

Abstract
The gut epithelium's large surface area, its direct exposure to ingested nutrients, its vast stem cell population and its immunotolerogenic environment make it an excellent candidate for therapeutic cells to treat diabetes. Thus, several attempts have been made to coax immature gut cells to differentiate into insulin‐producing cells by altering the expression patterns of specific transcription factors. Furthermore, because of similarities in enteroendocrine and pancreatic endocrine cell differentiation pathways, other approaches have used genetically engineered enteroendocrine cells to produce insulin in addition to their endogenous secreted hormones. Several studies support the utility of both of these approaches for the treatment of diabetes. Converting a patient's own gut cells into meal‐regulated insulin factories in a safe and immunotolerogenic environment is an attractive approach to treat and potentially cure diabetes. Here, we review work on these approaches and indicate where we feel further advancements are required.
Keywords: Cell differentiation, Enterocytes, Enteroendocrine cells
Introduction
Most patients with type 1 diabetes are dependent on daily insulin injections for survival. Despite the life‐saving impact of insulin in patients with this disease, insulin delivery regimens are far from optimal. Diabetic patients require intensive disease management, as they deal with a wide range of complications resulting from inaccurate insulin dosing, from the potentially deadly consequences of hypoglycemic episodes, to chronic complications, such as neuropathy and nephropathy as a result of prolonged hyperglycemia. The wide fluctuations in glucose levels observed throughout the day in the typical patient with diabetes stem from the difficulties of attempting to mimic the tightly regulated and nutrient‐responsive insulin secretion seen in normal physiology, using bolus insulin injections.
To circumvent the challenges and shortcomings of insulin injections, numerous strategies are being explored that could provide a more physiological insulin delivery to treat diabetes. Islet transplantation is a promising approach as a replacement for insulin therapy that is already in clinical use and can successfully restore tight glucose control without the need for insulin injections1, 2. However, the limited supply of donor pancreases, the requirement for immunosuppressive drugs and the failure of transplanted islets in most patients over the first 5 years have significantly limited the wide application of this approach in patients with diabetes. Hence, to overcome these limitations, there is tremendous need to find alternate strategies to restore the natural rhythm of insulin production seen in normal physiology. Recent advances using differentiated human embryonic stem cells are promising, and have opened a brand new avenue for the treatment of diabetes3, 4. The feasibility, side‐effects, and efficiency of this approach need to be determined and are already under investigation, with clinical trials recently initiated by ViaCyte, Inc., San Diego, CA, USA. In parallel, the gut is considered a potential source of stem cells to differentiate into endogenous insulin‐secreting cells. The gut contains one of the largest populations of stem cells in the body, potentially making it a replenishable source of insulin‐producing cells. The gut also contains enteroendocrine cells that express molecules involved in glucose responsiveness and processing of prohormones. Furthermore, as the pancreas and the gut both originate from the endoderm and have many differentiation pathways in common, this opens the possibility of reprogramming a patient's own gut cells to produce insulin without the need for immunosuppressants. Here, we review advances in utilizing and engineering gut cells to treat diabetes.
Gut as a unique candidate site for insulin replacement
The gut epithelium has a well‐defined architecture with a high rate of cell renewal. Its epithelial layer is folded for maximal surface area in order to facilitate the efficient absorption of nutrients, with a surface area over 2,000 square feet and a length of approximately 6 m5. Epithelial cells migrate from the crypts of Leiberkühn to the villi, where they are differentiated into one of three different cell lineages: goblet cells, enterocytes and enteroendocrine cells (Figure 1). The fourth type of differentiated cell, the Paneth cell, remains at the base of the crypt and releases antimicrobial molecules into the lumen. It is believed that cells in the epithelial layer show minimal differentiation, and turn over every 3–5 days by undergoing apoptosis or shedding from the tip of the villus into the gut lumen6. This amounts to an estimated turnover of approximately 100 billion cells in the adult human intestine every day6. The gut mucosa has the remarkable ability to maintain gut cell types and numbers, in addition to the ability to adjust the cell type and number to adapt to changes in diet composition. More importantly, gut stem cells can regenerate gut epithelium after acute damage, and it has been shown that an entire crypt can be regenerated from a single surviving stem cell7. The gut stem cells also have genome‐protective mechanisms; that is, the ability to retain old error‐free deoxyribonucleic acid template strands in times of mitosis. If any replication errors do occur in daughter cells, they will be short‐lived, as these cells will be discarded a few days later from the villus tip8. Also, in gut stem cells, deoxyribonucleic acid repair pathways, which can be error‐prone, are not active; therefore, when any deoxyribonucleic acid damage is detected, these cells undergo programmed cell death7, 9. These features of gut cells are attractive in the context of gene therapy approaches, as the risk and consequences of errors would be minimized compared with other targets.
Figure 1.
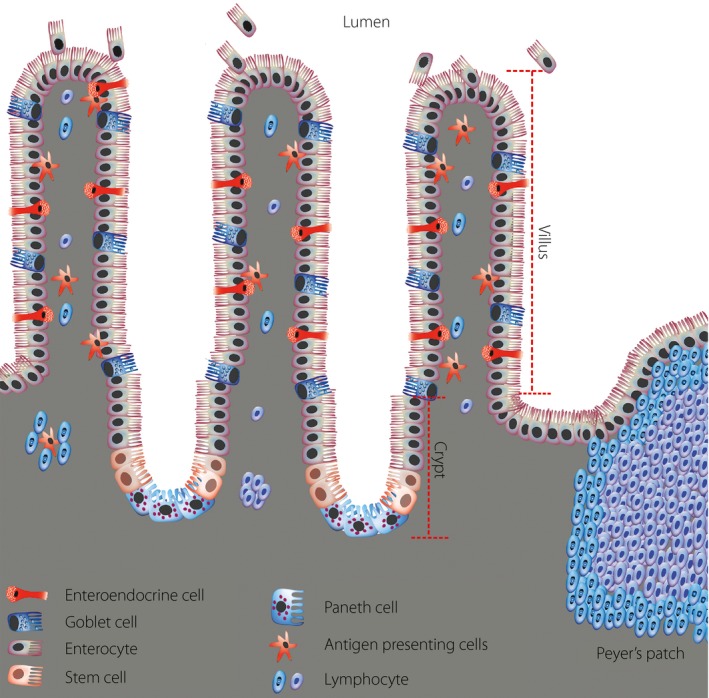
Cytoarchitecture of the intestinal epithelium. The intestinal epithelial cells constitute a single cell layer separating the intestinal lumen from the underlying lamina propria (grey region). The diagram illustrates the finger‐like structures of intestinal epithelium, the villi, which project into the lumen of the intestine. The epithelium at the base of the villi invaginates downward to form tube‐shaped in‐folds called crypts of Lieberkühn, the sites for production of new epithelial cells. The stem cell clusters lie near the base of the crypt just above the Paneth cell cluster. Transitory progenitor cells derived from stem cells migrate bidirectionally along the crypt–villus axis, with the large majority moving upward. These transitory cells divide rapidly, with an average cell‐cycle time of approximately 12 h. The upward moving progenitor cells ultimately differentiate into three of the four lineages of functional epithelial cells (goblet, enteroendocrine and absorptive enterocyte cells). The time required for migration of these cells to the villus tip takes approximately 3–5 days, after which the cells are shed into the intestinal lumen. Progenitor cells that are destined to become Paneth cells migrate to the base of the crypt. The lamina propria, which lies under the epithelial layer, contains large numbers of immune cells, including lymphocytes and antigen presenting cells. The intestinal epithelial cells play an important role in the gut immune response by delivering samples of foreign antigens from the lumen of the intestine to the underlying lymphoid tissue, which include the lymphoid nodules known as Peyer's patches.
Another potentially beneficial feature of the gut for insulin replacement strategies is its vast immune system. The gut constitutes the largest source of immune cells in the body10, and under normal conditions, suppresses immune reactions against dietary antigens and commensal bacteria, a process called oral tolerance11. Exfoliation of intestinal epithelial cells into the gut lumen is also an important defense strategy against pathogens, and it is enhanced during infection to rid the body of infected enterocytes12, 13, 14. In humans, several reports suggest a link between the gut immune system and type 1 diabetes15, 16, 17. Interestingly, it has been shown that tolerance generated to an antigen in the mucosa can promote systemic tolerance to the antigen18. Exploiting this attribute could have great clinical use in the treatment of autoimmune disorders, such as type 1 diabetes. In support of this, in the non‐obese diabetic (NOD) mouse, a model of human type 1 diabetes, oral delivery of insulin to the intestinal mucosa was able to prevent disease when given before the onset of autoimmunity19, 20, 21, 22. Thus, not only could insulin produced from gut cells provide a surrogate source of insulin, but it could potentially promote immunotolerance toward insulin produced in the pancreas, and thereby reduce autoimmune attack of β‐cells.
Converting gut cells into insulin‐secreting cells
Several attempts have been made to convert immature gut cells into insulin‐secreting cells by taking advantage of similarities between enteroendocrine and pancreatic endocrine cell differentiation pathways. The IEC‐6 (rat small intestine epithelioid) cell line23 was shown to produce and secrete insulin after transfection with the β‐cell transcription factor pancreatic and duodenal homeobox protein 1 (PDX1) after treatment with betacellulin (an epidermal growth factor) or after transplantation in diabetic rats24. A similar approach showed that overexpression of PDX1 along with islet‐1 in IEC‐6 cells resulted in insulin production and secretion without the need for betacellulin25. In both studies, the transformed IEC‐6 cells were able to improve blood glucose levels when transplanted into rats, but they did not display glucose‐regulated insulin secretion. Musculoaponeurotic fibrosarcoma oncogene homolog A (MAfA), another critical transcription factor in the development of pancreatic β‐cells that is particularly important for β‐cell maturation and glucose‐responsiveness, was overexpressed in rat intestinal cells through oral administration of an adenovirus containing the MafA gene, resulting in the differentiation of intestinal epithelial cells into insulin‐producing cells26. Mice with streptozotocin (STZ)‐induced diabetes administered with this adenovirus vector had reduced hyperglycemia and increased plasma insulin levels; however, the insulin secretion was not regulated by oral glucose26. A recent study reported that the transient intestinal expression of three β‐cell transcription factors, Pdx1, MafA and neurogenin 3 (Ngn3), promoted the conversion of intestinal crypt cells into insulin‐producing endocrine cells with ultrastructural characteristics of β‐cells. The intestinal islet‐like clusters were glucose responsive and could reverse STZ‐induced diabetes in mice. Interestingly, lineage‐tracing strategies showed that some of the insulin‐positive cells were the progeny of Ngn3+ endocrine progenitor cells27. However, this lineage‐tracing study did not fully exclude the possibility that some insulin‐positive cells were enteroendocrine cells27.
A series of elegant studies from the Accili group showed that Ngn3‐Cre‐driven knockout of the transcription factor, forkhead box O1 (FOXO1), in mice caused Ngn3+ progenitor cells in the gut to differentiate into cells that released insulin in response to glucose28. This FOXO1 ablation increased the expression of β‐cell transcription factors including Pdx1, Ngn3, MafA and NKX6.1, as well as prohormone convertase (PC) 2. Notably, although STZ resulted in initial loss of both pancreatic β‐cells and intestinal insulin‐producing cells, the latter rapidly regenerated in the gut and reversed hyperglycemia, showing that the intestinal insulin in these animals was bioactive and the insulin‐producing cells were replenishable28. The same group reported that in human gut organoids, both short hairpin ribonucleic acid‐mediated inhibition of FOXO1 and transduction with an adenovirus expressing a dominant negative mutant FOXO1, promoted the generation of insulin‐positive cells that released C‐peptide in response to glucose29. When the adenovirus‐transduced organoids were transplanted into mice, they maintained their epithelial structure and insulin‐immunoreactivity; however, limitations in the amount of transplantable tissue prevented the achievement of detectable circulating human c‐peptide levels29. Although these approaches are encouraging, a practical method of genetic manipulation of gut epithelium in a safe manner will be required to develop clinically relevant therapeutic strategies.
In addition to transcription factors, there is evidence that the peptide glucagon‐like peptide‐1(1‐37) (GLP‐1(1‐37)), derived from the preproglucagon gene that is expressed in intestinal L cells, pancreatic α‐cells and discrete brainstem neurons, can also promote insulin production in intestinal epithelial cells. This was shown in cultures of fetal intestines and was only observed with the full‐length peptide, GLP‐1(1‐37), but not the truncated forms that are typically secreted from L cells; that is, GLP‐1(7‐36) and GLP‐1(7‐37) 30. The GLP‐1(1‐37) treated cells showed glucose‐responsiveness in culture, formed islet‐like structures when transplanted into the intraperitoneal space of mice and were capable of reversing diabetes30. The authors also reported that intraperitoneal injection of GLP‐1(1‐37) in pregnant mice promoted differentiation of neonatal intestinal epithelial progenitors into insulin‐producing cells. To overcome the limitations as a result of the short biological half‐life of GLP‐1 plus difficulties with delivery of a bioactive compound to the luminal side of the gut, a recent study used human commensal bacteria engineered to secrete GLP‐1(1‐37) 31. These bacteria were able to convert rat and human intestinal epithelial cells into insulin‐secreting cells that expressed the β‐cell markers, PDX1, MafA and forkhead box protein A232. Diabetic rats fed the GLP‑1(1‐37)‐secreting bacteria daily for 50 days showed higher circulating insulin levels and had improved glucose tolerance compared to rats fed wild‐type bacteria. It remains to be determined whether such an approach would be effective in humans.
Targeting gut enteroendocrine cells
Genetically engineering gut endocrine cells to produce insulin is an intriguing approach for the treatment of diabetes. Although insulin production has been achieved in several tissues, such as muscle and the liver, the difficulty in generating meal‐responsive insulin secretion from these surrogate cells limits their potential for insulin replacement therapy. Approaches that target non‐endocrine cells and rely on transcriptional control of insulin production, with glucose‐responsive promoter elements, will never reproduce the rapid on and off insulin secretion kinetics of β‐cells. This issue might be overcome by using gut enteroendocrine cells that already show several features of β‐cells. Specifically, the G‐, K‐ and L cells, three types of enteroendocrine cell located in the epithelium of the small intestine producing gastrin, glucose‐dependent insulinotropic polypeptide (GIP) and GLP‐1, respectively, all carry sophisticated glucose/nutrient‐sensing machinery. Insulin production in G cells was achieved in transgenic mice harboring a chimeric gene consisting of a gastrin promoter fused to the human insulin gene33. Although the kinetics of insulin secretion from these cells was not discussed33, considering that G cells are primarily responsive to protein, but not glucose, we speculate that these cells might not be adequately glucose responsive and therefore less than optimal for development as surrogate β‐cells. Conversely, K‐ and L cells rapidly release GIP and GLP‐1, respectively, during meal ingestion or with glucose alone, and serve to enhance insulin secretion from β‐cells in a glucose‐dependent manner. Indeed, GIP and GLP‐1 have similar glucose‐induced secretion patterns as insulin34, raising the possibility of using these cells as surrogates for β‐cells to recapitulate meal‐regulated insulin release. Furthermore, enteroendocrine cells contain prohormone convertases that could process proinsulin into mature bioactive insulin and, like β‐cells, can store hormone products in secretory vesicles35, 36, 37, 38.
The ability of L cells to serve as surrogates for β‐cells has been studied in various GLP‐1‐secreting intestinal cell lines. NCI‐H716 human intestinal cells were engineered to produce insulin driven by a cytomegalovirus promoter, using a recombinant adeno‐associated virus; insulin and GLP‐1 were co‐localized in the cells and released with similar dynamics39. Murine glucagon gene‐simian virus‐40 large T‐antigen cells transfected with a plasmid containing the human insulin gene driven by a cytomegalovirus promoter were capable of secreting both GLP‐1 and mature insulin in response to multiple secretagogues; however, the transfected cells did not increase insulin secretion when stimulated with different concentrations of glucose36. Furthermore, transplanted engineered glucagon gene‐simian virus‐40 large T‐antigen cells did not produce enough insulin to ameliorate diabetes in mice40. Murine STC‐1 cells, which produce a number of gut hormones including GLP‐1, were transfected with a plasmid containing the insulin gene driven by a proglucagon promoter. Although the transduced cells produced insulin, little in the way of glucose‐responsive insulin secretion was observed41. Studies with primary L cells will be required to determine if this cell population can produce insulin in a sufficiently robust glucose‐dependent manner to appropriately regulate blood glucose levels.
Compared with GLP‐1 secretion levels from L cells, K cells produce higher levels of GIP under both basal and stimulated conditions42. Therefore, our group rationalized that K cells might be more suitable surrogates for β‐cells. We and others evaluated the feasibility of using these cells both in vitro, using a K cell line, and in vivo, using various transgenic mouse models43, 44, 45. Moderately glucose‐responsive production of insulin was obtained from GIP‐expressing murine GTC‐1 cells (a derivative of STC‐1 cells) after transfection with a transgene comprised of a rat GIP promoter upstream of human preproinsulin43. In another study, STC‐1 cells engineered to produce mouse insulin reversed diabetes after transplant under the kidney capsule in diabetic immunodeficient mice45. However, by 4 weeks post‐transplantation, the mice developed progressive hypoglycemia, possibly as a result of overgrowth of the tumoral STC‐1 cells45. Another study with STC‐1 cells producing human insulin also reversed diabetes in mice by 3 weeks post‐transplant, but longer‐term tracking was not reported46. We generated a regulatable cell‐based system using GTC‐1 cells in which transcriptional control of insulin was inducible by mifepristone in a dose‐dependent manner47. When transplanted into mice, these cells were able to ameliorate STZ‐induced diabetes. Unfortunately, most transformed cell lines do not match the tightly regulated hormone secretion of enteroendocrine cells. Therefore, it is difficult to extrapolate these findings to engineered native K cells.
To explore the utility of endogenous K cells for insulin production, and avoid the issues encountered with transformed cell lines, our group generated transgenic mice in which human preproinsulin is coexpressed with GIP in gut K cells, and is secreted into the circulation in a meal‐dependent manner43. These mice were protected from developing STZ‐induced diabetes and maintained normal glucose tolerance in the absence of exogenous insulin43, 44. To assess whether insulin‐producing K cells would be subjected to autoimmune attack similar to that which targets β‐cells in type 1 diabetes, we generated transgenic mice expressing murine insulin in K cells in the NOD mouse model of autoimmune diabetes. These transgenic mice showed high levels of circulating insulin immunoreactivity, but normal insulin sensitivity, bodyweight and glucose tolerance. Notably, diabetes incidence was significantly reduced in these mice. We have also observed that these K cells not only escape immune destruction, but that K cell insulin production appears to reduce the expected assault on pancreatic β‐cells in NOD mice, likely through the induction of immune tolerance to insulin44.
Our transgenic mice with insulin producing K cells showed high levels of insulin and proinsulin immunoreactivity, yet C‐peptide levels were comparable with non‐transgenic littermates44. Further evaluation of the insulin produced by K cells, using insulin western blotting of intestinal lysates, suggested that the majority of the insulin immunoreactivity was proinsulin, although mature insulin was also produced (unpubl. data). Thus, K cell insulin production can improve glucose regulation in NOD mice, even if only a portion of the proinsulin produced is fully processed into mature insulin. PC1/3 and PC2 are important for efficient processing of proinsulin into mature insulin in mice48, 49. While mouse β‐cells express both PC1/3 and PC2, most K cells express PC1/3, but not PC235. This likely explains the higher proinsulin relative to mature insulin production in our transgenic mice that express insulin in K cells. To circumvent this less than optimal insulin processing, we are currently assessing the impact of modification of the preproinsulin gene in our transgene to promote more efficient proinsulin processing by PC1/3 alone in K cells.
Conclusions
Patients with type 1 diabetes are dependent on multiple daily injections of insulin to survive. Even when blood glucose levels are closely monitored, and insulin doses are carefully coordinated with meals and physical activity, ideal glucose levels are seldom achievable. Therefore, restoring appropriate endogenous insulin production is a desirable goal. One approach is to utilize a patient's own gut cells to reestablish nutrient‐regulated insulin secretion. To exploit the potential of the gut to either reprogram its stem cells into β‐cells, or engineer enteroendocrine cells, such as K cells, into insulin‐producing cells, advancements in the development of safe and efficient genetic modification approaches are required. The possibility that insulin production by the gut could also promote systemic immune tolerance to insulin is a potentially significant benefit to this tactic. Numerous human gene therapy clinical trials are currently in progress to inform the selection of a clinically acceptable method for gene delivery or gene silencing to the gut cells. However, further investigation and refinements are required, as any clinical approach targeting the intestines for insulin production would need to safeguard against unregulated overexpression of insulin and unintended downregulation of other important cellular products. The burgeoning diabetes population and promising advances in this field warrant continued investments and efforts to explore the utility of engineered gut cells in the treatment of diabetes.
Disclosure
TJK is a cofounder and shareholder of enGene, Inc. (Montreal, Canada), a biotechnology company developing gene delivery to gastrointestinal mucosal cells for the production of therapeutic proteins. The other authors declare no conflict of interest.
J Diabetes Investig 2016; 7: 87–93
This article is based on the presentations given by the authors at a symposium, Incretin 2015, July 29‐31, 2015, Vancouver, BC Canada.
References
- 1. Cure P, Pileggi A, Froud T, et al Improved metabolic control and quality of life in seven patients with type 1 diabetes following islet after kidney transplantation. Transplantation 2008; 85: 801–812. [DOI] [PubMed] [Google Scholar]
- 2. Shapiro AM, Lakey JR, Ryan EA, et al Islet transplantation in seven patients with type 1 diabetes mellitus using a glucocorticoid‐free immunosuppressive regimen. N Engl J Med 2000; 343: 230–238. [DOI] [PubMed] [Google Scholar]
- 3. Quiskamp N, Bruin JE, Kieffer TJ. Differentiation of human pluripotent stem cells into ß‐cells: Potential and challenges. Best Pract Res Clin Endocrinol Metab 2015; 29: 833–847. [DOI] [PubMed] [Google Scholar]
- 4. Bruin JE, Rezania A, Kieffer TJ. Replacing and safeguarding pancreatic beta cells for diabetes. Sci Transl Med 2015; 7: 316ps23. [DOI] [PubMed] [Google Scholar]
- 5. Scanoln VCST. Essential of Anatomy and Physiology. Philadelphia: FA Davis Co, 2003. [Google Scholar]
- 6. Barker N. Adult intestinal stem cells: critical drivers of epithelial homeostasis and regeneration. Nat Rev Mol Cell Biol 2014; 15: 19–33. [DOI] [PubMed] [Google Scholar]
- 7. Potten CS. Radiation, the ideal cytotoxic agent for studying the cell biology of tissues such as the small intestine. Radiat Res 2004; 161: 123–136. [DOI] [PubMed] [Google Scholar]
- 8. Potten CS, Booth C, Hargreaves D. The small intestine as a model for evaluating adult tissue stem cell drug targets. Cell Prolif 2003; 36: 115–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cairns J. Somatic stem cells and the kinetics of mutagenesis and carcinogenesis. Proc Natl Acad Sci USA 2002; 99: 10567–10570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bourlioux P, Koletzko B, Guarner F, et al The intestine and its microflora are partners for the protection of the host: report on the Danone Symposium “The Intelligent Intestine”, held in Paris, June 14, 2002. Am J Clin Nutr 2003; 78: 675–683. [DOI] [PubMed] [Google Scholar]
- 11. Solly NR, Honeyman MC, Harrison LC. The mucosal interface between ‘self’ and ‘non‐self’ determines the impact of environment on autoimmune diabetes. Curr Dir Autoimmun 2001; 4: 68–90. [DOI] [PubMed] [Google Scholar]
- 12. Chang SY, Lee SN, Yang JY, et al Autophagy controls an intrinsic host defense to bacteria by promoting epithelial cell survival: a murine model. PLoS ONE 2013; 8: e81095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sellin ME, Muller AA, Felmy B, et al Epithelium‐intrinsic NAIP/NLRC4 inflammasome drives infected enterocyte expulsion to restrict Salmonella replication in the intestinal mucosa. Cell Host Microbe 2014; 16: 237–248. [DOI] [PubMed] [Google Scholar]
- 14. Sellin JH, Wang Y, Singh P, et al beta‐Catenin stabilization imparts crypt progenitor phenotype to hyperproliferating colonic epithelia. Exp Cell Res 2009; 315: 97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Akerblom HK, Virtanen SM, Ilonen J, et al Dietary manipulation of beta cell autoimmunity in infants at increased risk of type 1 diabetes: a pilot study. Diabetologia 2005; 48: 829–837. [DOI] [PubMed] [Google Scholar]
- 16. Ziegler AG, Schmid S, Huber D, et al Early infant feeding and risk of developing type 1 diabetes‐associated autoantibodies. JAMA 2003; 290: 1721–1728. [DOI] [PubMed] [Google Scholar]
- 17. Wen L, Ley RE, Volchkov PY, et al Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature 2008; 455: 1109–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mayer L, Shao L. Therapeutic potential of oral tolerance. Nat Rev Immunol 2004; 4: 407–419. [DOI] [PubMed] [Google Scholar]
- 19. Takiishi T, Korf H, Van Belle TL, et al Reversal of autoimmune diabetes by restoration of antigen‐specific tolerance using genetically modified Lactococcus lactis in mice. J Clin Invest 2012; 122: 1717–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Daniel D, Wegmann DR. Protection of nonobese diabetic mice from diabetes by intranasal or subcutaneous administration of insulin peptide B‐(9‐23). Proc Natl Acad Sci USA 1996; 93: 956–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang ZJ, Davidson L, Eisenbarth G, et al Suppression of diabetes in nonobese diabetic mice by oral administration of porcine insulin. Proc Natl Acad Sci USA 1991; 88: 10252–10256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Carel JC, Bougneres P, Vardi P. Suppression of diabetes in nonobese diabetic mice by oral administration of porcine insulin. J Endocrinol Invest 1994; 17: 573–580. [DOI] [PubMed] [Google Scholar]
- 23. Quaroni A, Kirsch K, Weiser MM. Synthesis of membrane glycoproteins in rat small‐intestinal villus cells. Redistribution of L‐[1,5,6‐3H]fucose‐labelled membrane glycoproteins among Golgi, lateral basal and microvillus membranes in vivo. Biochem J 1979; 182: 203–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kojima H, Nakamura T, Fujita Y, et al Combined expression of pancreatic duodenal homeobox 1 and islet factor 1 induces immature enterocytes to produce insulin. Diabetes 2002; 51: 1398–1408. [DOI] [PubMed] [Google Scholar]
- 25. Yoshida S, Kajimoto Y, Yasuda T, et al PDX‐1 induces differentiation of intestinal epithelioid IEC‐6 into insulin‐producing cells. Diabetes 2002; 51: 2505–2513. [DOI] [PubMed] [Google Scholar]
- 26. Nomura S, Nakamura T, Hashimoto T, et al MafA differentiates rat intestinal cells into insulin‐producing cells. Biochem Biophys Res Commun 2006; 349: 136–143. [DOI] [PubMed] [Google Scholar]
- 27. Chen YJ, Finkbeiner SR, Weinblatt D, et al De novo formation of insulin‐producing “neo‐beta cell islets” from intestinal crypts. Cell Rep 2014; 6: 1046–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Talchai C, Xuan S, Kitamura T, et al Generation of functional insulin‐producing cells in the gut by Foxo1 ablation. Nat Genet 2012; 44: 406–412. S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bouchi R, Foo KS, Hua H, et al FOXO1 inhibition yields functional insulin‐producing cells in human gut organoid cultures. Nat Commun 2014; 5: 4242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Suzuki A, Nakauchi H, Taniguchi H. Glucagon‐like peptide 1 (1‐37) converts intestinal epithelial cells into insulin‐producing cells. Proc Natl Acad Sci USA 2003; 100: 5034–5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Duan F, Curtis KL, March JC. Secretion of insulinotropic proteins by commensal bacteria: rewiring the gut to treat diabetes. Appl Environ Microbiol 2008; 74: 7437–7438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Duan FF, Liu JH, March JC. Engineered commensal bacteria reprogram intestinal cells into glucose‐responsive insulin‐secreting cells for the treatment of diabetes. Diabetes 2015; 64: 1794–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhukova E, Afshar A, Ko J, et al Expression of the human insulin gene in the gastric G cells of transgenic mice. Transgenic Res 2001; 10: 329–341. [DOI] [PubMed] [Google Scholar]
- 34. Schirra J, Katschinski M, Weidmann C, et al Gastric emptying and release of incretin hormones after glucose ingestion in humans. J Clin Invest 1996; 97: 92–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fujita Y, Asadi A, Yang GK, et al Differential processing of pro‐glucose‐dependent insulinotropic polypeptide in gut. Am J Physiol Gastrointest Liver Physiol 2010; 298: G608–G614. [DOI] [PubMed] [Google Scholar]
- 36. Bara H, Sambanis A. Insulin‐secreting L‐cells for the treatment of insulin‐dependent diabetes. Biochem Biophys Res Commun 2008; 371: 39–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Buchan AM, Polak JM, Capella C, et al Electronimmunocytochemical evidence for the K cell localization of gastric inhibitory polypeptide (GIP) in man. Histochemistry 1978; 56: 37–44. [DOI] [PubMed] [Google Scholar]
- 38. Grimelius L, Capella C, Buffa R, et al Cytochemical and ultrastructural differentiation of enteroglucagon and pancreatic‐type glucagon cells of the gastrointestinal tract. Virchows Arch B Cell Pathol 1976; 20: 217–228. [DOI] [PubMed] [Google Scholar]
- 39. Tang SC, Sambanis A. Development of genetically engineered human intestinal cells for regulated insulin secretion using rAAV‐mediated gene transfer. Biochem Biophys Res Commun 2003; 303: 645–652. [DOI] [PubMed] [Google Scholar]
- 40. Bara H, Thule PM, Sambanis A. A cell‐based approach for diabetes treatment using engineered non‐beta cells. J Diabetes Sci Technol 2009; 3: 555–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rasouli M, Ahmad Z, Omar AR, et al Engineering an L‐cell line that expresses insulin under the control of the glucagon‐like peptide‐1 promoter for diabetes treatment. BMC Biotechnol 2011; 11: 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Seino Y, Fukushima M, Yabe D. GIP and GLP‐1, the two incretin hormones: Similarities and differences. J Diabetes Investig 2010; 1: 8–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cheung AT, Dayanandan B, Lewis JT, et al Glucose‐dependent insulin release from genetically engineered K cells. Science 2000; 290: 1959–1962. [DOI] [PubMed] [Google Scholar]
- 44. Mojibian M, Lam AW, Fujita Y, et al Insulin‐producing intestinal K cells protect nonobese diabetic mice from autoimmune diabetes. Gastroenterology 2014; 147: 162–171. e6. [DOI] [PubMed] [Google Scholar]
- 45. Han J, Lee HH, Kwon H, et al Engineered enteroendocrine cells secrete insulin in response to glucose and reverse hyperglycemia in diabetic mice. Mol Ther 2007; 15: 1195–1202. [DOI] [PubMed] [Google Scholar]
- 46. Zhang Y, Yao L, Shen K, et al Genetically engineered K cells provide sufficient insulin to correct hyperglycemia in a nude murine model. Acta Biochim Biophys Sin (Shanghai) 2008; 40: 149–157. [DOI] [PubMed] [Google Scholar]
- 47. Unniappan S, Wideman RD, Donald C, et al Treatment of diabetes by transplantation of drug‐inducible insulin‐producing gut cells. J Mol Med (Berl) 2009; 87: 703–712. [DOI] [PubMed] [Google Scholar]
- 48. Zhu X, Zhou A, Dey A, et al Disruption of PC1/3 expression in mice causes dwarfism and multiple neuroendocrine peptide processing defects. Proc Natl Acad Sci USA 2002; 99: 10293–10298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Furuta M, Yano H, Zhou A, et al Defective prohormone processing and altered pancreatic islet morphology in mice lacking active SPC2. Proc Natl Acad Sci USA 1997; 94: 6646–6651. [DOI] [PMC free article] [PubMed] [Google Scholar]