

Abstract
We reported that native incretins, liraglutide and dipeptidyl peptidase‐4 inhibitors (DPP‐4i) all confer an anti‐atherosclerotic effect in apolipoprotein E‐null (Apoe −/−) mice. We confirmed the anti‐atherogenic property of incretin‐related agents in the mouse wire injury model, in which the neointimal formation in the femoral artery is remarkably suppressed. Furthermore, we showed that DPP‐4i substantially suppresses plaque formation in coronary arteries with a marked reduction in the accumulation of macrophages in cholesterol‐fed rabbits. DPP‐4i showed an anti‐atherosclerotic effect in Apoe −/− mice mainly through the actions of glucagon‐like peptide‐1 and glucose‐dependent insulinotropic polypepide. However, the dual incretin receptor antagonists partially attenuated the suppressive effect of DPP‐4i on atherosclerosis in diabetic Apoe −/− mice, suggesting an incretin‐independent mechanism. Exendin‐4 and glucose‐dependent insulinotropic polypepide elicited cyclic adenosine monophosphate generation, and suppressed the lipopolysaccharide‐induced gene expression of inflammatory molecules, such as interleukin‐1β, interleukin‐6 and tumor necrosis factor‐α, in U937 human monocytes. This suppressive effect, however, was attenuated by an inhibitor of adenylate cyclase and mimicked by 8‐bromo‐cyclic adenosine monophosphate or forskolin. DPP‐4i substantially suppressed the lipopolysaccharide‐induced expression of inflammatory cytokines without affecting cyclic adenosine monophosphate generation or cell proliferation. DPP‐4i more strongly suppressed the lipopolysaccharide‐induced gene expression of inflammatory molecules than incretins, most likely through inactivation of CD26. Glucagon‐like peptide‐1 and glucose‐dependent insulinotropic polypepide suppressed oxidized low‐density lipoprotein‐induced macrophage foam cell formation in a receptor‐dependent manner, which was associated with the downregulation of acyl‐coenzyme A cholesterol acyltransferase‐1 and CD36, as well as the up‐regulation of adenosine triphosphate‐binding cassette transporter A1. Our studies strongly suggest that incretin‐related agents have favorable effects on macrophage‐driven atherosclerosis in experimental animals.
Keywords: Incretins, Atherosclerosis, Vascular inflammation
Introduction
It is well known that patients with type 2 diabetes mellitus are at increased risk of cardiovascular (CV) disease. Elevated plasma glucose levels lead to increased CV risk combined with associated comorbidities, such as obesity, hypertension and dyslipidemia. Incretin‐based therapy, such as glucagon‐like peptide‐1 (GLP‐1) receptor agonists (RA) and dipeptidyl peptidase‐4 inhibitors (DPP‐4i), have been widely used in clinical practice. Incretin‐based therapy is expected to provide CV benefits because of its amelioration of fasting and postprandial hyperglycemia without hypoglycemia or bodyweight gain1, 2. Furthermore, lipid‐lowering and hypotensive effects were reported3, 4, 5. There have been a number of animal studies showing that GLP‐1RA (GLP‐1 receptor agonists) or DPP‐4i exert an anti‐atherosclerotic effect in hypercholestetolemic mice6, 7, 8, 9, 10, 11, 12. Because these mice are not diabetic, direct prevention of the effect of GLP‐1RA or DPP‐4i on atherosclerosis beyond glucose has been implicated. Unlike animal studies, recent clinical trials have failed to show a favorable effect of DPP‐4i on the suppression of CV outcomes in type 2 diabetic patients13, 14, 15. However, the majority of subjects enrolled in these trials already had CV diseases and were being treated with cardioprotective drugs, such as statins. Additionally, the observation periods were only a few years, which make the evaluation of the anti‐atherogenic potential of DPP‐4 inhibitors difficult, especially in the early stages of atherosclerosis. In the present mini‐review, we introduce representative animal studies, including our own study, implicating the favorable effects of GLP‐1, glucose‐dependent insulinotropic polypepide (GIP) and DPP‐4i on atherosclerosis.
GLP‐1 and GIP Prevent the Development of Atherosclerosis in Apolipoprotein E Knockout Mice
Arakawa et al.16 reported for the first time that exendin(Ex)‐4, a GLP‐1RA, reduced monocyte adhesion to the endothelium of aorta and suppressed atherosclerotic lesions in apolipoprotein E knockout (Apoe −/−) mice. Our group subsequently reported that chronic infusion of native GLP‐1 significantly suppressed atherosclerotic lesions and macrophage infiltration in the aortic wall in Apoe −/− mice17. These effects were cancelled by co‐infusions with a specific antagonist for GLP‐1 receptors, including Exendin (9‐39; Ex‐9). Surprisingly, the infusion of native GIP also suppressed the development of atherosclerotic lesions in Apoe −/− mice as potently as GLP‐1 infusion17, 18. This suppression was completely reversed by co‐infusions with a specific antagonist for GIP receptors, Pro(3)GIP. These results suggest that both native incretins confer an anti‐atherosclerotic effect through their own receptors. Gaspari et al.19 reported that a GLP‐1RA, liraglutide, inhibited the progression of early onset, low‐burden atherosclerotic disease in the Apoe −/− mouse mode, whereas no significant effect of liraglutide on the progression of late onset, high‐burden atherosclerotic disease was observed. That study might reasonably explain why incretin‐based therapy failed to prevent secondary events in diabetic patients premorbid for CV diseases.
Anti‐Atherogenic Effect of DPP‐4i Inhibitors is Mainly Incretin‐Dependent
Given that GLP‐1 and GIP both have anti‐atherosclerotic effects, increases in both active GLP‐1 and GIP by DPP‐4i might synergistically suppress the development of atherosclerosis. Indeed, a number of publications have shown that DPP‐4i powerfully suppresses the development of atherosclerotic lesions in Apoe −/− mice6, 7, 8, 9, 10, 11. Endogenous GLP‐1 levels are increased by just two‐ to threefold by DPP‐4i; however, the suppressive effect of DPP‐4i on atherosclerotic lesions was comparable with that of liraglutide, providing a much higher concentration of GLP‐1 in the blood20. Therefore, increased GIP might participate in the anti‐atherogenic effects of DPP‐4i. It remains unknown, however, whether this anti‐atherogenic property of DPP‐4i can be credited to higher endogenous levels of the active incretins GLP‐1 and GIP. We attempted to determine whether the anti‐atherogenic property of DPP‐4 inhibitor is derived from increased levels of the endogenous active incretins GLP‐1 and GIP or from other mechanisms. To achieve this, we administered GLP‐1 and GIP receptor antagonists to Apoe −/− mice that were simultaneously treated with DPP‐4i, and examined how an incretin receptor blockade attenuated the anti‐atherosclerotic effect of the DPP‐4 inhibitor. The two receptor blockers in combination completely abolished the anti‐atherosclerotic effect of vildagliptin in non‐diabetic mice10. We subsequently attempted the same experiment in streptozotocin‐induced diabetic Apoe −/− mice showing further‐progressed atherosclerotic lesions. Unlike non‐diabetic Apoe −/− mice, infusions with dual antagonists incompletely abolish the suppressive effect of vildagliptin on atherosclerosis, implying that vildagliptin partly confers an anti‐atherogenic effect beyond that from the incretins in the diabetic animals10. We estimated that incretin‐dependent and incretin‐independent mechanisms equally contributed to the anti‐atherosclerotic effect of vildagliptin in the diabetic Apoe −/− mice. As we will mention later, much attention has been given to the new hypothesis that DPP‐4is confer anti‐atherogenic effects by blocking the pro‐inflammatory and pro‐atherogenic properties of DPP‐4/CD26. We speculate that vascular inflammation is largely enhanced in diabetic Apoe −/− mice; thus, vildagliptin could show remarkable anti‐inflammatory effects, particularly in diabetic mice, beyond the action of incretins.
Incretins and DPP‐4 Inhibitors Suppress Neointimal Hyperplasia in Experimental Restenosis Models
As previously mentioned, the anti‐atherogenic effects of GLP‐1, GIP and DPP‐4 inhibitors are supported by in vivo and in vitro studies, and these findings have raised a new question as to whether incretins and DPP‐4 inhibitors can protect against other vascular diseases. Restenosis is an exaggerated healing process that arises after arterial interventions, such as percutaneous coronary angioplasty with or without stenting, and leads to decreased blood flow by luminal narrowing21, ultimately requiring an additional intervention. The development of restenosis is mainly attributed to the hyperplasia of regenerated intima, which is known as neointima, and consists of vascular smooth muscle cells and extracellular matrix. Although the pathology of neointimal hyperplasia is different from that of atherosclerotic plaque formation, there are a number of similarities in their mechanisms. Among the mechanisms involved, the migration and proliferation of vascular smooth muscle cells (VSMCs) have been shown to play an important role22. Because GLP‐1 and GIP receptors are expressed abundantly in VSMCs, researchers have focused on the effects of incretins and DPP‐4 inhibitors on restenosis. Denudation of the endothelium and apoptosis of medial VSMCs by the mechanical stress of arterial interventions is the initiating event in neointimal formation. In preclinical restenosis models, these events are induced by withdrawing an inflated balloon catheter from the rat carotid artery or by inserting a guidewire into the mouse femoral artery23, 24. These models are useful to evaluate therapeutic effects on restenosis because of their resemblance to the pathological characteristics of restenosis in humans.
In wild‐type mice, exendin‐4(Ex‐4), a GLP‐1 receptor agonist, reduces neointimal hyperplasia after femoral artery wire injury without affecting the metabolic parameters, including glucose tolerability25, 26. Similarly, Ex‐4 showed a protective effect against neointimal hyperplasia with reduced VSMC proliferation in a rat model of restenosis27. Furthermore, in vitro studies show supportive data that Ex‐4 suppressed rat VSMC proliferation stimulated by platelet‐derived growth factor. This effect of Ex‐4 was completely canceled by the co‐administration of a protein kinase A (PKA) inhibitor26, suggesting that the GLP‐1 receptor‐cAMP‐PKA pathway is involved in the suppression of neointimal hyperplasia. DPP‐4 inhibitors have also been investigated using experimental restenosis models. In wild‐type mice, linagliptin suppresses neointimal hyperplasia after an arterial injury28. In addition, the effect of DPP‐4is was examined in Otsuka Long‐Evans Tokushima fatty rats, which is a model of type 2 diabetes with insulin resistance29. In that study, sitagliptin significantly improved glucose tolerability and suppressed neointimal hyperplasia after a carotid artery balloon injury in a dose‐dependent manner. This effect was accompanied with reduced cell proliferation and increased cell apoptosis in the neointima. Furthermore, sitagliptin reduced VSMC proliferation and migration in vitro, showing that the inhibition of DPP‐4 activity directly affects VSMCs, independent of the increased activity of incretins or other bioactive peptides. We investigated the effect of GIP on restenosis in a mouse model. GIP treatment significantly suppressed neointimal hyperplasia accompanied with reduced cell proliferation in the neointima compared with vehicle. In accordance with the in vivo findings, GIP suppressed VSMC proliferation stimulated by platelet‐derived growth factor in vitro, showing the possibility that the suppression of neointimal hyperplasia is attributed to the direct effects of GIP on VSMCs. Those studies and our data show that an incretin‐based therapy would be a novel and beneficial therapeutic approach for the treatment of arterial restenosis.
DPP‐4i Suppresses Atherosclerotic Lesions in the Aorta and Coronary Arteries by Decreasing Macrophage Infiltration in Cholesterol‐Fed Rabbits
It remains unknown whether DPP‐4is exert anti‐atherogenic effects in coronary arteries and the aorta. Rabbits are bigger than rodents, and several earlier publications have shown that hypercholesterolemic rabbits develop severe atherosclerotic lesions in their coronary arteries30, 31. We examined the effect of anagliptin, a DPP‐4i, on atherosclerosis development in the aorta and coronary arteries in high‐cholesterol diet‐fed rabbits. Dietary cholesterol intake markedly increased the serum total cholesterol levels, and the most striking increase was observed in a major lipoprotein, very low‐density lipoprotein. Anagliptin slightly decreased both total cholesterol and very low‐density lipoprotein cholesterol, as well as the cholesterol absorption markers sitosterol and campesterol, although not significantly. Severe hypercholesterolemia resulted in the development of atherosclerosis in the aorta. Anagliptin remarkably suppressed aortic atherosclerosis, which was comparable with the suppression observed in Apoe −/− mice. Atherosclerotic lesions were clearly observed in the coronary arteries, where the mean intima‐media area was enlarged and intimal formation developed. Anagliptin treatment attenuated the intima‐media area and the intimal area. Alpha‐SMA (smooth muscle actin)‐positive and macrophage‐positive areas in the coronary arteries were remarkably suppressed after anagliptin treatment. The aortic lesion ratio and the coronary intima area were correlated with each other, and each lesion correlated with total cholesterol in whole cholesterol‐fed rabbits. Gene expression of the pro‐inflammatory cytokines, tumor necrosis factor (TNF)‐αand interleukin (IL)‐6 in the carotid arteries were markedly reduced, and vascular DPP‐4 activity was reduced after anagliptin treatment. Because the rabbits were not diabetic and anagliptin treatment did not affect glucose metabolism, the anti‐atherosclerotic effect of anagliptin could be attributed to the pleiotropic effect of DPP‐4is beyond their glucose‐lowering effect. Vittone et al.32 reported that sitagliptin treatment did not reduce the atherosclerotic lesion area in Apoe −/− mice, although macrophage infiltration into the plaque was significantly suppressed. Ta et al.6 reported that alogliptin suppressed aortic plaque formation in diabetic Apoe −/− mice, but not in non‐diabetic Apoe −/− mice. These results might be based on the technical difficulty of measuring vessel lesions in small animals. The merit of using rabbits for atherosclerosis models is that atherosclerotic lesions in the aorta are easily visible without special staining. This simple approach for the identification of atherosclerotic lesions convinced us of a powerful anti‐atherosclerotic effect of the DPP‐4i. Nader et al.33 recently reported that sitagliptin suppressed atherosclerotic lesion development in 0.5% cholesterol and 1% methionine‐fed rabbits, which is in good agreement with our results, and suggests that the anti‐atherosclerotic effect of DPP‐4i is applicable to animals that are bigger than rodents.
Incretins and the DPP‐4i Independently Suppress the Expression of Pro‐Inflammatory Genes in Monocytes/Macrophages
Atherosclerosis is a chronic inflammatory disease in the vasculature, and monocytes/macrophages play a crucial role in atherogenesis. It has been hypothesized that DPP‐4, which is known as CD26, is an inflammatory cytokine that is involved in atherosclerosis; hence, DPP4i might suppress the progression of atherosclerosis by inhibiting CD26‐mediated inflammation34, 35, 36, 37. Ikeda et al.38 showed that soluble CD26 enhanced the expression of TNF‐a and IL‐6 messenger ribonucleic acid and protein in THP‐1 human monocytes In this context, the anti‐inflammatory effect of DPP4i would be totally incretin‐independent. Incretins exert their actions through their own receptors in various tissues, including pancreatic islets, and cyclic adenosine monophosphate (cAMP) acts as a second messenger of incretin receptors39. Panjwani et al.40 reported that classical GLP‐1 receptors were not present in mouse macrophages, and doubted the anti‐atherogenic effect of GLP‐1 receptor agonists. We showed that the GLP‐1 receptor and GIP receptor genes were present in mouse and human monocytes, but were dramatically downregulated when differentiated to macrophages or foam cells10, 17, 30. Thus far, it remains to be elucidated whether incretins exert anti‐inflammatory actions through their own receptors in monocytes. We studied the direct anti‐inflammatory effect of incretins and DPP4i in cultured monocytes. In U937 human monocytes, the active form of the incretin receptor agonists, Ex‐4, GIP(1‐42), and D‐(Ala2)GIP, elicited cAMP generation, whereas the inactive form, GLP‐1(9‐36)amide and GIP (3‐42), did not, suggesting that the incretins' signal enters monocytes through classical incretin receptors. 8‐Bromo‐cAMP (a cAMP mimetic) or forskolin (a cAMP stimulator) attenuated lipopolysaccharide (LPS)‐induced IL‐6 gene expression. Ex‐4 or GIP significantly suppressed the expression of IL‐1β and IL‐6 that was stimulated by LPS. The suppression with incretins were completely reversed by incretin receptor antagonists (Ex‐9 or [Pro3]GIP), an adenylate cyclase inhibitor (SQ22,536) or PKA inhibitor. Therefore, the anti‐inflammatory effect of incretins was mediated through the incretin receptors–cAMP–PKA pathway. Teneligliptin, a DPP‐4i, substantially suppressed the LPS‐induced gene expression of IL‐1β, IL‐6 and TNF‐α without affecting cell viability or proliferation. These results suggest that GLP‐1 and GIP have a similar anti‐inflammatory effect in human monocytes in a cAMP/PKA‐dependent‐manner, whereas DPP‐4i exerts an anti‐inflammatory effect independent of incretins. We compared the anti‐inflammatory effect between GIP, EX‐4 and teneligliptin at the same concentration. Teneligliptin more strongly suppressed the LPS‐induced gene expression of inflammatory molecules than incretins without affecting cell proliferation. Similar to DPP‐4i, the anti‐CD26 antibody or CD26 small inferring ribonucleic acid remarkably suppressed pro‐inflammatory cytokine expression. These results suggest that the anti‐inflammatory effect of teneligliptin is mediated by the inactivation of CD26. The mechanisms of the anti‐inflammatory effect of incretin‐related agents still remains largely unknown; however, experimental data have shown that GLP‐1 or DPP‐4 inhibitors could shift the polarization profile of macrophages from the M1 phenotype towards the M2 phenotype41, 42. Further study is expected to elucidate the role of incretin‐related agents in macrophage differentiation.
Incretins Suppresses Foam Cell Formation in Monocytes/Macrophages
The development of atherosclerosis is influenced by abnormalities in cellular cholesterol homeostasis, which are shown by the subendothelial accumulation of lipid‐laden macrophage foam cells. The accumulated foam cells in the subendothelial space create a lipid‐rich plaque, which is the initial process of atherosclerosis43. Foam cell formation is regulated by several factors, including scavenger receptors, such as CD3644; acyl‐coenzyme A:cholesterol acyltransferase 1, a rate‐limiting enzyme for the esterification of cholesterol46; and free‐cholesterol efflux, which is mediated by adenosine triphosphate‐binding cassette transporter A145. We focused on the effect of GIP with regard to the development of aortic atherosclerotic lesions and macrophage foam cell formation, as well as related molecules in mice. Mouse peritoneal macrophages were obtained from ascites by injection of thioglycolate. Foam cell formation was determined by the incorporation of (3H)‐oleate into cholesteryl‐oleate, which was stimulated by oxidized‐low‐density lipoprotein. We previously reported that the infusions of GLP‐1 and GIP for 4 weeks significantly suppressed foam cell formation in Apo E‐null mice17. Similar to Apo E null mice, GIP infusion for 4 weeks significantly suppressed foam cell formation in C57BL/6 mice. Co‐infusion with Pro3GIP abolished the suppressive effect of GIP on foam cell formation. Pro3GIP infusion alone significantly stimulated foam cell formation. We carried out a similar experiment using GIPR‐knockout mice that were a kind gift from Professor Yamada from Akita University. As expected, the suppressive effect of GIP on foam cell formation was not observed in the GIPR (GIP receptors)‐KO mice. GLP‐1 and GIP significantly stimulated adenosine triphosphate‐binding cassette transporter A1 expression, and suppressed acyl‐coenzyme A:cholesterol acyltransferase 1 and CD 36 expression in human monocyte‐derived macrophages20. Recently, Dai et al.47 reported that DPP‐4i directly repress foam cell formation through the inhibition of CD36 and a scavenger receptor, LOX‐1. We confirmed the direct suppressive effect of DPP4i on foam cell formation in mouse peritoneal macrophages obtained from db/db diabetic mice. Foam cell formation is enhanced by the co‐presence of macrophage inflammation. Therefore, it is highly likely that the anti‐inflammatory property of incretin‐related agents is associated with the suppressive effect on macrophage foam cell formation.
Figure 1 shows the possible mechanisms of the anti‐atherosclerotic properties of incretin‐related agents based on animal studies. GLP‐1, GIP and DPP‐4 inhibitors all confer anti‐atherosclerotic effects in experimental animals through the suppression of inflammation and foam cell formation in monocytes/macrophages. Our studies strongly suggest that incretin‐related agents have favorable effects on atherosclerosis.
Figure 1.
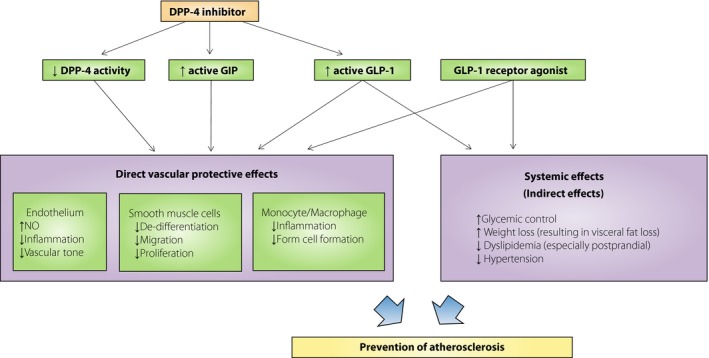
Possible mechanisms of the exerted anti‐atherosclerotic properties of incretin‐related agents.
Disclosure
The authors declare no conflict of interest.
Acknowledgments
The authors thank Michishige Terasaki, Munenori Hiromura, Masaharu Nagashima, Kyoko Kohashi, Masako Tomoyasu, Kyoko Shinmura and Hideki Kushima at Showa University, and Takuya Watanabe at Tokyo University Pharmacy and Life Sciences for their excellent work on this manuscript. The authors received financial support from Merck Sharp & Dohme (MSD) K.K., AstraZeneca K.K., Mitsubishi Tanabe Pharma Corporation, Takeda Pharmaceutical Company, Kowa Pharmaceutical Company, Eli Lilly Japan K.K., Novo Nordisk Pharma and Sanwa Kagaku Kenkyusho Co.
J Diabetes Investig 2016; 7: 80–86
This article is based on the presentations given by the authors at a symposium, Incretin 2015, July 29‐31, 2015, Vancouver, BC Canada.
References
- 1. Ussher JR, Drucker DJ. Cardiovascular biology of the incretin system. Endocr Rev 2012; 33: 187–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhong J, Gong Q, Goud A, et al Recent advances in dipeptidyl‐peptidase‐4 inhibition therapy: lessons from the bench and clinical trials. J Diabetes Res 2015; 2015: 606031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Matikainen N, Mänttäri S, Schweizer A, et al Vildagliptin therapy reduces postprandial intestinal triglyceride‐rich lipoprotein particles in patients with type 2 diabetes. Diabetologia 2006; 49: 2049–2057. [DOI] [PubMed] [Google Scholar]
- 4. Sun F, Wu S, Guo S, et al Impact of GLP‐1 receptor agonists on blood pressure, heart rate and hypertension among patients with type 2 diabetes: a systematic review and network meta‐analysis. Diabetes Res Clin Pract 2015; 110: 26–37. S0168‐8227(15)00341‐1 [DOI] [PubMed] [Google Scholar]
- 5. Lovshin JA, Zinman B. Blood pressure‐lowering effects of incretin‐based diabetes therapies. Can J Diabetes 2014; 38: 364–371. [DOI] [PubMed] [Google Scholar]
- 6. Ta NN, Schuyler CA, Li Y, et al DPP‐4 (CD26) inhibitor alogliptin inhibits atherosclerosis in diabetic apolipoprotein E‐deficient mice. J Cardiovasc Pharmacol 2011; 58: 157–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Matsubara J, Sugiyama S, Koichi S, et al A dipeptidyl peptidase‐4 inhibitor, des‐fluoro‐sitagliptin, improves endothelial function and reduces atherosclerotic lesion formation in apolipoprotein E‐deficient mice. J Am Coll Cardiol 2012; 59: 265–276. [DOI] [PubMed] [Google Scholar]
- 8. Ervinna N, Mita T, Yasunari E, et al Anagliptin, a DPP‐4 inhibitor, suppresses proliferation of vascular smooth muscles and monocyte inflammatory reaction and attenuates atherosclerosis in male apo E‐deficient mice. Endocrinology 2013; 154: 1260–1270. [DOI] [PubMed] [Google Scholar]
- 9. Terasaki M, Nagashima M, Watanabe T, et al Effects of PKF275‐055, a dipeptidyl peptidase‐4 inhibitor, on the development of atherosclerotic lesions in apolipoprotein E‐null mice. Metab Clin Exp 2012; 61: 974–979. [DOI] [PubMed] [Google Scholar]
- 10. Terasaki M, Nagashima M, Nohtomi K, et al Preventive effect of dipeptidyl peptidase‐4 inhibitor on atherosclerosis is mainly attributable to incretin's actions in nondiabetic and diabetic apolipoprotein E‐null mice. PLoS One 2013; 8: e70933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zeng Y, Li C, Guan M, et al The DPP‐4 inhibitor sitagliptin attenuates the progress of atherosclerosis in apolipoprotein‐E‐knockout mice via AMPK‐ and MAPK‐dependent mechanisms. Cardiovasc Diabetol 2014; 13: 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shah Z, Kampfrath T, Deiuliis JA, et al Long‐term dipeptidyl‐peptidase 4 inhibition reduces atherosclerosis and inflammation via effects on monocyte recruitment and chemotaxis. Circulation 2011; 124: 2338–2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Scirica BM, Bhatt DL, Braunwald E, et al SAVOR‐TIMI 53 Steering Committee and Investigators. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N Engl J Med 2013; 369: 1317–1326. [DOI] [PubMed] [Google Scholar]
- 14. White WB, Cannon CP, Heller SR, et al Alogliptin after acute coronary syndrome in patients with type 2 diabetes. N Engl J Med 2013; 369: 1327–1335. [DOI] [PubMed] [Google Scholar]
- 15. Green JB, Bethel MA, Armstrong PW, et al Effect of sitagliptin on cardiovascular outcomes in type 2 diabetes. N Engl J Med 2015; 373: 232–242. [DOI] [PubMed] [Google Scholar]
- 16. Arakawa M, Mita T, Azuma K, et al Inhibition of monocyte adhesion to endothelial cells and attenuation of atherosclerotic lesion by a glucagon‐like peptide‐1 receptor agonist, exendin‐4. Diabetes 2010; 59: 1030–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nagashima M, Watanabe T, Terasaki M, et al Native incretins prevent the development of atherosclerotic lesions in apolipoprotein E knockout mice. Diabetologia 2011; 54: 2649–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nogi Y, Nagashima M, Terasaki M, et al Glucose‐dependent insulinotropic polypeptide prevents the progression of macrophage‐driven atherosclerosis in diabetic apolipoprotein E‐null mice. PLoS One 2012; 7: e35683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gaspari T, Welungoda I, Widdop RE, et al The GLP‐1 receptor agonist liraglutide inhibits progression of vascular disease via effects on atherogenesis, plaque stability and endothelial function in an ApoE(‐/‐) mouse model. Diab Vasc Dis Res 2013; 10: 353–360. [DOI] [PubMed] [Google Scholar]
- 20. Tashiro Y, Sato K, Watanabe T, et al A glucagon‐like peptide‐1 analog liraglutide suppresses macrophage foam cell formation and atherosclerosis. Peptides 2014; 54: 19–26. [DOI] [PubMed] [Google Scholar]
- 21. Lespérance J, Bourassa MG, Schwartz L, et al Definition and measurement of restenosis after successful coronary angioplasty: implications for clinical trials. Am Heart J 1993; 125: 1394–1408. [DOI] [PubMed] [Google Scholar]
- 22. Orford JL, Selwyn AP, Ganz P, et al The comparative pathobiology of atherosclerosis and restenosis. Am J Cardiol 2000; 86: 6H–11H. [DOI] [PubMed] [Google Scholar]
- 23. Clowes AW, Schwartz SM. Significance of quiescent smooth muscle migration in the injured rat carotid artery. Circ Res 1985; 56: 139–145. [DOI] [PubMed] [Google Scholar]
- 24. Sata M, Maejima Y, Adachi F, et al A mouse model of vascular injury that induces rapid onset of medial cell apoptosis followed by reproducible neointimal hyperplasia. J Mol Cell Cardiol 2000; 32: 2097–2104. [DOI] [PubMed] [Google Scholar]
- 25. Goto H, Nomiyama T, Mita T, et al Exendin‐4, a glucagon‐like peptide‐1 receptor agonist, reduces intimal thickening after vascular injury. Biochem Biophys Res Commun 2011; 405: 79–84. [DOI] [PubMed] [Google Scholar]
- 26. Hirata Y, Kurobe H, Nishio C, et al Exendin‐4, a glucagon‐like peptide‐1 receptor agonist, attenuates neointimal hyperplasia after vascular injury. Eur J Pharmacol 2013; 699: 106–111. [DOI] [PubMed] [Google Scholar]
- 27. Eriksson L, Saxelin R, Röhl S, et al Glucagon‐like peptide‐1 receptor activation does not affect re‐endothelialization but reduces intimal hyperplasia via direct effects on smooth muscle cells in a nondiabetic model of arterial injury. J Vasc Res 2015; 52: 41–52. [DOI] [PubMed] [Google Scholar]
- 28. Terawaki Y, Nomiyama T, Kawanami T, et al Dipeptidyl peptidase‐4 inhibitor linagliptin attenuates neointima formation after vascular injury. Cardiovasc Diabetol 2014; 13: 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lim S, Choi SH, Shin H, et al Effect of a dipeptidyl peptidase‐IV inhibitor, des‐fluoro‐sitagliptin, on neointimal formation after balloon injury in rats. PLoS One 2012; 7: e35007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shiomi M, Koike T, Ito T. Contribution of the WHHL rabbit, an animal model of familial hypercholesterolemia, to elucidation of the anti‐atherosclerotic effects of statins. Atherosclerosis 2013; 231: 39–47. [DOI] [PubMed] [Google Scholar]
- 31. Wang Y, Niimi M, Nishijima K, et al Human apolipoprotein A‐II protects against diet‐induced atherosclerosis in transgenic rabbits. Arterioscler Thromb Vasc Biol 2013; 33: 224–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vittone F, Liberman A, Vasic D, et al Sitagliptin reduces plaque macrophage content and stabilises arteriosclerotic lesions in Apoe(‐/‐) mice. Diabetologia 2012; 55: 2267–2275. [DOI] [PubMed] [Google Scholar]
- 33. Nader MA. Sitagliptin ameliorates lipid profile changes and endothelium dysfunction induced by atherogenic diet in rabbits. Naunyn Schmiedebergs Arch Pharmacol 2014; 387: 433–444. [DOI] [PubMed] [Google Scholar]
- 34. Zhong J, Rao X, Rajagopalan S. An emerging role of dipeptidyl peptidase 4 (DPP4) beyond glucose control: potential implications in cardiovascular disease. Atherosclerosis 2013; 226: 305–314. [DOI] [PubMed] [Google Scholar]
- 35. Fadini GP, Avogaro A. Cardiovascular effects of DPP‐4 inhibition: beyond GLP‐1. Vascul Pharmacol 2011; 55: 10–16. [DOI] [PubMed] [Google Scholar]
- 36. Lamers A, de Kreutzenberg S, Fadini G. Dipeptidyl‐peptidase 4 inhibition: linking metabolic control to cardiovascular protection. Curr Pharm Des 2014; 20: 2387–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lamers D, Famulla S, Wronkowitz N, et al Dipeptidyl peptidase 4 is a novel adipokine potentially linking obesity to the metabolic syndrome. Diabetes 2011; 60: 1917–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ikeda T, Kumagai E, Iwata S, et al Soluble CD26/dipeptidyl peptidase IV enhances the transcription of IL‐6 and TNF‐α in THP‐1 cells and monocytes. PLoS One 2013; 8: e66520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Holst JJ. The physiology of glucagon‐like peptide 1. Physiol Rev 2007; 87: 1409–1439. [DOI] [PubMed] [Google Scholar]
- 40. Panjwani N, Mulvihill EE, Longuet C, et al GLP‐1 receptor activation indirectly reduces hepatic lipid accumulation but does not attenuate development of atherosclerosis in diabetic male ApoE(‐/‐) mice. Endocrinology 2013; 154: 127–139. [DOI] [PubMed] [Google Scholar]
- 41. Shiraishi D, Fujiwara Y, Komohara Y, et al Glucagon‐like peptide‐1 (GLP‐1) induces M2 polarization of human macrophages via STAT3 activation. Biochem Biophys Res Commun 2012; 425: 304–308. [DOI] [PubMed] [Google Scholar]
- 42. Brenner C, Franz WM, Kühlenthal S, et al DPP‐4 inhibition ameliorates atherosclerosis by priming monocytes into M2 macrophages. Int J Cardiol 2015; 199: 163–169. [DOI] [PubMed] [Google Scholar]
- 43. Ross R. Atherosclerosis – an inflammatory disease. N Engl J Med 1999; 340: 115–126. [DOI] [PubMed] [Google Scholar]
- 44. Huh HY, Pearce SF, Yesner LM, et al Regulated expression of CD36 during monocyte‐to‐macrophage differentiation: potential role of CD36 in foam cell formation. Blood 1996; 87: 2020–2028. [PubMed] [Google Scholar]
- 45. Chang TY, Li BL, Chang CCY, et al Acyl‐coenzyme A: cholesterol acyltransferases. Am J Physiol Endocrinol Metab 2009; 297: E1–E9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Oram JF, Vaughan AM. ATP‐binding cassette cholesterol transporters and cardiovascular disease. Circ Res 2006; 99: 1031–1043. [DOI] [PubMed] [Google Scholar]
- 47. Dai Y, Wang X, Ding Z, et al DPP‐4 inhibitors repress foam cell formation by inhibiting scavenger receptors through protein kinase C pathway. Acta Diabetol 2014; 51: 471–478. [DOI] [PubMed] [Google Scholar]