐dependent mechanisms. Epac2A has guanine nucleotide exchange factor activity toward Rap1, a small G protein. Binding of cAMP to Epac2A causes its conformational change, thereby releasing the catalytic region and enabling the binding and the activation of Rap113. A study of Epac2A knockout mice showed that Epac2A is essential in the potentiation of insulin granule exocytosis by cAMP, but primarily in the first phase of potentiation12. The following second phase potentiated by cAMP might involve protein kinase A signaling, but the precise mechanism is still unclear. In addition, it has recently been shown that Epac2A is a direct target of some of the antidiabetic sulfonylureas16, 17, drugs widely used for treatment of diabetes. Epac2A/Rap1 signaling also is required for augmentation of insulin secretion by a combination of incretin and sulfonylureas18.
Distinction between metabolomic profiles of MIN6‐K8 and ‐K20 β‐cells
Various β‐cell lines for studies of β‐cell biology have been established, including hamster pancreatic β‐cell line19, rat insulinoma cell lines (RINm5F and INS‐1)20, 21, 22, mouse insulinoma cell lines (beta TC and MIN6)23, 24 and human pancreatic β‐cell lines (betalox5, EndoC‐βH1 and EndoC‐βH2)25, 26, 27. These cell lines often show different insulin secretory profiles from those of native pancreatic β‐cells or islets28. In fact, many of these cell lines lack incretin responsiveness, and so are unsuitable for investigation of the mechanism of incretin/cAMP signaling. Incretin‐responsive and ‐unresponsive pancreatic β‐cell lines (designated MIN6‐K8 and MIN6‐K20 β‐cells, respectively) have recently been established by cloning of β‐cells from the insulinoma‐bearing IT6 mice29. MIN6‐K8 β‐cells secrete insulin in response to both glucose and incretin stimulations, whereas MIN6‐K20 β‐cells respond only to glucose, but not to incretin stimulation. These cell lines are useful tools to clarify the mechanism of incretin responsiveness in insulin secretion.
As incretin‐induced insulin secretion occurs in a glucose‐dependent manner, the difference in incretin responsiveness between MIN6‐K8 and ‐K20 β‐cells might well be due to differences in glucose metabolism of the two cell lines. In fact, metabolome analysis of MIN6‐K8 and ‐K20 β‐cells under glucose‐stimulated condition showed distinct metabolomic profiles of the two cell lines. In addition, contents of glucose 6‐phosphate, fructose 6‐phosphate, fructose 1,6‐bisphosphate, nicotinamide adenine dinucleotide, glutamate and aspartate are higher in MIN6‐K8 β‐cells than those in MIN6‐K20 β‐cells, indicating that both glycolysis and the MA shuttle are enhanced in incretin‐responsive MIN6‐K8 β‐cells in comparison with those in incretin‐unresponsive MIN6‐K20 β‐cells (Figure 1)30.
Figure 1.
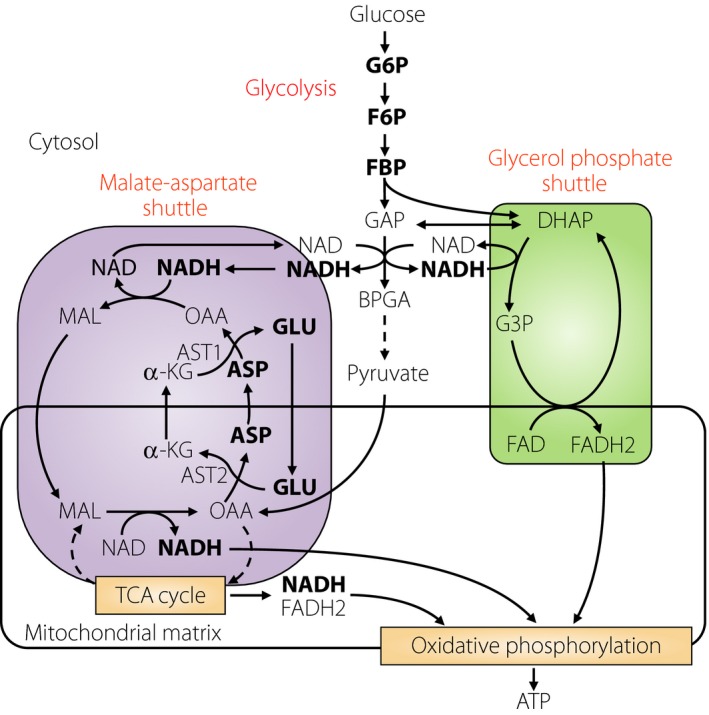
Nicotinamide adenine dinucleotide (NADH) shuttles. Two NADH shuttles (malate‐aspartate shuttle and glycerol phosphate shuttle) links to glycolysis are shown. Metabolites showing higher contents in MIN6‐K8 cells than those in MIN6‐K20 cells are shown in bold. α‐KG, α‐ketoglutarate; ASP, aspartate; AST1and 2, aspartate aminotransferase 1 and 2; BPGA, 1,3‐bisphosphoglycerate; DHAP, dihydroxyacetone phosphate; F6P, fructose 6‐phophate; FBP, fructose 1,6‐bisphosphate; G3P, glycerol 3‐phosphate; G6P, glucose 6‐phosphate; GAP, glyceraldehyde 3‐phosphate; GLU, glutamate; MAL, malate; OAA, oxaloacetate.
Glutamate produced through the malate‐aspartate shuttle is a critical signal in incretin‐induced insulin secretion
Glutamate in β‐cells is produced through three metabolic pathways: (i) conversion from α‐ketoglutarate by glutamate dehydrogenase 1 in the mitochondria; (ii) conversion from glutamine by glutaminase in the mitochondria; and (iii) transamination of α‐ketoglutarate by aspartate aminotransferase 1 associated with the MA shuttle in cytosol. Mitochondrial glutamate was previously proposed as a signal in GIIS31, 32, 33, but the notion has been controversial34, 35. Metabolic flux analysis is a useful technique to determine isotopomers, as well as altered metabolic rate in the cells. Using stable isotope‐labeled [U‐¹³C]‐glucose, glutamate isotopomers can be measured to learn whether or not the glutamate is produced from glucose. There are six glutamate isotopomers (M [no substitution with ¹³C] and M + 1 to M + 5 [one to five substitutions with ¹³C, respectively]). M and M + 1 isotopomers are the naturally occurring glutamate in cells; M + 2 to M + 5 isotopomers represent glutamate derived from [U‐¹³C]‐glucose (Figure 2). M + 2 to M + 5 glutamate isotopomers were increased in cytosol, as assessed by metabolic flux analysis, indicating that glutamate is produced from glucose. Inhibition of the MA shuttle by aminooxyacetate, an inhibitor of the MA shuttle36, 37, decreased production of M + 2 to M + 5 glutamate isotopomers in cytosol, indicating that cytosolic glutamate is produced through the MA shuttle. In addition, application of glutamate in the presence of cAMP into primary mouse β‐cells dose‐dependently stimulated exocytosis, as assessed by capacitance measurement. Total internal reflection fluorescence microscopy analysis showed that dimethyl‐glutamate, a membrane‐permeable glutamate precursor that is converted to glutamate by esterase within the cells, amplifies both the first and second phases of glucose‐induced insulin granule exocytosis30. Together, these findings strongly suggest that cellular glutamate acts as a critical amplifying signal in incretin‐induced insulin secretion.
Figure 2.
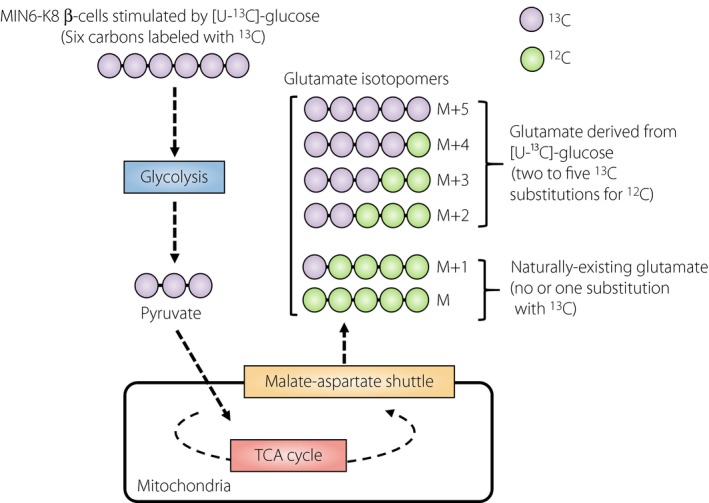
Determination of glucose‐derived glutamate isotopomers by mass spectrometry. [U‐13C]‐glucose: stable isotope labeled glucose. M and M + 1: naturally‐existing glutamate isotopomers in the cell (no or one substitution with13C). M + 2 to M + 5: glutamate isotopomers derived from [U‐¹³C]‐glucose (two to five13C substitutions for12C).
Transport of glutamate into insulin granules
Glutamate in the cytosol is known to be transported into secretory vesicles through vesicular glutamate transporters (VGLUTs)38. VGLUT1 is expressed in mouse pancreatic β‐cells, and various mouse and rat β‐cell lines; for example, MIN6‐K8, βTC6, RIN‐m and INS‐1E cells30, 39, 40. VGLUT2 is expressed in both pancreatic α‐cell lines (αTC1‐9 cells) and β‐cell lines (MIN6‐K8, βTC6 and RIN‐m cells)30, 39, 40. VGLUT3 is also expressed in mouse pancreatic α‐ and β‐cells40. Inhibition of glutamate transport into insulin granules by Evans Blue, an inhibitor of glutamate transporters, or knockdown of VGLUT1 did not affect GIIS, but reduced incretin‐induced insulin secretion in MIN6‐K8 β‐cells, suggesting that glutamate transport into insulin granules through VGLUT1 is required for incretin‐induced insulin secretion30. Glutamate transport through VGLUTs is regulated by electrical potential (Δϕ) and pH gradient (ΔpΗ) across the vesicular membrane38. Glutamate flux into insulin granules is involved in the generation of Δϕ and ΔpΗ across the insulin granule membrane40. A proton pump, V‐ATPase, in insulin granules also contributes to generation of Δϕ and ΔpΗ across the insulin granule membrane41, a process in which Cl− flux is required42, 43. In addition, it was reported that glutamate efflux through excitatory amino acid transporters was involved in the regulation of Δϕ and ΔpΗ in insulin granules40. Thus, glutamate flux through insulin granules is regulated by a complex mechanism. Interestingly, impaired glutamate efflux was found to decrease insulin granule exocytosis40, further suggesting a critical role of glutamate in insulin secretion. Although the precise mechanism by which glutamate in insulin granules stimulates insulin granule exocytosis remains to be elucidated, total internal reflection fluorescence microscopy analysis showed that dimethyl‐glutamate induces exocytosis of insulin granules, suggesting that glutamate in insulin granules might promote recruitment towards and/or fusion of the insulin granules with the plasma membrane (Figure 3)30.
Figure 3.
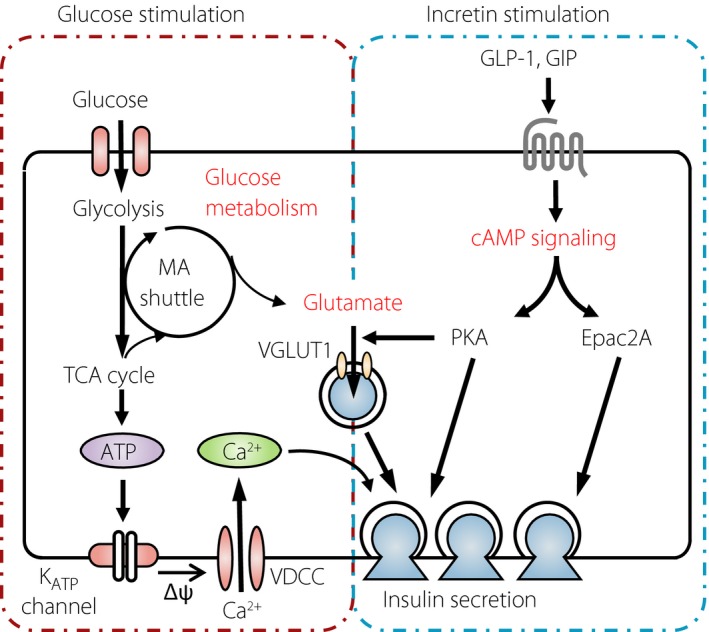
Model of cellular mechanism of incretin‐induced insulin secretion. Glutamate produced through the MA shuttle links glucose metabolism and cyclic adenosine monophosphate (cAMP) signaling to amplify insulin secretion. GLP‐1, glucagon‐like peptide‐1; GIP, glucose‐dependent insulinotropic polypeptide; KATP channel, adenosine triphosphate‐sensitive K+ channel; MA shuttle, malate‐aspartate shuttle; PKA, protein kinase A; TCA, tricarboxylic acid; VDCC, voltage‐dependent Ca2+ channel.
Glutamate production in pancreatic islets of diabetic and obese rat models
Impaired potentiation of insulin secretion by incretins is found in diabetes in clinical settings44. Animal models are useful for studying the pathogenesis and pathophysiology of diabetes. The Goto–Kakizaki rat is a non‐obese type 2 diabetes model with defective insulin secretion associated with impaired glucose metabolism in pancreatic β‐cells45. GIIS is markedly decreased in these rats compared with normal control Wistar rats, but the amplification effect by GLP‐1 is somewhat retained. Glucose‐stimulated glutamate production was increased slightly, but significantly, in the islets of Goto–Kakizaki rats. In contrast, the Zucker fatty rat, an obesity model as a result of mutation in the leptin receptor gene, shows a higher level of basal insulin secretion and insulin response to glucose. However, these rats showed no potentiation of insulin secretion in response to incretin stimulation. Glucose did not increase glutamate production in the islets of Zucker fatty rats. Dimethyl‐glutamate was able to amplify insulin secretion in Zucker fatty rats, mimicking the effect of incretins. Thus, impaired production of glutamate in the pancreatic β‐cells is closely associated with impaired incretin‐induced insulin secretion30.
Conclusions
A metabolomics‐based approach showed that glutamate acts as a key cell signal linking glucose metabolism to incretin/cAMP action to amplify insulin secretion. Unresponsiveness to endogenous incretins and unresponsiveness to exogenously administered incretin‐related drugs (incretin non‐responders) are found among patients with type 2 diabetes; abnormalities in β‐cell glutamate signaling might therefore underlie the unresponsiveness to incretins. Clarification of glutamate signaling could contribute to the development of novel therapies, as well as our understanding of the pathophysiology of diabetes and diabetic β‐cells. Metabolomics is a powerful approach to unveil metabolic signaling pathways.
Disclosure
The authors declare no conflict of interest.
Acknowledgments
We are grateful to the members of our laboratories who were involved in the studies cited in this review. The studies of our laboratories were supported by a CREST grant from the Japan Science and Technology Agency, and Grants‐in‐Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan. This study was also supported in part by a research grant from MSD K.K.
J Diabetes Investig 2016; 7: 38–43
This article is based on the presentations given by the authors at a symposium, Incretin 2015, July 29–31, 2015, Vancouver, BC Canada.
References
- 1. Wollheim CB, Sharp GW. Regulation of insulin release by calcium. Physiol Rev 1981; 61: 914–973. [DOI] [PubMed] [Google Scholar]
- 2. Malaisse WJ, Malaisse‐Lagae F. The role of cyclic AMP in insulin release. Experientia 1984; 40: 1068–1074. [DOI] [PubMed] [Google Scholar]
- 3. Prentki M, Matschinsky FM. Ca2 +, cAMP, and phospholipid‐derived messengers in coupling mechanisms of insulin secretion. Physiol Rev 1987; 67: 1185–1248. [DOI] [PubMed] [Google Scholar]
- 4. Prentki M, Matschinsky FM, Madiraju SR. Metabolic signaling in fuel‐induced insulin secretion. Cell Metab 2013; 18: 162–185. [DOI] [PubMed] [Google Scholar]
- 5. Seino S, Shibasaki T, Minami K. β‐cell biology of insulin secretion In: DeFronzo RA, Ferrannini E, Alberti KGMM, Zimmet P. (eds). International Textbook of Diabetes Mellitus, Chapter 7, 4th edn UK: Wiley, 2015; 96–107 [Google Scholar]
- 6. Ahrén B. Autonomic regulation of islet hormone secretion: implications for health and disease. Diabetologia 2000; 43: 393–410. [DOI] [PubMed] [Google Scholar]
- 7. Drucker DJ. Incretin action in the pancreas: potential promise, possible perils, and pathological pitfalls. Diabetes 2013; 62: 3316–3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Henquin JC. Regulation of insulin secretion: a matter of phase control and amplitude modulation. Diabetologia 2009; 52: 739–751. [DOI] [PubMed] [Google Scholar]
- 9. Jitrapakdee S, Wutthisathapornchai A, Wallace JC, et al Regulation of insulin secretion: role of mitochondrial signalling. Diabetologia 2010; 53: 1019–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ahrén B. Islet G protein‐coupled receptors as potential targets for treatment of type 2 diabetes. Nat Rev Drug Discov 2009; 8: 369–385. [DOI] [PubMed] [Google Scholar]
- 11. Kashima Y, Miki T, Shibasaki T, et al Critical role of cAMP GEFII‐Rim2 complex in incretin‐potentiated insulin secretion. J Biol Chem 2001; 276: 46046–46053. [DOI] [PubMed] [Google Scholar]
- 12. Shibasaki T, Takahashi H, Miki T, et al Essential role of Epac2/Rap1 signaling in regulation of insulin granule dynamics by cAMP. Proc Natl Acad Sci USA 2007; 104: 19333–19338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gloerich M, Bos JL. Epac: defining a new mechanism for cAMP action. Annu Rev Pharmacol Toxicol 2010; 50: 355–375. [DOI] [PubMed] [Google Scholar]
- 14. Schmitz‐Peiffer C, Biden TJ. Protein kinase C function in muscle, liver, and β‐cells and its therapeutic implications for type 2 diabetes. Diabetes 2008; 57: 1774–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fujimoto W, Miki T, Ogura T, et al Niflumic acid‐sensitive ion channels play an important role in the induction of glucose‐stimulated insulin secretion by cyclic AMP in mice. Diabetologia 2009; 52: 863–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang CL, Katoh M, Shibasaki T, et al The cAMP sensor Epac2 is a direct target of antidiabetic sulfonylurea drugs. Science 2009; 325: 607–610. [DOI] [PubMed] [Google Scholar]
- 17. Takahashi T, Shibasaki T, Takahashi H, et al Antidiabetic sulfonylureas and cAMP cooperatively activate Epac2A. Sci Signal 2013; 6: ra94. [DOI] [PubMed] [Google Scholar]
- 18. Takahashi H, Shibasaki T, Park JH, et al Role of Epac2A/Rap1 signaling in interplay between incretin and sulfonylurea in insulin secretion. Diabetes 2015; 64: 1262–1272. [DOI] [PubMed] [Google Scholar]
- 19. Santerre RF, Cook RA, Crisel RM, et al Insulin synthesis in a clonal cell line of simian virus 40‐transformed hamster pancreatic beta cells. Proc Natl Acad Sci USA 1981; 78: 4339–4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gazdar AF, Chick WL, Oie HK, et al Continuous, clonal, insulin‐and somatostatin‐secreting cell lines established from a transplantable rat islet cell tumor. Proc Natl Acad Sci USA 1980; 77: 3519–3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Praz GA, Halban PA, Wollheim CB, et al Regulation of immunoreactive‐insulin release from a rat cell line (RINm5F). Biochem J 1983; 210: 345–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Asfari M, Janjic D, Meda P, et al Establishment of 2‐mercaptoethanol‐dependent differentiated insulin‐secreting cell lines. Endocrinology 1992; 130: 167–178. [DOI] [PubMed] [Google Scholar]
- 23. Efrat S, Linde S, Kofod H, et al Beta‐cell lines derived from transgenic mice expressing a hybrid insulin gene‐oncogene. Proc Natl Acad Sci USA 1988; 85: 9037–9041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Miyazaki J, Araki K, Yamato E, et al Establishment of a pancreatic β cell line that retains glucose‐inducible insulin secretion: special reference to expression of glucose transporter isoforms. Endocrinology 1990; 127: 126–132. [DOI] [PubMed] [Google Scholar]
- 25. Halvorsen TL, Leibowitz G, Levine F. Telomerase activity is sufficient to allow transformed cells to escape from crisis. Mol Cell Biol 1999; 19: 1864–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ravassard P, Hazhouz Y, Pechberty S, et al A genetically engineered human pancreatic β cell line exhibiting glucose‐inducible insulin secretion. J Clin Invest 2011; 121: 3589–3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Carlier G, Maugein A, Cordier C, et al Human fucci pancreatic Beta cell lines: new tools to study Beta cell cycle and terminal differentiation. PLoS ONE 2014; 9: e108202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hauge‐Evans AC, Squires PE, Persaud SJ, et al Pancreatic beta‐cell‐to‐beta‐cell interactions are required for integrated responses to nutrient stimuli: enhanced Ca2 + and insulin secretory responses of MIN6 pseudoislets. Diabetes 1999; 48: 1402–1408. [DOI] [PubMed] [Google Scholar]
- 29. Iwasaki M, Minami K, Shibasaki T, et al Establishment of new clonal pancreatic β‐cell lines (MIN6‐K) useful for study of incretin/cyclic adenosine monophosphate signaling. J Diabetes Investig 2010; 1: 137–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gheni G, Ogura M, Iwasaki M, et al Glutamate acts as a key signal linking glucose metabolism to incretin/cAMP action to amplify insulin secretion. Cell Rep 2014; 9: 661–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Maechler P, Wollheim CB. Mitochondrial glutamate acts as a messenger in glucose‐induced insulin exocytosis. Nature 1999; 402: 685–689. [DOI] [PubMed] [Google Scholar]
- 32. Hoy M, Maechler P, Efanov AM, et al Increase in cellular glutamate levels stimulates exocytosis in pancreatic beta‐cells. FEBS Lett 2002; 531: 199–203. [DOI] [PubMed] [Google Scholar]
- 33. Casimir M, Lasorsa FM, Rubi B, et al Mitochondrial glutamate carrier GC1 as a newly identified player in the control of glucose‐stimulated insulin secretion. J Biol Chem 2009; 284: 25004–25014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. MacDonald MJ, Fahien LA. Glutamate is not a messenger in insulin secretion. J Biol Chem 2000; 275: 34025–34027. [DOI] [PubMed] [Google Scholar]
- 35. Bertrand G, Ishiyama N, Nenquin M, et al The elevation of glutamate content and the amplification of insulin secretion in glucose‐stimulated pancreatic islets are not causally related. J Biol Chem 2002; 277: 32883–32891. [DOI] [PubMed] [Google Scholar]
- 36. MacDonald MJ. Evidence for the malate aspartate shuttle in pancreatic islets. Arch Biochem Biophys 1982; 213: 643–649. [DOI] [PubMed] [Google Scholar]
- 37. Eto K, Tsubamoto Y, Terauchi Y, et al Role of NADH shuttle system in glucose‐induced activation of mitochondrial metabolism and insulin secretion. Science 1999; 283: 981–985. [DOI] [PubMed] [Google Scholar]
- 38. Omote H, Miyaji T, Juge N, et al Vesicular neurotransmitter transporter: bioenergetics and regulation of glutamate transport. Biochemistry 2011; 50: 5558–5565. [DOI] [PubMed] [Google Scholar]
- 39. Bai L, Zhang X, Ghishan FK. Characterization of vesicular glutamate transporter in pancreatic alpha ‐ and beta ‐cells and its regulation by glucose. Am J Physiol Gastrointest Liver Physiol 2003; 284: G808–G814. [DOI] [PubMed] [Google Scholar]
- 40. Gammelsaeter R, Coppola T, Marcaggi P, et al A role for glutamate transporters in the regulation of insulin secretion. PLoS ONE 2011; 6: e22960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Aspinwall CA, Brooks SA, Kennedy RT, et al Effects of intravesicular H+ and extracellular H+ and Zn2 + on insulin secretion in pancreatic beta cells. J Biol Chem 1997; 272: 31308–31314. [DOI] [PubMed] [Google Scholar]
- 42. Xie XS, Stone DK, Racker E. Determinants of clathrin‐coated vesicle acidification. J Biol Chem 1983; 258: 14834–14838. [PubMed] [Google Scholar]
- 43. Xie XS, Crider BP, Stone DK. Isolation and reconstitution of the chloride transporter of clathrin‐coated vesicles. J Biol Chem 1989; 264: 18870–18873. [PubMed] [Google Scholar]
- 44. Holst JJ, Knop FK, Vilsbøll T, et al Loss of incretin effect is a specific, important, and early characteristic of type 2 diabetes. Diabetes Care 2011; 34 (Suppl. 2): S251–S257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ostenson CG, Khan A, Abdel‐Halim SM, et al Abnormal insulin secretion and glucose metabolism in pancreatic islets from the spontaneously diabetic GK rat. Diabetologia 1993; 36: 3–8. [DOI] [PubMed] [Google Scholar]