

Abstract
Precise control of blood glucose is dependent on adequate β‐cell mass and function. Thus, reductions in β‐cell mass and function lead to insufficient insulin production to meet demand, and result in diabetes. Recent evidence suggests that paracrine signaling in the islet might be important in obesity, and disruption of this signaling could play a role in the pathogenesis of diabetes. For example, we recently discovered a novel islet incretin axis where glucagon‐like peptide‐1 regulates β‐cell production of another classic gut hormone, cholecystokinin. This axis is stimulated by obesity, and plays a role in enhancing β‐cell survival. In the present review, we place our observations in the wider context of the literature on incretin regulation in the islet, and discuss the potential for therapeutic targeting of these pathways.
Keywords: Cholecystokinin, Glucagon‐like peptide‐1, Pancreatic islet
Introduction
The endocrine pancreas tightly regulates hormone secretion to maintain normoglycemia. Under conditions of stress and increased demand, such as the inflammation and insulin resistance observed in obesity, adaptation of the pancreatic islet is necessary for continued maintenance of blood glucose. This compensatory adaptation could include β‐cell proliferation and enhanced β‐cell survival, and can allow increased insulin production to maintain euglycemia. Failure to maintain adequate functional β‐cell mass results in diabetes. Therefore, understanding the pathways that control compensatory adaptation of the islet in non‐diabetic obesity can provide insight into β‐cell mass regulation. Drug development targeting these pathways could provide important new therapies for diabetes, as none of the current diabetes medications directly target β‐cell mass in humans.
The β‐cell does not exist in isolation, but is intimately associated with a host of other cell types in the pancreatic islet, including: α‐cells, δ‐cells, PP‐cells, endothelial cells (from the islet capillaries), and neurons. Although less well understood, communication between these various cell types in the form of paracrine signaling is integral to the function of the islet as a micro‐organ that can respond both acutely and chronically to stress and increased demand. The importance of cell–cell communication on inflammatory stress response in the human islet was recently implicated by work showing that the islet as a whole responds differently to cytokine‐induced stress than transformed β‐cells in isolation1. This suggests that perturbations in paracrine signaling could impair the adaptive response to obesity, and contribute to the pathophysiology of diabetes in humans.
We and others have recently discovered that classic gut‐derived peptide hormones, including glucagon‐like peptide‐1 (GLP‐1) and cholecystokinin (CCK), are produced in and secreted from islet cells under various stress conditions, including obesity2, 3, 4, 5. The production of incretin hormones in the pancreatic islet might be an adaptive mechanism to improve β‐cell function and survival under conditions of stress. Both GLP‐1 and CCK have positive effects on the β‐cell, as they can stimulate insulin secretion, and might promote β‐cell proliferation or protection from apoptosis6, 7. In the present review, we discuss the production of GLP‐1 and CCK, and how they might regulate one another in the pancreatic islet. It is not well understood how the production of these peptides is regulated in the islet, but identification of these regulatory pathways might allow us to find ways to enhance this beneficial adaptive response.
Mechanisms of GLP‐1 and CCK regulation
GLP‐1 is a peptide hormone, well studied as a product of neuroendocrine cells in the intestine and brain. It functions as an incretin to stimulate insulin secretion, but also has important roles in stimulating satiety and delaying gastric emptying8. GLP‐1 is cleaved from the precursor protein proglucagon by prohormone convertase 1/3 (PC1/3). The enzyme dipeptidyl peptidase‐4 degrades circulating GLP‐1 within minutes9, 10, making it unlikely that postprandial GLP‐1 secretion from the intestine can provide a sustained endocrine signal directly to the islet. A recent study suggests that the effects of GLP‐1 on the β‐cell are not as a result of gut‐derived hormone, but perhaps neuronal or even paracrine action8. In the pancreatic islet, the α‐cell normally produces prohormone convertase 2 to cleave proglucagon into glucagon. However, PC1/3 and GLP‐1 are expressed in α‐cells under conditions of islet stress including pregnancy, the prediabetic non‐obese diabetic mouse, and the ob/ob and db/db models of obesity and diabetes2. Neither diabetes nor obesity is associated with a significant change in serum GLP‐1 levels in humans11. However, we have shown that GLP‐1 is secreted from human islets, with enhanced secretion from islets from obese donors5. This suggests that intra‐islet paracrine function of GLP‐1 might be one of the adaptive responses to stress that improves islet function and survival.
CCK is also a peptide hormone, and is best known for its role in digestion and satiety. CCK is post‐translationally processed into a variety of isoforms, with the most bioactive form being sulfated CCK‐8. It is secreted by intestinal cells to stimulate gallbladder contraction and pancreatic exocrine secretion, and is found in the brain to modulate satiety, anxiety and memory12. At supraphysiological levels, CCK can act as an insulin secretagogue13. CCK is also expressed in the pancreatic β‐cell14, and is the most highly upregulated islet gene in response to obesity3. The upregulation of islet Cck occurs in multiple models of insulin resistance, including ob/ob, Agouti and high‐fat diet (HFD)‐fed mice. Therefore, CCK production might be part of a general adaptive islet response to insulin resistance or β‐cell stress. However, until recently, the mechanisms that underlie endogenous CCK regulation and function in the β‐cell were unknown.
There is evidence that the Cck promoter is regulated by cyclic adenosine monophosphate (cAMP) signaling in other cell types. Activation of adenylate cyclase with forskolin stimulates Cck transcription in teratocarcinoma cells15. This transcriptional activation is mediated by cAMP response element binding protein (CREB) that binds to the CCK promoter in intestinal L cells and cells of neuronal origin15, 16, 17. Transient CREB overexpression in teratocarcinoma cells can increase Cck promoter activity and conversely, deletion of a cAMP response element in the Cck promoter or treatment with a dominant negative CREB dramatically reduces forskolin‐mediated Cck transcription15. Based on these data, we hypothesized that a similar mechanism of regulation might occur in the β‐cell to stimulate Cck transcription during obesity. Indeed, we observe recruitment of CREB to the Cck promoter in cultured β‐cells treated with cAMP5.
The GLP‐1 receptor is coupled to Gαs proteins, and can stimulate adenylate cyclase and cAMP production in response to GLP‐1 binding. Therefore, GLP‐1 could regulate CCK transcription in the islet. In support of this, we find that islets from ob/ob mice secrete active GLP‐1 and also transcribe the Cck gene5. In cultured β‐cells, GLP‐1 can stimulate Cck transcription through direct targeting by CREB. In vivo, CREB occupancy at the mouse islet Cck promoter increases as a function of obesity. As α‐cell GLP‐1 secretion and β‐cell Cck transcription are both increased in obesity, locally produced GLP‐1 might be responsible for CREB activation of β‐cell Cck in vivo (Figure 1).
Figure 1.
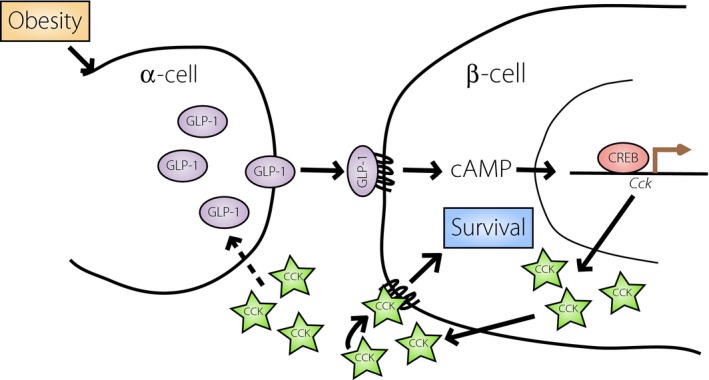
Model for glucagon‐like peptide‐1 (GLP‐1)/cholecystokinin (CCK)‐mediated β‐cell survival in obesity. Our recent data suggest that in obesity, GLP‐1 produced from the α‐cell rapidly stimulates production and secretion of CCK through direct targeting in the β‐cell. We hypothesize that β‐cell CCK mediates survival through autocrine/paracrine signaling and could also further stimulate α‐cell production of GLP‐1 to create a positive feedback loop. cAMP, cyclic adenosine monophosphate; CREB, cyclic adenosine monophosphate response element binding protein.
Notably, the stimulation of CCK by GLP‐1 or cAMP does not depend on insulin production, and occurs equally well in low glucose, suggesting a direct effect (unpublished observations and Linnemann et al.5). This is unique from the glucose‐dependent mechanism of GLP‐1 action as an insulin secretagogue. This also suggests that CCK secretion from the β‐cell is regulated by a mechanism that is distinct from that of insulin, and CCK is not packaged in the insulin secretory granule. As intestinal cells show regulated secretion of CCK18, 19, we suspect that CCK secretion from the islet is also regulated. It is likely that CCK is packaged in dense‐core vesicles or synaptic‐like microvesicles. It still remains to be determined whether the hyperglycemic conditions observed in diabetes affect islet secretion of CCK.
GLP‐1 and CCK might also regulate one another in a feed‐forward mechanism to maximize their production in a stressful environment, such as obesity. In rat islets, GLP‐1 treatment increases PC1/3 and GLP‐1 production through a positive feedback mechanism20. Treatment with a stable CCK analog can also increase α‐cell GLP‐1 and PC1/3 expression in obese mice21. Reinforcement of a GLP‐1/CCK islet signaling axis in vivo through a positive feedback loop could amplify the signals leading to compensatory adaptation to obesity.
Determining the function of GLP‐1 and CCK in the obese islet
Despite compelling evidence that GLP‐1 plays a role in apoptosis protection in rodent islet and cell lines22, 23, there have been a relatively small number of studies suggesting that GLP‐1 can directly protect human islets from β‐cell apoptosis. Human islets in culture have reduced basal rates of cell death when cultured in the presence of GLP‐124. Cytokines are elevated in obesity, and contribute to β‐cell apoptosis in both type 1 and type 2 diabetes25. Human islets treated ex vivo with GLP‐1 have reduced β‐cell apoptosis in response to cytokine treatment26. However, another group found that combination treatment with both the GLP‐1 receptor agonist, exendin‐4, and the growth factor, betacellulin, was required to protect human islets from cytokine‐mediated apoptosis27. This combined treatment resulted in increased/preserved expression of active Akt (protein kinase B), active CREB and the pro‐survival protein, BCL2 (B‐cell lymphoma‐2). The anti‐apoptotic effects of GLP‐1 are dependent on CREB, as they were reduced when a dominant negative version of CREB was expressed27. Many studies using rodent islets or cultured β‐cells have also shown that GLP‐1 has anti‐apoptotic effects, including when produced locally in the islet28.
Islet‐derived CCK also plays a role in protecting β‐cells from apoptosis. Loss of CCK in ob/ob mice causes a reduction in islet size, decreased β‐cell mass and increased β‐cell death leading to hyperglycemia3. Overexpression of CCK in these obese knockout islets or in a β‐cell line can rescue from apoptosis3, 29. Additionally, transgenic overexpression of CCK in the β‐cell confers protection from streptozotocin (STZ)‐induced apoptosis29. Considering our observation that GLP‐1 stimulates β‐cell production and secretion of CCK5, we hypothesize that GLP‐1‐stimulated production of CCK in the islet is important for β‐cell survival in obesity. This adaptive response might also contribute to diabetes prevention through increased β‐cell numbers, and thus increased capabilities for insulin production.
The mechanism by which CCK signals to protect β‐cells from apoptosis is not well understood. CCK can signal through two G‐protein coupled receptors, the CCK A receptor (CCKAR or CCK 1 receptor) and CCK B receptor (CCKBR or CCK 2 receptor) to activate a number of different signal transduction pathways30. The expression pattern of the CCK receptors within the islet is not clear in the literature, partly because of problems with quality and specificity of antibodies against these G‐protein coupled receptors proteins30, 31, 32, 33. Receptor expression in models of obesity and diabetes has not been studied, and these states might alter receptor levels. We used a non‐selective CCK receptor antagonist to show that autocrine CCK signaling in the β‐cell is required for GLP‐1‐mediated apoptosis protection5. However, specific blockade of the CCKAR inhibited downstream CCK signaling pathways in a β‐cell line and mouse islets34, supporting an important role for CCKAR in the β‐cell.
We propose a model wherein GLP‐1 secreted from obese islets acts in a paracrine manner to stimulate β‐cell CCK production (Figure 1). The islet‐derived CCK can act locally to protect the β‐cell from apoptosis through autocrine regulation. In light of evidence that exogenous CCK treatment enhances α‐cell GLP‐1 production under HFD feeding21, there might also be a positive feedback loop that amplifies this pathway in vivo.
Disruptions in islet GLP‐1 and CCK signaling
Disruptions in GLP‐1 and CCK signaling could contribute to the pathophysiology of type 2 diabetes. A reduction in GLP‐1 levels could be a factor in the clinical presentation of diabetes, although evidence is mixed and collectively there appears to be no difference in plasma GLP‐1 levels in patients with diabetes11. Therefore, GLP‐1 production or signaling might be perturbed in other ways that contribute to diabetes pathogenesis, including at the level of the islet. Interestingly, islets from donors with a history of type 2 diabetes secrete more GLP‐1 than non‐diabetic islets (Marchetti35 and unpublished observations) and mice with STZ‐induced diabetes similarly have increased pancreatic α‐cell production of GLP‐136. Thus, it is likely that an intra‐islet defect in GLP‐1 signaling exists downstream of α‐cell GLP‐1 production. Indeed, human type 2 diabetic islets have decreased GLP‐1 receptor (GLP1R) expression37. Induction of diabetes with low‐dose STZ in GLP1R knockout mice also leads to a more severe phenotype with increased levels of islet apoptosis36. Furthermore, GLP1R null mice show abnormalities in islet adenylate cyclase activity and increased sensitivity to β‐cell injury38, 39.
We find that GLP‐1 can stimulate β‐cell CCK production and prevent β‐cell apoptosis in vitro, and this protection is at least partially dependent on CCK receptor signaling5. In line with this, we find that CCK is both necessary and sufficient to protect β‐cells from apoptosis in vivo 3, 29. While specific downstream signaling pathways are implicated in GLP‐1‐mediated protection40, 41, CCK production and signaling through the CCK receptors can provide an alternate explanation for activation of these same signaling molecules. It is plausible that in vivo β‐cell protection mediated by GLP‐1 depends on CCK production and paracrine signaling. Therefore, we propose that the increased susceptibility to β‐cell apoptosis with GLP1R loss might be at least partially as a result of the inability to upregulate CCK production. It is intriguing to consider that GLP‐1 and CCK signaling pathways overlap in the islet such that when one pathway is perturbed, they might compensate for one another. For example, islets from GLP‐1 receptor/glucagon receptor double knockout mice have significantly increased expression of Cckar transcript and sensitivity to CCK42, and this enhanced CCK signaling could minimize the impact of the genotype on blood glucose homeostasis.
Further support for the importance of CCK signaling in maintenance of β‐cell mass comes from the Otsuka Long‐Evans Tokushima Fatty (OLETF) rat. This rat has a naturally occurring mutation in the CCKAR, and develops obesity and diabetes. The OLETF rat shows increased β‐cell apoptosis and ultimately reduced β‐cell mass26, 28. There is also some evidence that CCKBR signaling might be relevant in protection from β‐cell apoptosis. Gastrin, which predominantly signals through the CCKBR, can reduce β‐cell apoptosis after partial pancreatectomy43. Combined therapy with gastrin and GLP‐1 can also reduce β‐cell apoptosis44, 45, but these studies are confounded by the fact that treated animals have improved glycemia and therefore less β‐cell stress. Regardless of the receptor involved in CCK signaling, evidence suggests that GLP‐1 and CCK signaling are both clearly important for β‐cell survival. Although the mechanism is still unclear in vivo, it is likely that GLP‐1 regulation of β‐cell CCK in the intact islet is a major contributor to stress adaptation and enhanced β‐cell survival.
Current and future status of incretin based therapies to treat diabetes
The use of GLP‐1 mimetics and dipeptidyl peptidase‐4 inhibitors for the treatment of type 2 diabetes is increasing rapidly, and several recent clinical trials have also examined their efficacy in the treatment of type 1 diabetes46, 47. Although GLP‐1‐based therapy seems to reduce dependence on insulin in type 1 diabetics, it remains to be seen whether long‐term use in either type 1 or type 2 diabetes can directly impact β‐cell mass to prevent further decline. It is clear that higher‐powered and longer‐term studies will be required, and examination of ongoing registered clinical trials suggests that we will see more of these types of studies in the near future.
We have described a novel mechanism whereby GLP‐1‐directed therapies might exert protective effects on the β‐cell through activation of CCK production. We also provide evidence that activation of intra‐islet GLP‐1 signaling is a natural physiological response, or adaptation, to obesity5. It is of note that our proposed intra‐islet incretin axis is stimulated by obesity. This shows that one or more obesity driven factors is regulating this system. Future work will be necessary to determine the factor(s) responsible and the mechanism by which they activate an anti‐apoptotic adaptation to obesity. Many questions remain regarding if or how GLP‐1 mimetics could contribute to β‐cell mass preservation in individuals with diabetes.
It was recently shown that acute treatment of mice on a HFD with either a combination of CCK and GLP‐1 or a novel CCK/GLP‐1 hybrid peptide can lower blood glucose and improve glucose tolerance48, 49. Synergy between CCK/gastrin and GLP‐1 has also been shown elsewhere in HFD and ob/ob mouse models50. Given what we now know regarding the ability of both GLP‐1 and CCK to regulate one another and prevent β‐cell death, we propose that long‐term treatment with these hormones might impact islet peptide production to allow maintenance of β‐cell mass. Examination of how GLP‐1 and CCK specifically signal in the islet might also provide new therapeutic targets that could minimize non‐islet side effects. Importantly, putting this work into the context of disrupted signaling events in diabetes will provide critical mechanistic insight for future development of targeted receptor agonists as potential therapies.
Disclosure
The authors declare no conflict of interest.
Acknowledgments
This work was carried out using facilities and resources from the William S Middleton Memorial Veterans Hospital. This work does not represent the views of the Department of Veterans Affairs or the USA government. AKL is supported by the National Institutes of Health, National Institute of Diabetes, Digestive and Kidney Diseases (NIH‐NIDDK) Grant 1K01DK102492. DBD is supported by the United States Department of Veterans Affairs Grant 1101BX001880 and the University of Wisconsin.
J Diabetes Investig 2016; 7: 44–49
This article is based on the presentations given by the authors at a symposium, Incretin 2015, July 29–31, 2015, Vancouver, BC Canada.
References
- 1. Oleson BJ, McGraw JA, Broniowska KA, et al Distinct differences in the responses of the human pancreatic β‐cell line EndoC‐βH1 and human islets to proinflammatory cytokines.. Am J Physiol Regul Integr Comp Physiol 2015; 309: R525–R534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kilimnik G, Kim A, Steiner DF, et al Intraislet production of GLP‐1 by activation of prohormone convertase 1/3 in pancreatic α‐cells in mouse models of β‐cell regeneration. Islets 2010; 2: 149–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lavine JA, Raess PW, Stapleton DS, et al Cholecystokinin is up‐regulated in obese mouse islets and expands β‐cell mass by increasing β‐cell survival. Endocrinology 2010; 151: 3577–3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ellingsgaard H, Hauselmann I, Schuler B, et al Interleukin‐6 enhances insulin secretion by increasing glucagon‐like peptide‐1 secretion from L cells and alpha cells. Nat Med 2011; 17: 1481–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Linnemann AK, Neuman JC, Battiola TJ, et al Glucagon‐like peptide‐1 Regulates Cholecystokinin Production in beta‐cells to Protect from Apoptosis. Mol Endocrinol 2015; 7: 978–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rehfeld JF. Incretin physiology beyond glucagon‐like peptide 1 and glucose‐dependent insulinotropic polypeptide: cholecystokinin and gastrin peptides. Acta Physiol 2011; 201: 405–411. [DOI] [PubMed] [Google Scholar]
- 7. Lavine JA, Attie AD. Gastrointestinal hormones and the regulation of β‐cell mass. Ann N Y Acad Sci 2010; 1212: 41–58. [DOI] [PubMed] [Google Scholar]
- 8. Donath MY, Burcelin R. GLP‐1 effects on islets: hormonal, neuronal, or paracrine? Diabetes Care 2013; 2: S145–S148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Deacon CF, Johnsen AH, Holst JJ. Degradation of glucagon‐like peptide‐1 by human plasma in vitro yields an N‐terminally truncated peptide that is a major endogenous metabolite in vivo. J Clin Endocrinol Metab 1995; 80: 952–957. [DOI] [PubMed] [Google Scholar]
- 10. Hansen L, Deacon CF, Orskov C, et al Glucagon‐like peptide‐1‐(7‐36)amide is transformed to glucagon‐like peptide‐1‐(9‐36)amide by dipeptidyl peptidase IV in the capillaries supplying the L cells of the porcine intestine. Endocrinology 1999; 140: 5356–5363. [DOI] [PubMed] [Google Scholar]
- 11. Calanna S, Christensen M, Holst JJ, et al Secretion of glucagon‐like peptide‐1 in patients with type 2 diabetes mellitus: systematic review and meta‐analyses of clinical studies. Diabetologia 2013; 56: 965–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dockray GJ. Cholecystokinin. Curr Opin Endocrinol Diabetes Obes 2012; 19: 8–12. [DOI] [PubMed] [Google Scholar]
- 13. Sandberg E, Ahren B, Tendler D, et al Cholecystokinin (CCK)‐33 stimulates insulin secretion from the perfused rat pancreas: studies on the structure‐activity relationship. Pharmacol Toxicol 1988; 63: 42–45. [DOI] [PubMed] [Google Scholar]
- 14. Keller MP, Choi Y, Wang P, et al A gene expression network model of type 2 diabetes links cell cycle regulation in islets with diabetes susceptibility. Genome Res 2008; 18: 706–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hansen TV. Cholecystokinin gene transcription: promoter elements, transcription factors and signaling pathways. Peptides 2001; 22: 1201–1211. [DOI] [PubMed] [Google Scholar]
- 16. Deavall DG, Raychowdhury R, Dockray GJ, et al Control of CCK gene transcription by PACAP in STC‐1 cells. Am J Physiol Gastrointest Liver Physiol 2000; 279: G605–G612. [DOI] [PubMed] [Google Scholar]
- 17. Nielsen FC, Pedersen K, Hansen TV, et al Transcriptional regulation of the human cholecystokinin gene: composite action of upstream stimulatory factor, Sp1, and members of the CREB/ATF‐AP‐1 family of transcription factors. DNA Cell Biol 1996; 15: 53–63. [DOI] [PubMed] [Google Scholar]
- 18. Wang Y, Chandra R, Samsa LA, et al Amino acids stimulate cholecystokinin release through the Ca2 + ‐sensing receptor. Am J Physiol Gastrointest Liver Physiol 2011; 300: G528–G537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chandra R, Wang Y, Shahid RA, et al Immunoglobulin‐like domain containing receptor 1 mediates fat‐stimulated cholecystokinin secretion. J Clin Invest 2013; 123: 3343–3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Irwin N, Frizelle P, Montgomery IA, et al Beneficial effects of the novel cholecystokinin agonist (pGlu‐Gln)‐CCK‐8 in mouse models of obesity/diabetes. Diabetologia 2012; 55: 2747–2758. [DOI] [PubMed] [Google Scholar]
- 21. Irwin N, Montgomery IA, Moffett RC, et al Chemical cholecystokinin receptor activation protects against obesity‐diabetes in high fat fed mice and has sustainable beneficial effects in genetic ob/ob mice. Biochem Pharmacol 2013; 85: 81–91. [DOI] [PubMed] [Google Scholar]
- 22. Bregenholt S, Møldrup A, Blume N, et al The long‐acting glucagon‐like peptide‐1 analogue, liraglutide, inhibits β‐cell apoptosis in vitro. Biochem Biophys Res Commun 2005; 330: 577–584. [DOI] [PubMed] [Google Scholar]
- 23. Buteau J, Spatz ML, Accili D. Transcription factor FoxO1 mediates glucagon‐like peptide‐1 effects on pancreatic β‐cell mass. Diabetes 2006; 55: 1190–1196. [DOI] [PubMed] [Google Scholar]
- 24. Farilla L, Bulotta A, Hirshberg B, et al Glucagon‐like peptide 1 inhibits cell apoptosis and improves glucose responsiveness of freshly isolated human islets. Endocrinology 2003; 144: 5149–5158. [DOI] [PubMed] [Google Scholar]
- 25. Eizirik DL, Grieco FA. On the immense variety and complexity of circumstances conditioning pancreatic β‐cell apoptosis in type 1 diabetes. Diabetes 2012; 61: 1661–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rondas D, Bugliani M, D'Hertog W, et al Glucagon‐like peptide‐1 protects human islets against cytokine‐mediated β‐cell dysfunction and death: a proteomic study of the pathways involved. J Proteome Res 2013; 12: 4193–4206. [DOI] [PubMed] [Google Scholar]
- 27. Sarkar SA, Gunter J, Bouchard R, et al Dominant negative mutant forms of the cAMP response element binding protein induce apoptosis and decrease the anti‐apoptotic action of growth factors in human islets. Diabetologia 2007; 50: 1649–1659. [DOI] [PubMed] [Google Scholar]
- 28. Wideman RD, Yu ILY, Webber TD, et al Improving function and survival of pancreatic islets by endogenous production of glucagon‐like peptide 1 (GLP‐1). Proc Natl Acad Sci USA 2006; 103: 13468–13473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lavine JA, Kibbe CR, Baan M, et al Cholecystokinin expression in the β‐cell leads to increased β‐cell area in aged mice and protects from streptozotocin‐induced diabetes and apoptosis. Am J Physiol Endocrinol Metab 2015; 309: E819–E828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dufresne M, Seva C, Fourmy D. Cholecystokinin and gastrin receptors. Physiological reviews. Am Physiol Soc 2006; 86: 805–847. [DOI] [PubMed] [Google Scholar]
- 31. Bourassa J, Lainé J, Kruse ML, et al Ontogeny and species differences in the pancreatic expression and localization of the CCK(A) receptors. Biochem Biophys Res Commun 1999; 260: 820–828. [DOI] [PubMed] [Google Scholar]
- 32. Morisset J, Julien S, Laine J. Localization of cholecystokinin receptor subtypes in the endocine pancreas. J Histochem Cytochem 2003; 51: 1501–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Konno K, Takahashi‐Iwanaga H, Uchigashima M, et al Cellular and subcellular localization of cholecystokinin (CCK)‐1 receptors in the pancreas, gallbladder, and stomach of mice. Histochem Cell Biol 2014; 143: 301–312. [DOI] [PubMed] [Google Scholar]
- 34. Ning S, Zheng W, Su J, et al Different downstream signaling of CCKAR regulates distinct functions of CCK in pancreatic β cells. Br J Pharmacol 2015; 172: 5050–5067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Marchetti P, Lupi R, Bugliani M, et al A local glucagon‐like peptide 1 (GLP‐1) system in human pancreatic islets. Diabetologia 2012; 55: 3262–3272. [DOI] [PubMed] [Google Scholar]
- 36. Vasu S, Moffett RC, Thorens B, et al Role of Endogenous GLP‐1 and GIP in beta cell compensatory responses to insulin resistance and cellular stress. PLoS ONE 2014; 9: e101005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shu L, Matveyenko AV, Kerr‐Conte J, et al Decreased TCF7L2 protein levels in type 2 diabetes mellitus correlate with downregulation of GIP‐ and GLP‐1 receptors and impaired beta‐cell function. Hum Mol Genet 2009; 18: 2388–2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Flamez D, Gilon P, Moens K, et al Altered cAMP and Ca2 + signaling in mouse pancreatic islets with glucagon‐like peptide‐1 receptor null phenotype. Diabetes 1999; 48: 1979–1986. [DOI] [PubMed] [Google Scholar]
- 39. Li Y. Glucagon‐like peptide‐1 receptor signaling modulates beta cell apoptosis. J Biol Chem 2002; 278: 471–478. [DOI] [PubMed] [Google Scholar]
- 40. Quoyer J, Longuet C, Broca C, et al GLP‐1 mediates antiapoptotic effect by phosphorylating Bad through a β‐arrestin 1‐mediated ERK1/2 activation in pancreatic β‐cells. J Biol Chem 2010; 285: 1989–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shin S, Le Lay J, Everett LJ, et al CREB mediates the insulinotropic and anti‐apoptotic effects of GLP‐1 signaling in adult mouse. Mol Metab 2014; 3: 803–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ali S, Lamont BJ, Charron MJ, et al Dual elimination of the glucagon and GLP‐1 receptors in mice reveals plasticity in the incretin axis. J Clin Invest 2011; 121: 1917–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kim A, Miller K, Jo J, et al Islet architecture: a comparative study. Islets 2009; 1: 129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Suarez‐Pinzon WL, Power RF, Yan Y, et al Combination therapy with glucagon‐like peptide‐1 and gastrin restores normoglycemia in diabetic NOD mice. Diabetes 2008; 57: 3281–3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dalboge LS, Almholt DLC, Neerup TSR, et al The novel GLP‐1‐gastrin dual agonist ZP3022 improves glucose homeostasis and increases β‐cell mass without affecting islet number in db/db mice. J Pharmacol Exp Ther 2014; 350: 353–360. [DOI] [PubMed] [Google Scholar]
- 46. Pieber TR, Deller S, Korsatko S, et al Counter‐regulatory hormone responses to hypoglycaemia in people with type 1 diabetes after 4 weeks of treatment with liraglutide adjunct to insulin: a randomized, placebo‐controlled, double‐blind, crossover trial. Diabetes Obes Metab 2015; 17: 742–750. [DOI] [PubMed] [Google Scholar]
- 47. Schopman JE, Hoekstra JBL, Frier BM, et al Effects of sitagliptin on counter‐regulatory and incretin hormones during acute hypoglycaemia in patients with type 1 diabetes: a randomized double‐blind placebo‐controlled crossover study. Diabetes Obes Metab 2015; 17: 546–553. [DOI] [PubMed] [Google Scholar]
- 48. Irwin N, Hunter K, Montgomery IA, et al Comparison of independent and combined metabolic effects of chronic treatment with (pGlu‐Gln)‐CCK‐8 and long‐acting GLP‐1 and GIP mimetics in high fat‐fed mice. Diabetes Obes Metab 2013; 15: 650–659. [DOI] [PubMed] [Google Scholar]
- 49. Irwin N, Pathak V, Flatt PR. A Novel CCK‐8/GLP‐1 Hybrid Peptide Exhibiting Prominent Insulinotropic, Glucose‐Lowering, and Satiety Actions With Significant Therapeutic Potential in High‐Fat–Fed Mice. Diabetes 2015; 64: 2996–3009. [DOI] [PubMed] [Google Scholar]
- 50. Trevaskis JL, Sun C, Athanacio J, et al Synergistic metabolic benefits of an exenatide analogue and cholecystokinin in diet‐induced obese and leptin‐deficient rodents. Diabetes Obes Metab 2015; 17: 61–73. [DOI] [PubMed] [Google Scholar]