

ABSTRACT
Preterm birth is the leading cause of infant morbidity and mortality. Necrotizing enterocolitis (NEC) is an inflammatory bowel disease affecting primarily premature infants, which can be lethal. Microbial intestinal colonization may alter epigenetic signatures of the immature gut establishing inflammatory and barrier properties predisposing to the development of NEC. We hypothesize that a crosstalk exists between the epigenome of the host and the initial intestinal colonizing microbiota at critical neonatal stages. By exposing immature enterocytes to probiotic and pathogenic bacteria, we showed over 200 regions of differential DNA modification, which were specific for each exposure. Reciprocally, using a mouse model of prenatal exposure to dexamethasone we demonstrated that antenatal treatment with glucocorticoids alters the epigenome of the host. We investigated the effects on the expression profiles of genes associated with inflammatory responses and intestinal barrier by qPCR-based gene expression array and verified the DNA modification changes in 5 candidate genes by quantitative methylation specific PCR (qMSP). Importantly, by 16S RNA sequencing-based phylogenetic analysis of intestinal bacteria in mice at 2 weeks of life, we showed that epigenome changes conditioned early microbiota colonization leading to differential bacterial colonization at different taxonomic levels. Our findings support a novel conceptual framework in which epigenetic changes induced by intrauterine influences affect early microbial colonization and intestinal development, which may alter disease susceptibility.
KEYWORDS: Antenatal glucocorticoid treatment, developmental origin of disease, DNA modification profiling, epigenome, intrauterine influence, microbiome, transcriptomic profiling
Introduction
Neonatal necrotizing enterocolitis (NEC) is an inflammatory bowel disease affecting primarily premature infants. The etiology of NEC is not completely understood. Preterm birth, microbial colonization and enteral feeding are the primary risk factors associated with this disease (reviewed in ref. 1). It has been shown that the absence of beneficial commensal bacteria and increases in potentially pathogenic bacteria in the preterm intestine play a role in the development of NEC; 2,3 however, a causal pathogen has not been identified. We have previously demonstrated that in the healthy preterm infant there is a temporal course to microbiome development with key clustering prior to 2 weeks of life.2 Furthermore, we have shown that a decrease in Firmicutes accompanied by a bloom of Gammaproteobacteria precedes the development of NEC.2 We thus suggest that an optimal early microbial intestinal colonization rather than a later pathogenic pattern may be key.
Epigenetic processes regulate early cellular differentiation through interactions between genes and environment leading to stable changes in cellular phenotype.4 Gestational epigenetic changes (DNA and histone modifications, as well as expression of non-coding RNAs) can have long-term consequences in the phenotype of the offspring.5,6 Microbiota dynamic progression begins prior to birth and stabilizes in early childhood. In addition, it has been shown that colonizing microbiota can induce epigenetic changes in the host.7-9 This interaction between colonizing microbiota and the epigenome of the host is a potential explanation for disease etiology.10,11
Relevant to preterm infants, at a molecular level, epigenetic processes constitute a major mechanism by which environmental factors may establish a new phenotypic trait during the plastic neonatal interval,12,13 Recent studies have suggested that pre- and post-natal alterations to intestinal DNA methylation patterns contribute to high NEC susceptibility in preterm neonates.14 An optimal early gut colonization may alter epigenetic signatures to establish inflammatory and barrier properties that protect against later insults that trigger NEC. It has also been shown that the epigenome of the host can be altered prior to exposure to early microbiota by gestational environmental exposures (e.g., nutrition, infection, drugs) or due to transgenerational inheritance.15,16 We thus additionally postulate that host epigenome may condition the normal progression of microbiota upon early exposure after birth, thus introducing another influence to disease risk in the neonatal period or later in life.
In the present study, we hypothesize that a dynamic crosstalk exists between the epigenome of the host and the initial colonizing microbiota at critical neonatal stages. We propose 2 complementary mechanisms by which the epigenome and microbiome may interact to affect health: i) the initial colonizing microbiota may induce epigenetic alterations in the immature intestinal epithelial tissue leading to phenotypic change, modeled in vitro using probiotics vs. pathogenic bacterial interaction with intestinal epithelial cells, and ii) epigenetic changes induced during fetal life by the intrauterine environment may influence the composition of the early intestinal microbiota itself, modeled in vivo by dexamethasone administration to pregnant mouse dams. These early epigenome-microbiome interactions may impact the establishment of an optimal microbiome as well as host responses, along with subsequent susceptibility to or protection against disease.
Results
Differential DNA modification patterns in fetal and adult epithelial cells exposed to probiotics or pathogenic bacteria
To determine the effect of direct bacteria-enterocyte interaction on the alteration of DNA modification patterns, we exposed human intestinal epithelial cell lines H4 (fetal, immature) and NCM460 (adult, mature) to i) probiotics (Lactobacillus acidophilus and Bifidobacterium infantis, L+BH4 and L+Bad groups) or ii) Klebsiella spp., a gram-negative bacteria representative of the Gammaproteobacteria dominance of preterm infants (KSSH4 and KSSad groups).17 A different pattern of DNA modification was observed between the treated group (exposed to either L+BH4 or KSSH4 group) and unexposed controls (CTRH4 group).
As shown in Fig. 1A, in immature intestinal epithelial cells (H4), we identified significant DNA modification differences in 92 ROIs (44 and 48 with increased and decreased DNA modification, respectively) in response to L+B exposure, and 180 ROIs (73 and 107 with increased and decreased DNA modification, respectively) in response to KSS exposure (P < 0.05, Bonferroni corrected t-test) (Fig. 1A, Supplementary Table S1). ROIs were associated with 114 and 222 unique RefSeq genes for the L+BH4 and KSSH4 groups, respectively. More than 30% of the differentially modified probes (n = 33 and n = 56 for the L+BH4 and KSSH4 groups, respectively) were associated with the gene transcriptional start site (TSS), suggesting a role for epigenetic regulation of gene expression.
Figure 1.

Epigenome variations in immature intestinal cells exposed to probiotic and pathogenic bacterial communities. A) Differential DNA modification in H4 cells exposed to probiotic (L+BH4 group, left plot) or pathogenic (KSSH4 group, right plot). Ninety-two and 180 ROIs showed significant DNA modification differences (Bonferroni corrected P-value < 0.05) for the L+BH4 and KSSH4 groups compared with the CTRH4 group, respectively. Microarray signal intensities (averaged β value, ABV) for ROIs with significant differential DNA modification are depicted in the heatmap in a color scale from yellow (ABV = 0.0) over green (ABV = 0.5) to blue (ABV = 1). B) GO analysis of the regions with differential DNA modifications identified in both comparisons. Red and green bars represent the FCE values in GO term for the genes with increased and decreased DNA modification, respectively. C) Partial overlap of regions with differential DNA modification identified by comparing the epigenomes of human H4 cells exposed to probiotic (L+BH4 group, yellow circle) or pathogenic (KSSH4 group, blue circle) bacteria with the unexposed cell cultures (CTRH4 group), respectively. Distinctive GO profiles for regions with differential DNA modification detected only by comparing L+BH4 (upper panel) and KSSH4 (lower panel) groups with the CTRH4 groups. Blue bars represent the FCE values for each particular GO terms in the overlapped genes.
Gene ontology (GO) terms associated with transcriptional regulation were overrepresented in genes with increased and decreased DNA modification in the L+BH4 group and decreased DNA modification in the KSSH4 group, when compared to the CTRH4 group (Fig. 1B). In the KSSH4 group, genes associated with cytoskeleton/actin remodeling and cell adhesion functions were also overrepresented in ROIs with decreased DNA modification compared to the CTRH4 group (Fig. 1B). Twenty transcript-associated ROIs showed significant differential DNA modification in both groups (L+BH4 and KSSH4) when compared to the CTRH4 group (Fig. 1C). GO analysis revealed that genes associated with nucleotide binding mechanisms were overrepresented in these ROIs (Fig. 1C).
The DNA modification profiles from the adult NCM460 cells were completely different from those from the fetal H4 cells. Even without exposure to any bacterial community (Supplementary Fig. S1), we detected more than 200,000 regions with differential DNA modifications (P < 0.05, Bonferroni corrected t-test) between the 2 groups. By increasing the stringency of the cutoff values (absolute log2FC > 5 and P < 0.001, Bonferroni corrected t-test, Supplementary Table S2) we identified 107 regions with differential DNA modifications (16 and 91 with increased and decreased DNA modification in NCM460 cells, respectively) that distinguished H4 from NCM460 cells based only on the DNA modification profiles. (Supplementary Fig. S1). Exposure to probiotic (L+Bad group) or pathogenic bacteria (KSSad group) resulted in 96 (53 increased and 43 decreased DNA modification) and 117 (44 increased and 73 decreased DNA modification) ROIs compared to the non-exposed controls (CTRad group), respectively (P < 0.05, Bonferroni corrected t-test) (Supplementary Fig. S2A, Supplementary Table S3). ROIs were associated with 65 and 89 unique RefSeq genes for the L+Bad and KSSad groups, respectively. GO terms analyses showed that genes associated to glycosylation of proteins and lysosomes were overrepresented among those showing increased and decreased DNA modification after exposure to probiotic bacterial communities (L+Bad group). In turn, genes associated to chromatin organization were overrepresented among those showing decreased DNA modification after exposure to pathogenic bacterial communities (KSSad group). In genes showing increased DNA modification in the KSSad group, there was no enrichment of GO terms (Supplementary Fig. S2B). Thirty-seven transcript-associated ROIs showed differential DNA modification in both groups (L+Bad and KSSad) when compared to CTRad. Overrepresented GO terms in those ROIs were associated to chromatin organization (Supplementary Fig. S2C). The proportion of ROI-associated transcripts showing differential DNA modification in both groups was significantly higher in NCM460 cells (37/336 transcripts) than in H4 cells (20/154 transcripts) (P = 1.03 × 10−5; OR = 6.81, 95%CI: 3.62–13.11; Fisher's Exact test).
Pathogenic bacteria effects on cytoskeleton in H4 cells
Since the KSSH4 group demonstrated a specific decreased DNA modification of genes associated with cytoskeleton/actin remodeling and cell adhesion functions, we investigated actin/cytoskeletal function in H4 cells exposed to L+B or KSS and in comparison to unexposed H4 controls (CTRH4 group). As shown in Fig. 2, we identified an upregulation of F-actin in the KSSH4 group with dense actin bundles assembled into stress fibers (white arrows). The α-tubulin expression pattern in the KSSH4 group was also altered. As shown in Fig. 2B, cells in the KSSH4 group had less α-tubulin and less organized microtubules, resulting in a more scattered pattern of cell growth.
Figure 2.
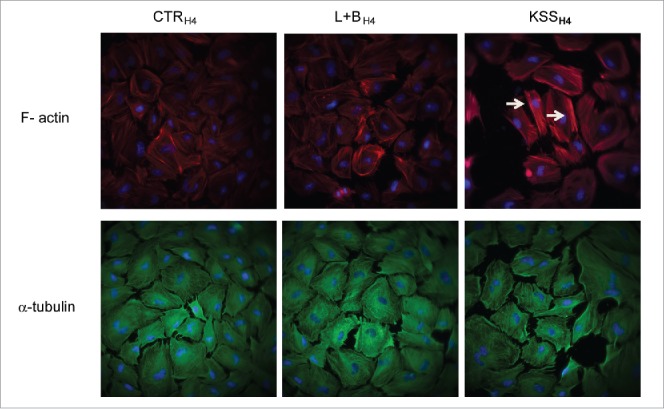
Immunohistochemistry study of actin/cytoskeleton change in immature intestinal cells exposed to probiotic and pathogenic bacterial communities. A) F-actin was analyzed by fluorescent staining with Alexa 594 labeled phalloidin in H4 cells treated with media (CTRH4 group), probiotics (L+BH4 group) or pathogenic bacteria (KSSH4 group). Images were captured using Olympus TIRF microscope and analyzed using Slidebook 6.0 software Representative areas are shown (magnification 400x). White arrows indicate increased expression of actin bundle (stress fiber) in KSSH4 treated cells. B) Alpha-tubulin was analyzed by immunohistochemistry in H4 cells (CTRH4, L+BH4 or KSSH4 group) using a monoclonal antibody. Representative areas (n = 2/per group) are shown (magnification 400x).
Glucocorticoid effects on DNA modification in H4 cells
It is known that the natural glucocorticoid surge in late gestation is associated with changes in DNA methylation of the fetal epigenome, and antenatal synthetic glucocorticoid treatment for premature labor can prematurely initiate epigenetic changes in the fetal genome.18,19 To evaluate the role of the synthetic glucocorticoid, dexamethasone, in modulating DNA modification patterns in immature enterocytes, H4 cells were exposed to dexamethasone for 48 h, and the global pattern of the modification change was examined (Supplementary Fig. S3). Compared to untreated cells, we detected 147 regions with differential DNA modifications with 75 and 72 regions with increased and decreased DNA modifications, respectively. The gene ontology analysis revealed that in the genes with increased DNA modification, the top overrepresented GO terms corresponded mainly to MHC antigen presentation and to poly/monosaccharide metabolism. GO terms enriched in genes with decreased DNA modification corresponded to cell adhesion and morphogenesis.
Antenatal dexamethasone treatment leads to alteration of gene expression and DNA modification profiles in the gut of the offspring
Since dexamethasone is administered to pregnant women at risk of preterm delivery, and dexamethasone treatment elicited significant changes in DNA modification in immature enterocytes in vitro, we next examined how early exposure to dexamethasone may lead to alterations in the epigenome of the offspring in vivo. We used a mouse model of prenatal exposure to dexamethasone and investigated the effects of antenatal dexamethasone treatment on the expression profiles of genes associated with inflammatory responses and intestinal barrier function in the ileal tissue of 2-week-old mouse pups. As a control group for epigenome variation, we treated pregnant mice with a known inhibitor of DNA modification (5-azacytidine) in a non-lethal dose. We used a qPCR array platform to assess 84 genes in parallel for the TLR and TJ functional groups, respectively. The changes in gene expression levels (fold change enrichment, FCE, values) between each treatment and the unexposed control pups (CTR group) are listed in Supplementary Table S4.
In the TJ array, we identified 50 differentially expressed genes (FCE ≥ ± 1.2; 31 and 19 up- and down-regulated, respectively) for the offspring of dexamethasone treated mothers (DEX group), and 82 differentially expressed genes (FCE ≥ ± 1.2; 79 and 19 genes up- and down-regulated, respectively) for the offspring of 5-azacytidine treated mothers (AZA group), when compared to the offspring of untreated mothers (CTR group). Bioinformatics analysis showed that pathways related to immune cell migration (e.g., leukocyte extravasation signaling –log(P-value): 34, granulocyte adhesion and diapedesis –log(P-value): 19.6, and granulocyte adhesion and diapedesis –log(P-value):19.1), as well as epithelial barrier formation (epithelial adherens junction signaling –log(P-value):4.71) were among the canonical pathways overrepresented in the TJ functional group (Supplementary Table S5, Fig. 3A).
Figure 3.
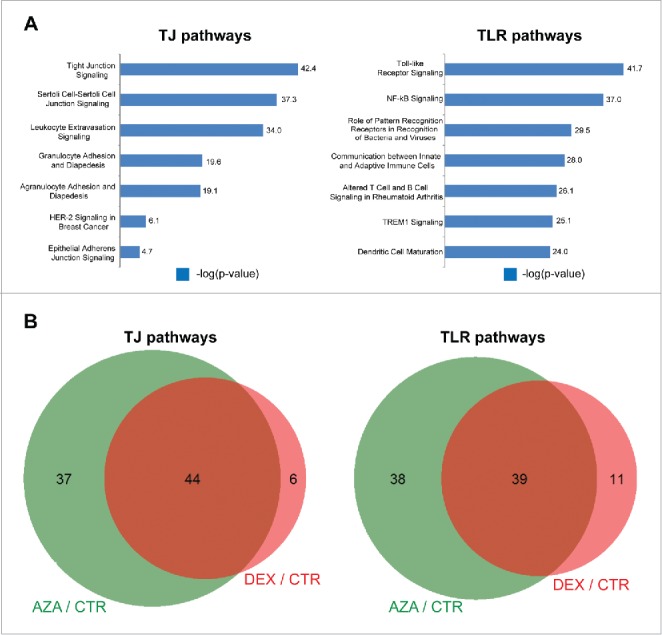
Gene expression analysis: Transcriptional profile variation in the offspring upon antenatal dexamethasone treatment. A) Canonical pathways affected in the TJ (left plots) and TLR (right plots) functional groups by antenatal dexamethasone treatment. Blue bars represent the significance of the enrichment for each pathway expressed as –log(P-value). B) Overlap between differentially expressed regions upon exposure to dexamethasone (DEX group, red circles) and the DNA modification inhibitor 5-azacytidine (AZA group, green circles) compared to the offspring of untreated mothers (CTR group).
We also identified 50 differentially expressed genes in the TLR array (FCE ≥ 1.2; 26 and 24 up- and down-regulated, respectively) in the DEX group, and 79 differentially expressed genes (FCE ≥ 1.2; 57 and 22 genes up- and down-regulated, respectively) in the AZA group. Among the canonical pathways overrepresented in the TLR functional group, we identified inactivated pathways related to immune response (e.g., NF-κB signaling –log(P-value): 37, role of pattern recognition receptors in recognition of bacteria and viruses –log(P-value): 29.5, and TREM1 signaling –log(P-value):25.1) (Supplementary Table S5, Fig. 3A). In particular, for the NF-κB pathway, our results predict inactivation of the inflammatory response through the IKK complex and the activation of an alternate pathway for NF-κB activation through proteasomal processing resulting in transcriptional activation, lymphogenesis, and B-cell maturation (Supplementary Fig. S4).
The majority of the genes showing differential expression between the DEX and CTR groups also showed differential expression between the AZA and CTR groups (44 out of 50 genes and 39 out of 50 genes for the TJ and TLR arrays, respectively; Fig. 3B), suggesting that antenatal dexamethasone treatment alters DNA modification profiles of the offspring. We selected 5 candidate genes from the TJ [tight junction protein 1 (Tjp1, also known as ZO-1), and cingulin (Cgn)] and TLR pathways [myeloid differentiation primary response gene 88 (Myd88), toll interacting protein (Tollip), and Toll-like receptor 2 (Tlr2)] and assessed the DNA modification status in their promoter regions in ileal tissue samples from the CTR, DEX and AZA groups (n = 8 mice/group) (Table 1). Out of the 5 gene-associated regions evaluated by qMSP (Table 1), 3 showed significant differential DNA modification between the DEX and CTR groups: Cgn (FCEDNA modification = −39.4; P = 1.95 × 10−5), Tollip (FCEDNA modification = −1.93; P = 6.15 × 10−8) and Tlr2 (FCEDNA modification = −1.91; P = 0.002). In addition, we observed that decreased DNA modification in 3 out of the 5 studied regions correlated with increased expression of the cognate gene: Tjp1 (FCEDNA modification = −1.20; FCEmRNA expression = 2.93), Cgn (FCEDNA modification = −39.4; FCEmRNA expression = 1.21), and Tollip (FCEDNA modification = −1.91; FCEmRNA expression = 1.03).
Table 1.
DNA modification and mRNA expression in candidate genes for DEX and AZA groups compared to CTR.
| 2 weeks offspring |
E18 fetuses |
||||||
|---|---|---|---|---|---|---|---|
| DEX/CTR1 | P-value* | AZA/CTR1 | P-value* | DEX/CTR1 | P-value* | ||
| Tjp1 | DNA modification | −1.20 | 0.955 | −1.43 | 0.186 | 3.29 | 0.160 |
| mRNA expression | 2.93 | 2.55 | 1.71 | ||||
| Cgn | DNA modification | −39.40 | 1.95 × 10−5 | −2.17 | 0.909 | −4.77 | 0.655 |
| mRNA expression | 1.21 | 2.09 | 3.19 | ||||
| Myd88 | DNA modification | −1.25 | 0.957 | 4.38 | 1.000 | −0.24 | 0.299 |
| mRNA expression | −1.37 | 1.25 | −1.51 | ||||
| Tollip | DNA modification | −1.93 | 6.15 × 10−8 | −1.21 | 0.002 | −3.27 | 0.031 |
| mRNA expression | 1.03 | 1.77 | 2.07 | ||||
| Tlr2 | DNA modification | −1.91 | 0.002 | 1.49 | 0.028 | −0.45 | 0.745 |
| mRNA expression | −10.51 | −2.40 | 2.28 | ||||
FCE values. Positive and negative values represent increased and decreased DNA modification or mRNA expression values upon treatment (DEX or AZA groups) compared to untreated (CTR group), respectively.
2-tailed t-test for DNA modification differences. Significant values (P < 0 0.05) are in bold.
Antenatal dexamethasone treatment leads to alteration of gene expression and DNA modification profiles in the fetus
To confirm that the epigenetic alterations seen in utero occurred in response to dexamethasone, we next examined whether antenatal dexamethasone exposure leads to alteration of gene expression and DNA modification profiles in the fetus. We used the same qPCR array platform corresponding to the TLR and TJ functional groups. The changes in gene expression levels (FCE values) between treatment and control pups (CTR group) are listed in Supplementary Table S4. In the TJ array, we identified 80 differentially expressed genes (FCE ≥ ± 1.2; 77 and 3 up- and down-regulated, respectively) for the fetus of dexamethasone treated mothers (DEX group), when compared to that untreated mothers (CTR group). We also identified 72 differentially expressed genes in the TLR array (FCE ≥ 1.2; 62 and 10 up- and downregulated, respectively) in the DEX group when compared to CTR group. We selected the same 5 candidate genes from TJ pathways (Tjp1 and Cgn) and TLR pathways (Myd88, Tollip, and Tlr2) and assessed the DNA modification status in their promoter regions in the full fetus from CTR and DEX groups (n = 3/group) (Table 1). Out of the 5 gene-associated regions evaluated by qMSP (Table 1), 3 showed same trend as seen in 2 week-old small intestinal tissues. Moreover, Tollip showed significant differential DNA modification between the DEX and CTR groups.
Epigenetic alterations induced by antenatal dexamethasone treatment are associated with altered composition of the gut microbiome in the offspring
We have shown that antenatal dexamethasone treatment modified the DNA and gene expression profiles of TLR-, as well as TJ-signaling pathways in the small intestine of the offspring. Since TLRs and TJs are likely to be key signaling pathways in cross-species homeostatic regulation we next sought to study whether dexamethasone-induced epigenetic changes are associated with changes in the intestinal microbiome. Fecal samples from dams and their respective pups were collected and analyzed by 16S rRNA-based sequencing. We compared the microbiota composition in the offspring of the DEX and CTR group (n=6/group). We also studied the microbiomes of offspring from the AZA group (n=6), which served as a control for a known modified epigenome of the host. The sequencing results for all the studied samples are provided in Supplementary Table S7.
Antenatal dexamethasone treatment led to changes in the microbiota composition at different taxonomic levels in the offspring at 2 weeks of life (Fig. 4), and also in the mothers (Supplementary Fig. S5). At the phyla level, we detected a slight increase in Firmicutes (FCE = 1.16) and a slight decrease in Bacteroidetes (FCE = −1.20) (Fig. 4A). At the class level, we detected an increase in the Clostridia class within the phylum Firmicutes (FCE = 2.3). In turn, within the Proteobacteria phylum we observed a slight increase in Alphaproteobacteria (FCE = 1.44) and a slight decrease in Betaproteobacteria (FCE = −1.45) (Fig. 4B). At the genus level, we detected a slight decrease in Bacteroides (FCE = −1.19) within the Bacteroidia class, a decrease in Streptococcus (FCE = −1.71) within the class Bacilli, a strong increase in Candidatus arthromitus (FCE = 82.31) together with a decrease in Anaerotruncus (FCE = −1.86), Clostridium (FCE = −1.71), and Ruminococcus (FCE = −1.65) within the class Clostridia, a decrease in Anaplasma (FCE = −1.23) in the Alphaproteobacteria class, a decrease in Parasutterella within the Betaproteobacteria class, and an increase in Proteus (FCE = 2.22) in the Gammaproteobacteria class (Fig. 4C). At the species level, we detected the biggest changes within the genus Clostridium, with an increase in C. aminophilum (FCE = 14.5), C. hathewayi (FCE = 1.42), and C. orbisciendens (FCE = 1.20), as well as a decrease in C. cocleatum (FCE = −11.96) and C. lactatifermentans (FCE = −8.78) (Fig. 4D). Antenatal 5-azacytidine treatment also led to changes in the microbiota composition of the mothers (Supplementary Fig. S5) and offspring at 2 weeks of life (Fig. 4) Thus, antenatal dexamethasone and 5-AZA exposure were associated with altered gut microbial communities in the offspring.
Figure 4.
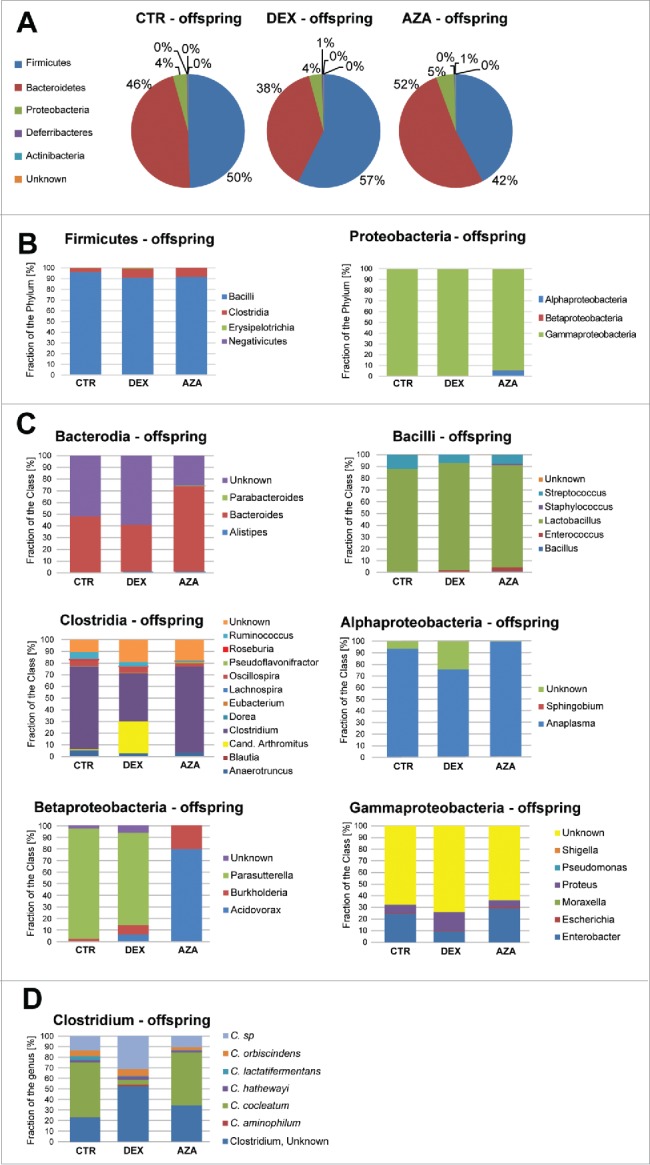
Microbiome variation at different taxonomical levels observed in the offspring upon antenatal dexamethasone treatment. A) Microbiome distribution in offspring of mothers treated with dexamethasone (DEX), 5-azacytidine (AZA) or untreated (CTR) at the phyla (panel A), class (panel B) and genus (panel C) levels. Panel D highlights the species variation within the genus Clostridium, where the highest variation was observed.
Discussion
Gut microbial communities play an important role in maintaining homeostasis and modulating the host's immune system. They are also crucial for host development and physiology, including organ development, morphogenesis, and host metabolism. However, the underlying molecular mechanisms of host–microorganism interactions remain largely unknown. In this study, we present a novel concept in which a dynamic crosstalk exists between the epigenome of the intestine and the colonizing microbiota at neonatal stages. We aimed to comprehensively show the existence of such crosstalk; however, further investigation is warranted to unravel the molecular mechanisms involved.
We demonstrate that exposure to probiotic and pathogenic bacteria induces different epigenetic effects in immature intestinal epithelial cells. Less than 10% of regions with differential DNA modifications were shared between the probiotic (L+BH4 group) and pathogenic (KSSH4 group) bacteria (Fig. 1) treated cells. Our findings specifically suggest that whereas bacterial exposure in general will trigger transcriptional cascades through epigenetic regulation, there might be a specific epigenetic (mis)regulation of the cytoskeleton of epithelial cells and cell adhesion after exposure to gram-negative bacteria (e.g., Klebsiella spp.) leading to alterations of intestinal barrier function. Moreover, our results suggest that fetal epithelial cells are more sensitive to such pathogenic-specific changes than adult epithelial cells, in line with the existence of a pre- and neo-natal plastic interval for epigenetic-mediated stable reprogramming of transcriptional profiles predisposing to disease later in life.15 Epigenome variations in the host may be induced by the microbiota through 3 main mechanisms: i) alterations in the availability of chemical donors for DNA or histone modifications, which depend on nutrition and the metabolic activities expressed by the microbiota (reviewed in ref. 20); ii) DNA modifications caused by the mechanisms triggered by the incorporation of foreign genetic material into the genome of the host 21-24; and iii) direct interaction with enzymes responsible for DNA or histone modifications, such as DNA methyltransferases or histone deacetylases.25-27 Further studies are needed to determine the relevant mechanisms for the developing gut.
We additionally postulated that gestational exposure to environmental agents would lead to altered epigenomes in the offspring, which would condition the microbial colonization patterns upon early exposure after birth, with the potential to subsequently alter disease susceptibility in the neonatal period or later in life. Other studies have described how genomic variation could condition the composition of the colonizing bacterial communities. Le et al. identified a genetic locus, the commensal colonization factors (ccf) in Bacteroidetes, which is fundamental in establishing symbiosis with the host.28 It has been also shown that L. rhamnosus strains carrying particular genomic alterations will likely possess an increased niche-specific fitness.29 In addition, it has been recently shown that allelic variants of bacterial proteins in the Salmonella enterica serovar Typhimurium likely contribute to pathoadaption to diverse hosts.30 On the other hand, studying how variations in the host genome will predispose to colonization by specific bacterial strands will also be of interest for understanding the etiology of disease. We focused our work on epigenetic variations since they are more dynamic than genetic variations. Therefore, they can i) explain how environmental variation, such as antenatal dexamethasone treatment or exposure to other factors in utero, will result in altered microbiota colonization at birth, and ii) be reversed or induced as a therapeutic intervention.
In our model, a pathogenic microbiome composition acts as the “effector” in the triggering of the diseases, whereas the epigenome of the host acts as the “selective agent” for the pathogenic composition of the microbiome and as the “bridge” across generations. As a clinically relevant example for testing our model, we sought to determine whether antenatal dexamethasone administration would lead to epigenetic changes in the offspring that altered gene expression profiles and microbiome composition. Steroids are routinely administered to women in preterm labor due to known improved outcomes in preterm infants, especially in accelerating lung maturation.31 Glucocorticoids have been shown to alter DNA methylation,32,33 thus representing a putative modifier of the offspring epigenome that may explain the long–term consequences of the antenatal glucocorticoid exposure on neurologic, cardiovascular, and metabolic function.19
In our studies, treating pregnant mice with dexamethasone resulted in major gene expression changes in the tight junction and toll-like receptor associated pathways of the offspring, associated with alterations in the microbiome. We recognize that glucocorticoids have effects beyond epigenetic modification, including functioning as a known intestinal trophic factor and potent anti-inflammatory agent that could also affect microbial colonization patterns. However, since antenatal administration of 5-AZA, an agent known to demethylate DNA globally without the other effects of glucocorticoids, also altered expression changes in the majority of the studied genes and the microbiota composition of the offspring, there remains evidence that host epigenetic alterations influence colonization patterns. Specific association between gene silencing and DNA modification at the TSS-surrounding regions of the differentially expressed genes for both dexamethasone and 5-AZA treatment needs further confirmation (Table 1).
Previous studies have focused on explaining how different microbiomes induce epigenetic modifications in the host, 7-9,20-27,34 but the possibility of a modulation of the colonizing microbiome composition by the host's epigenome has not been previously explored. The composition of the offspring's microbiome can be conditioned by several pre- and post-natal factors, such as delivery method,35 antibiotic treatment,36,37 and nutrition.38,39 In turn, epigenomes can be also responsible for modulating the response to environmental factors.40-46,47 In the modulation of microbiota colonization, we theorize that epigenetically regulated mechanisms may be responsible for differential immune responses, the establishment of more or less permeable epithelial barriers, and bacterial metabolite processing, which will favor the colonization of particular bacterial communities.
Our data demonstrate that for the epigenetic alterations in the offspring's gut caused by antenatal dexamethasone treatment, the greatest changes in the microbiome were detected at species level within the genus Clostridium (Fig. 4C). Recent studies of microbiota progression showed a gradual progression to Clostridial abundance for children born most prematurely. Accumulating evidence suggest that Clostridia are involved in the maintenance of overall gut function.48 Clostridia, in close contact with intestinal cells, play crucial roles in modulating physiologic, metabolic and immune processes in the gut, and are necessary for the welfare of maintaining normal gut immune homeostasis.48 Our data indicate that antenatal dexamethasone exposure alters Clostridial abundance in the offspring when compared to the control pups.
Our findings strongly support the idea of a reciprocal crosstalk in which epigenetic changes induced by intrauterine influences affect microbial colonization, potentially changing disease susceptibility later in life. Further studies will be required to unravel the precise molecular mechanisms and processes ruling the epigenome/microbiome interaction. This novel conceptual framework is expected to yield insights at multiple levels into how preterm infant microbiota affects normal intestinal development and function, and how intrauterine epigenomic reprogramming affects gut microbiota composition and function to thus influence health and disease outcomes.
Materials and methods
Animals and treatments
C57BL/6J mice were maintained in the gnotobiotic facility of the Digestive Disease Research Core Center (DDRCC) at the University of Chicago. Mice were housed in the animal care facilities under specific pathogen free environment (SPF) conditions. All groups of mice were allowed ad libitum access to Harlan Teklad 7012 (SPF) chow. Pregnant C57Bl/6 dams received dexamethasone (0.4 µg/g) (Sigma-Aldrich, Cat. No. D1756) 49 or 5-azacytidine (100 µg/kg) (Sigma-Aldrich, Cat. No. A2385) at E16 to induce epigenetic changes. For prenatal expression and DNA modification analyses, dams with or without dexamethasone exposure were sacrificed at E18 and fetuses were harvested. All experimental procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Chicago.
Cell cultures and bacterial exposure
Human fetal (H4) and adult (NCM460) primary intestinal epithelial cells were cultured as detailed elsewhere.50 Upon confluence, cells were inoculated with 107 CFU of Lactobacillus acidophilus and Bifidobacterium infantis (L+BH4 and L+Bad groups), representing probiotic organisms, or 5 × 105 Klebsiella spp cultures representing the Gamma proteobacteria dominant in the microbiota of preterm infants (KSSH4 and KSSad groups).2 Cell cultures not receiving any bacteria served as control (CTRH4 and CTRad group). After 24-hour exposure, genomic DNA from H4 and NCM460 was isolated and epigenetic profiles were studied.
Microarray-based DNA modification profiling
Since the methods for assessing DNA modification used in this study (based on sodium bisulfite conversion) do not differentiate 5-methylcytosine (5mC) from 5-hydroxylmetylcytosine (5hmC),51 we will use the term ‘DNA modification’ to describe our findings. Large-scale DNA modification profiles were studied using the Infinium HumanMethylation450 BeadChip (Illumina, Catalog No. WG-314-1003) according to the manufacturer's instructions. In brief, 500 ng of DNA isolated from H4 cells exposed to probiotic or pathogenic bacteria (L+BH4 and KSSH4 groups, respectively) or media alone (CTRH4 group) were treated with sodium bisulfite using the EZ DNA methylation kit (Zymo Research, Cat. No. D5001) and targets were prepared, labeled and hybridized using the kits and reagents indicated by the manufacturer [Infinium HD Methylation Assay Protocol Guide (15019519 B), Illumina]. The array allowed the interrogation of more than 485,000 modification sites per sample at single-nucleotide resolution. The array covers 99% of RefSeq genes, with an average of 17 cytosine-guanine dinucleotides (CpG) per gene region distributed across the promoter, 5′ untranslated region (5′UTR), first exon, gene body, and 3′UTR. It covers 96% of CpG islands, with additional coverage in island shores and the regions flanking them. Generated microarray data were analyzed using Genome Studio software v2011.1 (Illumina) with the analysis module Methylation v1.9.0. Data were normalized against the controls on the microarray and background subtracted. Differential DNA modification was assessed as differences between mean signals, and the P-values were calculated by t-test corrected for multiple hypotheses testing by the Benjamini-Hochberg method in combination with the Illumina custom false discovery rate (FDR) model. The cutoff for differential DNA modification was set at FDR-corrected P-value lower than 0.05. The detected regions of interest (ROIs) were then annotated for nearby genes by comparing genomic coordinates of the probes with RefSeq transcript annotation of human reference genome assembly mm8 (NCBI build 37). The function and ontology of the associated genes was studied using the Database for Annotation, Visualization and Integrated Discovery (DAVID) version 6.7.52 Microarray data will be deposited in the NCBI's Gene Expression Omnibus (GEO).
Gene expression analysis
Total RNA was isolated from the full fetuses at E18 and ileal tissue of 2-week-old offspring of mothers treated with dexamethasone (DEX group), 5-azacytidine (AZA group), or untreated mothers (CTR group) using the RNeasy kit (Qiagen, Cat. No. 74104), according to the manufacturer's instructions. Expression profiles in the tight-junction (TJ) and the toll-like receptor (TLR) functional groups were assessed using the 96-well plate RT2 Profiler PCR Arrays (Qiagen, Cat. No. PAMM-143Z and PAMM-018Z, respectively). Each array contained PCR primers for SYBR green-based RT-PCR quantification of expression of 84 genes belonging to the functional group. The relative expression was determined using the ΔΔCt method.53 Statistical differences between the groups were detected by t-test. Networks and pathways significantly enriched in the genes of interest were identified through Ingenuity Pathways Analysis (IPA) (Ingenuity® Systems, www.ingenuity.com).
Single locus DNA modification assessment
DNA modification in 5 candidate genes (Table 1) was studied using quantitative methylation-specific PCR (qMSP) analysis.54 DNA from the full fetuses at E18 and ileal tissue of 2-week-old offspring and dams from the DEX, AZA and CTR groups, as well as from H4 and NCM460 cells after L+B or KSS inoculation, was isolated using DNeasy Blood and Tissue Kit (Qiagen, Cat. No. 69504). Genomic DNA (500 ng) was bisulfite-treated using the Epitect kit (Qiagen, Cat. No. 59104). Bisulfite-treated DNA was subjected to locus-specific amplification using qMSP primers (Supplementary Table S7). The qMSP reaction consisted of 10 ng bisulfite-converted DNA, 1× ABI master mix containing Taq polymerase, dNTPs, SYBR green dye and ROX as passive dye (Thermo Fisher Scientific, Cat. No. 4309155) and 200 nM of specific primers. Amplification and analyses were performed using the 7500 System (Thermo Fisher Scientific). A fragment within the Actb locus, which did not contain any cytosine-guanine (CpG) dinucleotide, was used as a calibrator (ΔCtcalibrator). Fold change enrichment (FCE) was calculated using the equation: FCE = 2(ΔCttarget- ΔCtcalibrator)
Microbiome analysis. Microbiome analysis was performed as described elsewhere.55 DNA was extracted from stool samples using the QIAamp Stool DNA kit (Qiagen, Cat. No. 51104), according to manufacturer's instruction, and amplified using specific primers for the region of the 16S rRNA encoding gene (515F: 5′-GTGCCAGCMGCCGCGGTAA-3′ and 806R: 5′- GGACTACHVGGGTWTCTAAT-3′). Such primers contained Illumina 3′ adapter sequences as well as a 12 bp barcode 56 enabling library preparation for sequencing. Sequencing was performed by the Next Generation Sequencing service (Roche 454 GS FLX+ Amplicon Sequencing) at Research and Testing Laboratory (Lubbock, TX). For identifying operational taxonomical units (OTUs), sequences were trimmed and then analyzed using QIIME.56 OTUs were picked at 97% sequence identity using uclust and a representative sequence was then chosen for each OTU by selecting the most abundant sequence in that OTU for each group (DEX, AZA, or CTR). These representative sequences were classified and assigned a taxonomic string using the RDP Classifier.2
Fluorescence immunohistochemistry study of actin/cytoskeleton. H4 cells were harvested upon confluence and plated on tissue culture treated coverslips (18 mm) at 5 × 105 and cultured in H4 media (H4 cells were routinely maintained in Dubelco's modified Eagles medium (DMEM) supplemented with 10% fetal calf serum (FBS) and human recombinant insulin (0.5 unit/ml). When cells reached at least 75% confluency, H4 cells were incubated with 0, 2 × 106, and 5 × 106 probiotic L+B, or pathogenic KSS for 24 and 48. h After incubation, cells were washed with PBS and fixed with either methanol for tubulin staining or 4% paraformaldehyde for F-actin staining. Cells were then blocked with 10% goat serum in PBS containing 0.1% triton 100x. Primary antibody against α-tubulin (ab18251, Abcam, Cambridge, MA) was applied at 1 μg/ml to H4 cells. A FITC-conjugated secondary antibody was applied. For F-actin, an Alexa Fluor® 594-conjugated phalloidin was applied at 1 unit/ml. H4 cells on coverslips were then mounted and examined using an Olympus TIRF microscope (Olympus Corporation of the Americas, Center Valley, PA) and analyzed using Slidebook 6.0 software (Intelligent Imaging Innovations, Denver, CO).
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was funded by grants R01HD059123 and R01HD083481 from the National Institutes of Health to ECC and utilized the Digestive Disease Research Core Center of the University of Chicago which is funded by NIH grant P30DK42086.
References
- 1.Claud EC. Neonatal Necrotizing Enterocolitis -Inflammation and Intestinal Immaturity. Antiinflamm Antiallergy Agents Med Chem 2009; 8:248-59; PMID:20498729; http://dx.doi.org/ 10.2174/187152309789152020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang Y, Hoenig JD, Malin KJ, Qamar S, Petrof EO, Sun J, Antonopoulos DA, Chang EB, Claud EC. 16S rRNA gene-based analysis of fecal microbiota from preterm infants with and without necrotizing enterocolitis. ISME J 2009; 3:944-54; PMID:19369970; http://dx.doi.org/ 10.1038/ismej.2009.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berrington JE, Stewart CJ, Cummings SP, Embleton ND. The neonatal bowel microbiome in health and infection. Curr Opin Infect Dis 2014; 27:236-43; PMID:24751892; http://dx.doi.org/ 10.1097/QCO.0000000000000061 [DOI] [PubMed] [Google Scholar]
- 4.Bird A. DNA methylation patterns and epigenetic memory. Genes Dev 2002; 16:6-21; PMID:11782440; http://dx.doi.org/ 10.1101/gad.947102 [DOI] [PubMed] [Google Scholar]
- 5.Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, Heine-Suñer D, Cigudosa JC, Urioste M, Benitez J, et al.. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A 2005; 102:10604-9; PMID:16009939; http://dx.doi.org/ 10.1073/pnas.0500398102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cortese R, Khalyfa A, Bao R, Andrade J, Gozal D. Epigenomic profiling in visceral white adipose tissue of offspring of mice exposed to late gestational sleep fragmentation. Int J Obes (Lond) 2015; 39:1432; PMID:26347212; http://dx.doi.org/24286462 10.1038/ijo.2015.38. [DOI] [PubMed] [Google Scholar]
- 7.Kumar H, Lund R, Laiho A, Lundelin K, Ley RE, Isolauri E, et al. Gut microbiota as an epigenetic regulator: pilot study based on wholegenome methylation analysis. MBio 2014; 5:e02113-14; http://dx.doi.org/ 10.1128/mBio.02113-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stilling RM, Dinan TG, Cryan JF. Microbial genes, brain & behaviour - epigenetic regulation of the gut-brain axis. Genes Brain Behav 2014; 13:69-86; PMID:24286462; http://dx.doi.org/ 10.1111/gbb.12109 [DOI] [PubMed] [Google Scholar]
- 9.Yang T, Owen JL, Lightfoot YL, Kladde MP, Mohamadzadeh M. Microbiota impact on the epigenetic regulation of colorectal cancer. Trends Mol Med 2013; 19:714-25; PMID:24051204; http://dx.doi.org/ 10.1016/j.molmed.2013.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takahashi K, Sugi Y, Hosono A, Kaminogawa S. Epigenetic regulation of TLR4 gene expression in intestinal epithelial cells for the maintenance of intestinal homeostasis. J Immunol 2009; 183:6522-9; PMID:19846881; http://dx.doi.org/ 10.4049/jimmunol.0901271 [DOI] [PubMed] [Google Scholar]
- 11.Takahashi K, Sugi Y, Nakano K, Tsuda M, Kurihara K, Hosono A, Kaminogawa S. Epigenetic control of the host gene by commensal bacteria in large intestinal epithelial cells. J Biol Chem 2011; 286:35755-62; PMID:21862578; http://dx.doi.org/ 10.1074/jbc.M111.271007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gluckman PD, Hanson MA, Low FM. The role of developmental plasticity and epigenetics in human health. Birth Defects Res C Embryo Today 2011; 93:12-8; PMID:21425438; http://dx.doi.org/ 10.1002/bdrc.20198 [DOI] [PubMed] [Google Scholar]
- 13.Godfrey KM, Inskip HM, Hanson MA. The long-term effects of prenatal development on growth and metabolism. Semin Reprod Med 2011; 29:257-65; PMID:21769765; http://dx.doi.org/ 10.1055/s-0031-1275518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gao F, Zhang J, Jiang P, Gong D, Wang JW, Xia Y, ϕ stergaard MV, Wang J, Sangild PT. Marked methylation changes in intestinal genes during the perinatal period of preterm neonates. BMC Genomics 2014; 15:716; PMID:25163507; http://dx.doi.org/ 10.1186/1471-2164-15-716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dolinoy DC, Das R, Weidman JR, Jirtle RL. Metastable epialleles, imprinting, and the fetal origins of adult diseases. Pediatr Res 2007; 61:30R-7R; PMID:17413847; http://dx.doi.org/ 10.1203/pdr.0b013e31804575f7 [DOI] [PubMed] [Google Scholar]
- 16.Jirtle RL, Skinner MK. Environmental epigenomics and disease susceptibility. Nat Rev Genet 2007; 8:253-62; PMID:17363974; http://dx.doi.org/ 10.1038/nrg2045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Westerbeek EA, van den Berg A, Lafeber HN, Knol J, Fetter WP, van Elburg RM. The intestinal bacterial colonisation in preterm infants: a review of the literature. Clin Nutr 2006; 25:361-8; PMID:16677741; http://dx.doi.org/ 10.1016/j.clnu.2006.03.002 [DOI] [PubMed] [Google Scholar]
- 18.Crudo A, Petropoulos S, Suderman M, Moisiadis VG, Kostaki A, Hallett M, Szyf M, Matthews SG. Effects of antenatal synthetic glucocorticoid on glucocorticoid receptor binding, DNA methylation, and genome-wide mRNA levels in the fetal male hippocampus. Endocrinology 2013; 154:4170-81; PMID:24029241; http://dx.doi.org/ 10.1210/en.2013-1484 [DOI] [PubMed] [Google Scholar]
- 19.Moisiadis VG, Matthews SG. Glucocorticoids and fetal programming part 2: Mechanisms. Nat Rev Endocrinol 2014; 10:403-11; PMID:24863383; http://dx.doi.org/ 10.1038/nrendo.2014.74 [DOI] [PubMed] [Google Scholar]
- 20.Nagy-Szakal D, Kellermayer R. The remarkable capacity for gut microbial and host interactions. Gut Microbes 2011; 2:178-82; PMID:21646867; http://dx.doi.org/ 10.4161/gmic.2.3.16107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yao Y, Tao H, Park DI, Sepulveda JL, Sepulveda AR. Demonstration and characterization of mutations induced by Helicobacter pylori organisms in gastric epithelial cells. Helicobacter 2006; 11:272-86; PMID:16882331; http://dx.doi.org/ 10.1111/j.1523-5378.2006.00408.x [DOI] [PubMed] [Google Scholar]
- 22.Gutierrez MI, Siraj AK, Khaled H, Koon N, El-Rifai W, Bhatia K. CpG island methylation in Schistosoma- and non-Schistosoma-associated bladder cancer. Mod Pathol 2004; 17:1268-74; PMID:15154012; http://dx.doi.org/ 10.1038/modpathol.3800177 [DOI] [PubMed] [Google Scholar]
- 23.Miyazaki T, Murayama Y, Shinomura Y, Yamamoto T, Watabe K, Tsutsui S, Kiyohara T, Tamura S, Hayashi N. E-cadherin gene promoter hypermethylation in H. pylori-induced enlarged fold gastritis. Helicobacter 2007; 12:523-31; PMID:17760721; http://dx.doi.org/ 10.1111/j.1523-5378.2007.00519.x [DOI] [PubMed] [Google Scholar]
- 24.Ushijima T, Nakajima T, Maekita T. DNA methylation as a marker for the past and future. J Gastroenterol 2006; 41:401-7; PMID:16799880; http://dx.doi.org/ 10.1007/s00535-006-1846-6 [DOI] [PubMed] [Google Scholar]
- 25.Burgers WA, Blanchon L, Pradhan S, de Launoit Y, Kouzarides T, Fuks F. Viral oncoproteins target the DNA methyltransferases. Oncogene 2007; 26:1650-5; PMID:16983344; http://dx.doi.org/ 10.1038/sj.onc.1209950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee JO, Kwun HJ, Jung JK, Choi KH, Min DS, Jang KL. Hepatitis B virus X protein represses E-cadherin expression via activation of DNA methyltransferase 1. Oncogene 2005; 24:6617-25; PMID:16007161; http://dx.doi.org/ 10.1038/sj.onc.1208827 [DOI] [PubMed] [Google Scholar]
- 27.Uozaki H, Fukayama M. Epstein-Barr virus and gastric carcinoma–viral carcinogenesis through epigenetic mechanisms. Int J Clin Exp Pathol 2008; 1:198-216; PMID:18784828 [PMC free article] [PubMed] [Google Scholar]
- 28.Lee SM, Donaldson GP, Mikulski Z, Boyajian S, Ley K, Mazmanian SK. Bacterial colonization factors control specificity and stability of the gut microbiota. Nature 2013; 501:426-9; PMID:23955152; http://dx.doi.org/ 10.1038/nature12447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kant R, Rintahaka J, Yu X, Sigvart-Mattila P, Paulin L, Mecklin JP, Saarela M, Palva A, von Ossowski I. A comparative pan-genome perspective of niche-adaptable cell-surface protein phenotypes in Lactobacillus rhamnosus. PLoS One 2014; 9:e102762; PMID:25032833; http://dx.doi.org/ 10.1371/journal.pone.0102762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yue M, Han X, Masi LD, Zhu C, Ma X, Zhang J, Wu R, Schmieder R, Kaushik RS, Fraser GP, et al.. Allelic variation contributes to bacterial host specificity. Nat Commun 2015; 6:8754; PMID:26515720; http://dx.doi.org/ 10.1038/ncomms9754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roberts D, Dalziel S. Antenatal corticosteroids for accelerating fetal lung maturation for women at risk of preterm birth. Cochrane Database Syst Rev 2006:Cd004454; PMID:16856047; http://dx.doi.org/ 10.1002/14651858. [DOI] [PubMed] [Google Scholar]
- 32.Adcock IM, Ito K, Barnes PJ. Glucocorticoids: effects on gene transcription. Proc Am Thorac Soc 2004; 1:247-54; PMID:16113442; http://dx.doi.org/ 10.1513/pats.200402-001MS [DOI] [PubMed] [Google Scholar]
- 33.Robyr D, Wolffe P. Hormone action and chromatin remodelling. Cell Mol Life Sci 1998; 54:113-24; PMID:9539951; http://dx.doi.org/ 10.1007/s000180050130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chernov AV ea. Mycoplasma CG- and GATC-specific DNA methyltransferases selectively and efficiently methylate the host genome and alter the epigenetic landscape in human cells. Epigenetics. 2015; 10(4): 303–318; http://dx.doi.org/ 10.1080/15592294.2015.1020000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dominguez-Bello MG, Costello EK, Contreras M, Magris M, Hidalgo G, Fierer N, et al.. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci U S A. 2010; 107(26): 11971–11975; http://dx.doi.org/ 10.1073/pnas.1002601107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thanabalasuriar A, Kubes P. Neonates, antibiotics and the microbiome. Nat Med 2014; 20:469-70; PMID:24804751; http://dx.doi.org/ 10.1038/nm.3558 [DOI] [PubMed] [Google Scholar]
- 37.Deshmukh HS, Liu Y, Menkiti OR, Mei J, Dai N, O'Leary CE, Oliver PM, Kolls JK, Weiser JN, Worthen GS. The microbiota regulates neutrophil homeostasis and host resistance to Escherichia coli K1 sepsis in neonatal mice. Nature Medicine 2014; 20:524-30; PMID:24747744; http://dx.doi.org/ 10.1038/nm.3542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.O'Sullivan A, He X, McNiven EMS, Haggarty NW, Lönnerdal B, Slupsky CM. Early Diet Impacts Infant Rhesus Gut Microbiome, Immunity, and Metabolism. J Proteome Res. 2013; 12(6):2833-45; http://dx.doi.org/ 10.1021/pr4001702 [DOI] [PubMed] [Google Scholar]
- 39.T MMaP . More than just a gut instinct-the potential interplay between a baby's nutrition, its gut microbiome, and the epigenome.Am J Physiol Regul Integr Comp Physiol. 2013;304(12):R1065-9; http://dx.doi.org/ 10.1152/ajpregu.00551. [DOI] [PubMed] [Google Scholar]
- 40.Peters I, Dubrowinskaja N, Abbas M, Seidel C, Kogosov M, Scherer R, Gebauer K, Merseburger AS, Kuczyk MA, Grünwald V, et al.. DNA methylation biomarkers predict progression-free and overall survival of metastatic renal cell cancer (mRCC) treated with antiangiogenic therapies. PLoS One 2014; 9:e91440; PMID:24633192; http://dx.doi.org/ 10.1371/journal.pone.0091440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dubrowinskaja N, Gebauer K, Peters I, Hennenlotter J, Abbas M, Scherer R, Tezval H, Merseburger AS, Stenzl A, Grünwald V, Kuczyk MA, et al.. Neurofilament Heavy polypeptide CpG island methylation associates with prognosis of renal cell carcinoma and prediction of antivascular endothelial growth factor therapy response. Cancer Med 2014; 3:300-9; PMID:24464810; http://dx.doi.org/ 10.1002/cam4.181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Powell TR, Smith RG, Hackinger S, Schalkwyk LC, Uher R, McGuffin P, Mill J, Tansey KE. DNA methylation in interleukin-11 predicts clinical response to antidepressants in GENDEP. Transl Psychiatry 2013; 3:e300; PMID:24002086; http://dx.doi.org/ 10.1038/tp.2013.73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Iramaneerat K, Rattanatunyong P, Khemapech N, Triratanachat S, Mutirangura A. HERV-K hypomethylation in ovarian clear cell carcinoma is associated with a poor prognosis and platinum resistance. Int J Gynecol Cancer 2011; 21:51-7; PMID:21330831; http://dx.doi.org/ 10.1097/IGC.0b013e3182021c1a [DOI] [PubMed] [Google Scholar]
- 44.Kawakami K, Matsunoki A, Kaneko M, Saito K, Watanabe G, Minamoto T. Long interspersed nuclear element-1 hypomethylation is a potential biomarker for the prediction of response to oral fluoropyrimidines in microsatellite stable and CpG island methylator phenotype-negative colorectal cancer. Cancer Sci 2011; 102:166-74; PMID:21087350; http://dx.doi.org/ 10.1111/j.1349-7006.2010.01776.x [DOI] [PubMed] [Google Scholar]
- 45.Chaudhry P, Srinivasan R, Patel FD. Utility of gene promoter methylation in prediction of response to platinum-based chemotherapy in epithelial ovarian cancer (EOC). Cancer Invest 2009; 27:877-84; PMID:19548140; http://dx.doi.org/ 10.1080/07357900902849699 [DOI] [PubMed] [Google Scholar]
- 46.Shen L, Kondo Y, Ahmed S, Boumber Y, Konishi K, Guo Y, Chen X, Vilaythong JN, Issa JP. Drug sensitivity prediction by CpG island methylation profile in the NCI-60 cancer cell line panel. Cancer Res 2007; 67:11335-43; PMID:18056460; http://dx.doi.org/ 10.1158/0008-5472.CAN-07-1502 [DOI] [PubMed] [Google Scholar]
- 47.Zhong XB, Leeder JS. Epigenetic regulation of ADME-related genes: focus on drug metabolism and transport. Drug Metab Dispos 2013; 41:1721-4; PMID:23935066; http://dx.doi.org/ 10.1124/dmd.113.053942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lopetuso LR, Scaldaferri F, Petito V, Gasbarrini A. Commensal Clostridia: leading players in the maintenance of gut homeostasis. Gut Pathog 2013; 5:23; PMID:23941657; http://dx.doi.org/ 10.1186/1757-4749-5-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Solomon NS, Gartner H, Oesterreicher TJ, Henning SJ. Development of glucocorticoid-responsiveness in mouse intestine. Pediatr Res 2001; 49:782-8; PMID:11385138; http://dx.doi.org/ 10.1203/00006450-200106000-00012 [DOI] [PubMed] [Google Scholar]
- 50.Nanthakumar NN, Fusunyan RD, Sanderson I, Walker WA. Inflammation in the developing human intestine: A possible pathophysiologic contribution to necrotizing enterocolitis. Proc Natl Acad Sci U S A 2000; 97:6043-8; PMID:10823949; http://dx.doi.org/20569209 10.1073/pnas.97.11.6043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nestor C, Ruzov A, Meehan R, Dunican D. Enzymatic approaches and bisulfite sequencing cannot distinguish between 5-methylcytosine and 5-hydroxymethylcytosine in DNA. Biotechniques 2010; 48:317-9; PMID:20569209; http://dx.doi.org/ 10.2144/000113403 [DOI] [PubMed] [Google Scholar]
- 52.Jiao X, Sherman BT, Huang da W, Stephens R, Baseler MW, Lane HC, Lempicki RA. DAVID-WS: a stateful web service to facilitate gene/protein list analysis. Bioinformatics 2012; 28:1805-6; PMID:22543366; http://dx.doi.org/ 10.1093/bioinformatics/bts251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 2001; 29:e45; PMID:11328886; http://dx.doi.org/ 10.1093/nar/29.9.e45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci U S A 1996; 93:9821-6; PMID:8790415; http://dx.doi.org/ 10.1073/pnas.93.18.9821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lu L, Yu Y, Guo Y, Wang Y, Chang EB, Claud EC. Transcriptional modulation of intestinal innate defense/inflammation genes by preterm infant microbiota in a humanized gnotobiotic mouse model. PLoS One 2015; 10:e0124504; PMID:25928420; http://dx.doi.org/ 10.1371/journal.pone.0124504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M, et al.. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. Isme j 2012; 6:1621-4; PMID:22402401; http://dx.doi.org/ 10.1038/ismej.2012.8 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.