

ABSTRACT
Small cell prostate carcinoma (SCPC) morphology is rare at initial diagnosis but often emerges during prostate cancer progression and portends a dismal prognosis. It does not express androgen receptor (AR) or respond to hormonal therapies. Clinically applicable markers for its early detection and treatment with effective chemotherapy are needed. Our studies in patient tumor–derived xenografts (PDX) revealed that AR–negative SCPC (AR−SCPC) expresses neural development genes instead of the prostate luminal epithelial genes characteristic of AR–positive castration-resistant adenocarcinomas (AR+ADENO). We hypothesized that the differences in cellular lineage programs are reflected in distinct epigenetic profiles. To address this hypothesis, we compared the DNA methylation profiles of AR− and AR+ PDX using methylated CpG island amplification and microarray (MCAM) analysis and identified a set of differentially methylated promoters, validated in PDX and corresponding donor patient samples. We used the Illumina 450K platform to examine additional regions of the genome and the correlation between the DNA methylation profiles of the PDX and their corresponding patient tumors. Struck by the low frequency of AR promoter methylation in the AR−SCPC, we investigated this region's specific histone modification patterns by chromatin immunoprecipitation. We found that the AR promoter was enriched in silencing histone modifications (H3K27me3 and H3K9me2) and that EZH2 inhibition with 3-deazaneplanocin A (DZNep) resulted in AR expression and growth inhibition in AR−SCPC cell lines. We conclude that the epigenome of AR− is distinct from that of AR+ castration-resistant prostate carcinomas, and that the AR− phenotype can be reversed with epigenetic drugs.
KEYWORDS: Androgen receptor, DNA methylation, epigenetics, EZH2, histone methylation, neuroendocrine, prostate cancer, small-cell prostate carcinoma, xenograft
Introduction
Many castration-resistant prostate carcinomas (CRPC) remain sensitive to secondary hormone therapies as a result of androgen receptor (AR) activity.1,2 However, approximately 20% of men who die of CRPC have tumors that exhibit small cell prostate carcinoma (SCPC) features.3 SCPC is a morphological variant that does not express AR and is resistant to hormonal therapies 4 but is sensitive to platinum-based chemotherapy.5 Although rarely identified at the initial diagnosis of prostate cancer, SCPC frequently emerges during the castration-resistant progression of the disease.6 At present, a biopsy is required for its diagnosis but because SCPC is often found admixed with adenocarcinoma components, biopsies are frequently falsely negative and thus, appropriate chemotherapy treatment is delayed. Moreover, we and others have shown that morphology can be unlinked from the clinical behavior and therapy responsiveness of prostate cancer, such that the biology underlying the SCPC phenotype is likely to be shared by tumors that do not fully meet criteria to be called SCPC. Biomarkers that can be serially followed to identify the emergence of SCPC or its biological equivalents are needed to apply early chemotherapy and, more importantly, to develop novel, effective therapies that can improve the outcome of men afflicted by this aggressive prostate cancer variant. To this end, in a previous study, we developed patient prostate tumor–derived xenografts (PDX) of AR–negative SCPC (AR−SCPC) and compared the AR−SCPC gene expression profiles to those of AR–positive adenocarcinomas (AR+ADENO) derived from men with CRPC.7,8 In line with the findings of others,9,10 we found that AR−SCPC is characterized by a loss of prostate luminal epithelial markers and a gain of neural progenitor/stem cell markers. Because DNA methylation profiles reflect cellular differentiation,11 we hypothesized that the DNA methylation profiles of AR− and AR+ are distinct and that specific DNA methylation markers might be used to detect AR− disease, which is of therapeutic relevance. The low frequency of methylation observed at the AR promoter in the AR− prostate tumors led us to investigate histone modification patterns and the reversibility of AR silencing.
Materials and methods
Human prostate cancer xenografts and patient tumor samples
Available samples of human prostate cancer xenografts (fresh frozen) or in dimethyl sulfoxide (DMSO) established between December 1996 and March 2008 were developed in the laboratory of Dr. Nora Navone at The University of Texas MD Anderson Cancer Center, as previously described.12,13 Patients who donated the tissue from which the xenografts were developed (n = 24) provided written informed consent before sample acquisition, according to a protocol approved by MD Anderson's Institutional Review Board.
All animal experiments were conducted in accordance with the standards of the Institutional Animal Care and Use Committee of MD Anderson.
Normal human prostate epithelial cells (PrEC) culture, histology, and immunohistochemistry methods are described in the Supplementary Materials. The electronic medical records of the donor patients were reviewed retrospectively under MD Anderson's Institutional Review Board–approved protocol RCR06-1075 to extract relevant clinicopathological information.
We also searched the tissue bank files of the MD Anderson Department of Pathology to identify available fresh frozen and formalin-fixed paraffin-embedded biopsy or surgical specimens of CRPC and SCPC from patients who provided written informed consent allowing the use of their tissue for research (n = 20).
DNA was extracted from fresh frozen and DMSO-preserved samples via standard proteinase K and phenol-chloroform extraction and was quantified on a NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). DNA was extracted from formalin-fixed paraffin-embedded patient samples by using the Cold Spring Harbor protocol.14 Briefly, samples were subjected to deparaffinization with xylene (X5P-1GAL, Fisher Scientific, Pittsburgh, PA, USA) and then to protease digestion and DNA isolation using a RecoverAll Total Nucleic Acid Isolation Kit (AM1975, Ambion Life Technologies/Thermo Fisher Scientific).
Quantitative polymerase chain reaction of mouse and human β-globin
Quantitative polymerase chain reaction (qPCR) was performed on genomic DNA from each xenograft sample in 20-µL reactions using iTaq Universal with ROX (Bio-Rad Laboratories, Hercules, CA, USA) and TaqMan primers and probes specific for the human and mouse β-globin (repressed in prostate tissue) genes designed with Primer Express software (Thermo Fisher Scientific). All probes were labeled with 6-carboxyfluorescein fluorophore (6-FAM) and a custom-synthesized non-fluorescent minor groove binder (MGB) quencher from Applied Biosystems (Life Technologies, Carlsbad, CA, USA). Primer and probe sequences included (a) murine β-globin assay (Mu-bglo-239F, 5′-AGGCCCATGGCAAGAAAGT-3′; Mu-bglo-306R, 5′-GCCCTTGAGGCTGTCCAA-3′; and Mu-bglo-259T [MGB probe, FAM-labeled], 5′-ATAACTGCCTTTAACGATG-3′) and (b) human β-globin assay (hu-bglo-232F, 5′-TGAAGGCTCATGGCAAGAAA-3′; hu-bglo-285R, 5′-GGTGAGCCAGGCCATCAC-3′; and hu-bglo-253T [MGB probe, FAM-labeled], 5′-TGCTCGGTGCCTTT-3′). The primers were used at 900-nM concentrations, and the probes were used at 100-nM concentrations.
Known quantities of human and mouse DNA were used to construct a standard curve for both primer sets to determine the efficiency of each qPCR. Using the specific mouse β-globin gene, we determined the threshold cycle values (Ct) for each sample and a control containing 100% mouse DNA and the differences in Ct (ΔCt) using the Stratagene Mx3005P system (Agilent Technologies, Inc., Santa Clara, CA, USA). Relative amounts of mouse DNA contamination were calculated (2ΔCt) and then were converted to the overall percentage of mouse DNA found in each sample.
Methylated CpG island amplification and microarray and Illumina 450K analyses
A pool of genomic DNA extracted from normal male human peripheral blood mononuclear cells was used as a control for methylated CpG island (CGI) amplification (MCA) 15 coupled to CGI microarray (MCAM) analysis, as previously described.16,17 Details of MCA are provided in the Supplementary Materials. Briefly, following digestion with SmaI and XmaI (New England BioLabs, Inc., Ipswich, MA, USA), PDX DNA was ligated to RMCA PCR adapters and amplified. Amplicons from xenograft samples were labeled with cyanine5 (Cy5) dye and cohybridized against amplicons from peripheral blood mononuclear cells labeled with cyanine3 (Cy3) dye on Agilent Technologies 4 × 44K custom DNA microarrays. The 42,222 probes (corresponding to 8,321 unique NCBI Reference Sequence Database genes) on the array recognized SmaI/XmaI fragments predominantly located around gene transcription start sites (TSSs). Fluorescence signals were normalized with locally weighted scatterplot smoothing (LOWESS),18 and trimmed averages of normalized log2ratios were calculated for amplicons covered by multiple probes. Hypermethylation was defined as normalized log2 ratio of Cy5/Cy3fluorescence (M values) greater than 1.3 (equivalent to 2.5-fold or higher of xenografts/control signal intensity) on the basis of previous experimental data.17
Genomic DNA was sent to the University of Southern California Epigenome Center (Los Angeles, CA, USA) for Illumina Infinium Methylation 450K array analysis (San Diego, CA, USA).
Pyrosequencing
Methylation status of the candidate genes was validated by using bisulfite pyrosequencing methylation analysis.19 Details are provided in the Supplementary Materials, and primers are listed in Supplementary Table S1. The methylation percentage of each gene was computed as the average of 2 to 4 CpG sites.
Chromatin immunoprecipitation
Fresh xenograft tissues were enzymatically dissociated in Accumax Cell Aggregate Dissociation Medium (10 mL per gram of tissue; 00-4666-56, eBioscience, San Diego, CA, USA) and were incubated at 37°C for 30 min under constant agitation. The cells were strained, washed with phosphate-buffered saline solution, and counted for crosslinking. Chromatin immunoprecipitation (ChIP) assays were performed by treating the cells in culture and the xenograft cells with 1% formaldehyde to crosslink histones to DNA. The crosslinking was stopped by treating the samples with glycine (0.125 M) for 5 min, and then the samples were washed with cold phosphate-buffered saline solution containing a 38-protease inhibitor cocktail (Roche, Basel, Switzerland). The chromatin was then extracted and fragmented by sonication, and the lysate was subjected to immunoprecipitation using Dynabeads Protein A and G (Thermo Fisher Scientific, Grand Island, NY) magnetic beads and the following antibodies: H3K4me3 (17-614, EMD Millipore, Billerica, MA, USA); H3K9Ac (07-352, EMD Millipore); H3K9me (ab1220, Abcam, Cambridge, MA, USA); H3K27me3 (17-622, EMD Millipore); rabbit anti-histone H3 (ab1791-100, Abcam); and rabbit anti-mouse immunoglobulin G H&L (ab46540, Abcam). ChIP products were used for TaqMan qPCR with oligonucleotide primers covering 2 regions of the AR exon 1 (Fig.3A), and β-actin and human β-globin as controls). The fold enrichment of each histone modification to histone H3 was calculated using the ΔCt method.
Cell culture and drug treatment
NCI-H660, LNCaP, PC3, and DU145 were purchased from American Type Culture Collection (ATCC, Manassas, VA). C42b was obtained from the MD Anderson Cell Line Core Facility. MDA 144-13 cell line was derived from the MDA 144-13 xenograft line developed in the Prostate Cancer Xenograft Bank at The University of Texas MD Anderson Cancer Center. All cell lines were grown in RPMI-1640 medium with 5% fetal bovine serum and penicillin/streptomycin solution (100 μg/mL). Cells were seeded (300,000 per 10-cm dish) 72 h before treatment. Cells were then treated with DZNep (0.1 μmol/L, 1 μmol/L, or 5 μmol/L) diluted in DMSO or DMSO alone for 72 h. The cells were harvested and counted using the Vi-CELL Series Cell Viability Analyzer (Beckman Coulter, Brea, CA, USA).
Western blot experiments
Protein extracts were prepared by homogenizing the tissues and cells in lysis buffer supplemented with complete protease inhibitor cocktail tablets (Roche). Soluble proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and were transferred to polyvinylidene fluoride membranes (Bio-Rad). Membranes were incubated with mouse anti-KMT6/EZH2 antibody (3827-1, Epitomics, Burlingame, CA, USA); rabbit anti-H3K27me3 (9733s, Cell Signaling Technology, Danvers, MA, USA); rabbit anti-glyceraldehyde 3-phosphate dehydrogenase (2118s, Cell Signaling Technology); rabbit anti-AR (n-20, Santa Cruz Biotechnology, Dallas, TX, USA); and rabbit anti-histone H3 (ab1791-100, Abcam). The antigen-antibody complexes were detected with Luminata Western HRP substrate (EMD Millipore).
Statistical analyses
Descriptive statistics (means, medians, ranges, and percentages) were used to describe the xenograft samples and were computed on GraphPad Prism software (GraphPad Software, Inc.., La Jolla, CA, USA). LOWESS normalization and analysis of microarray data have been described previously.20 To identify potential subgroups among the samples we performed unsupervised hierarchical clustering on GENE-E software (Broad Institute, Cambridge, MA, USA) using Ward linkage and Euclidean distance methods for continuous values (the normalized log2 ratio of Cy5/Cy3 signals from MCAM) and Jaccard index and complete linkage methods for categorical values (β-values from the Illumina Infinium Methylation 450K array). The Marker Selection tool (GENE-E) was used to analyze differentially methylated genes between subgroups that were obtained in the clustering analysis.
Results
Xenograft and donor patient characteristics
For the MCAM studies, we analyzed DNA extracted from mouse subcutaneous tissue, from cultured PrECs, and from 34 human prostate cancer xenograft tissues derived from the tumors of 24 donor patients. For the Illumina 450K analysis, we analyzed DNA extracted from an additional four xenografts and the corresponding donor patient tumor tissues. The MDA 79, MDA 117, MDA 118b, MDA 144, MDA 146, MDA 155, MDA 170, and MDA 180 xenograft lines and sublines have been previously described.7-8,21
Of the 38 samples, 14 included two to six xenograft sublines derived from the same donor tumor (e.g., MDA 146-10 and MDA 146-12) and two biologic replicates (i.e., the same xenograft subline but grown in a different mouse; e.g., MDA 144-4R) (Table 1). Twelve of the 38 samples displayed SCPC morphology, one showed mixed ADENO and SCPC features, and three had large cell neuroendocrine carcinoma (LCNEC) morphology. LCNEC is a rare variant that has similarities to SCPC and is thought to represent a transitional form between ADENO and SCPC.7 Additional AR–negative samples included one with squamous cell carcinoma morphology, another with mixed adenocarcinoma and sarcomatoid features, and five that did not meet SCPC criteria. Paraffin-embedded tissues were not available for xenografts MDA 46 and MDA 102, so the morphology of these xenografts is unknown. The remaining 11 samples had ADENO morphology and expressed AR on immunohistochemical analysis.
Table 1.
Xenograft Sample Description.
| Patient Donor | Tumor Site | Xenograft Line-Subline | DMSO or FF | DNA Quantity (ug) | 260/280 Ratio | DNA-EP Smear | Morphology | AR (%cells) | % Mouse DNA Avg | % Mouse DNA SD |
|---|---|---|---|---|---|---|---|---|---|---|
| 31 | Liver | MDA-31 | D | 828 | 1.94 | ADCA | 85 ± 5 | 1.11 | 0 | |
| 40 | Liver | MDA-40 | D | 435 | 1.97 | ADCA | 0 ± 0 | 1.19 | 0.22 | |
| 43 | Adrenal | MDA-43 | D | 1596 | 1.98 | ADCA | 80 ± 0 | 1.3 | 0.25 | |
| 44 | SQ nodule | MDA-44 | D | 369 | 1.88 | SCPC | 0 ± 0 | 2 | 1.16 | |
| 46 | Pleural fluid | MDA-46 | D | 654 | 1.89 | na | na | 0.71 | 0.26 | |
| 51 | Liver | MDA-51 | D | 229 | 1.94 | ADCA | 0 ± 0 | 6.83 | 0.46 | |
| 62 | Ascitic fluid | MDA-62 | D | 570 | 1.93 | Mixed ADCA & SARC | 0 ± 0 | 37.15 | 14.73 | |
| 66 | Pelvic tumor | MDA-66 | D | 289 | 1.94 | ADCA | 0 ± 0 | 3.79 | 3.69 | |
| 75 | Brain | MDA-75 | D | 51 | 1.96 | ADCA | 90 ± 0 | 1.54 | 0.05 | |
| 76 | Pelvic tumor | MDA-76 | D | 1250 | 1.9 | Yes | ADCA | 86 ± 12 | 14.47 | 3.02 |
| 79 | Pelvic tumor | MDA-79 | D | 522 | 1.92 | Yes | ADCA | 89 ± 6 | 57.41 | 51.61 |
| 80 | Pleural fluid | MDA-80 | D | 111 | 1.94 | ADCA | 90 ± 4 | 0.97 | 0.06 | |
| 91 | Liver | MDA-91A | D | 450 | 1.96 | SCPC | 0 ± 0 | 1.92 | 0.67 | |
| MDA-91B | D | 704 | 1.89 | SCPC | 0 ± 0 | 1.69 | 0 | |||
| 94 | Pleural fluid | MDA-94 | D | 277 | 1.96 | ADCA | 0 ± 0 | 3.56 | 2.81 | |
| 100 | Pelvic tumor | MDA-100 | D | 190 | 1.93 | SQ CELL CA | 0 ± 0 | 9.32 | 3.1 | |
| 101 | Liver | MDA-101 | D | 1422 | 1.94 | Yes | ADCA | 63 ± 20 | 7.92 | 4.11 |
| 102 | Pelvic tumor | MDA-102 | D | 909 | 1.93 | Yes | na | na | 12.84 | 6.87 |
| 117 | Pelvic tumor | MDA-117-9 | FF | 554 | 1.9 | ADCA | 96 ± 2 | 1.28 | 0.41 | |
| 118 | Bone | MDA-118b | FF | 308 | 1.91 | ADCA | 0 ± 0 | 57.38 | 21 | |
| 122 | Adrenal | MDA-122 | FF | 880 | 1.97 | ADCA | 90 ± 0 | 11.58 | 2.89 | |
| 137 | RPLN | MDA-137 | FF | 763 | 1.89 | ADCA | 100 ± 0 | 10.64 | 1.69 | |
| 144 | Pelvic tumor | MDA-144-11 | FF | 540 | 1.94 | SCPC | 0 ± 0 | 5.07 | 0.28 | |
| MDA-144-13 | FF | 576 | 1.95 | SCPC | 0 ± 0 | 1.84 | 0.32 | |||
| MDA-144-13R | FF | 876 | 1.89 | SCPC | 0 ± 0 | 1.69 | 0.6 | |||
| MDA-144-20 | FF | 1415 | 1.98 | SCPC | 0 ± 0 | 0.78 | 0.05 | |||
| MDA-144-23 | FF | 831 | 1.96 | SCPC | 0 ± 0 | 1.24 | 0.03 | |||
| MDA-144-4 | FF | 1122 | 1.98 | LCNEC | 0 ± 0 | 1.51 | 0.03 | |||
| MDA-144-4R | FF | 808 | 1.92 | LCNEC | 0 ± 0 | 1.16 | 0.78 | |||
| MDA-144-6 | FF | 1398 | 1.95 | LCNEC | 1 ± 2 | 2.53 | 2.6 | |||
| 146 | Pelvic tumor | MDA-146-10 | FF | 970 | 1.91 | SCPC | 0 ± 0 | 0.66 | 0.53 | |
| MDA-146-12 | FF | 1051 | 2.04 | Mixed ADCA & SCPC | 97 ± 6 | 3.35 | 0.28 | |||
| 155 | Pelvic tumor | MDA-155-12 | FF | 884 | 1.95 | SCPC | 0 ± 0 | 1.01 | 0.01 | |
| MDA-155-2 | FF | 1132 | 1.96 | SCPC | 0 ± 0 | 1.35 | 0.36 |
Abbreviations: MDA, MD Anderson; DMSO or D, stored in dimethyl sulfoxide; FF, fresh frozen; EP, electropphoresis; ADCA, adenocarcinoma; na, not available; SCPC, small cell prostate sarcomatoid; SQ CELL CA, squamous cell carcinoma; LCNEC, large cell neuroendocrinandrogen receptor.
Four of the thirty-eight DNA samples exhibited a smear on gel electrophoresis, indicating significant DNA degradation; however, a strong high molecular-weight band was still present in all four samples. Seven samples had greater than 10% mouse DNA contamination (Table 1, Supplementary Fig. S1). Neither DNA degradation nor mouse DNA contamination affected the quality of the arrays.
The charts of the 24 donor patients (4 of whose tumors yielded >1 xenograft line) were reviewed retrospectively. All patients had received androgen deprivation therapy, and 20 had received at least one line of chemotherapy (often with more than one agent) before the xenografts were established. One patient (the donor of MDA 43) remains alive following bilateral adrenalectomy 16.1 y after diagnosis.
DNA methylation profiles of patient tumor–derived CRPC xenografts
Unsupervised hierarchical clustering (Ward linkage, Euclidean distance) using all the MCAM M values for the 16,621 SmaI sites after LOWESS normalization showed that, for the most part, the xenograft lines derived from the same patient clustered together (Fig. 1A): only one of the MDA 144 sublines (MDA 144-11) did not cluster with the other seven MDA 144 samples. All other xenograft sublines derived from a single patient tumor clustered together (MDA 146-10 with MDA 146-12, MDA 155-2 with MDA 155-12, and MDA 91A with MDA 91B).
Figure 1.
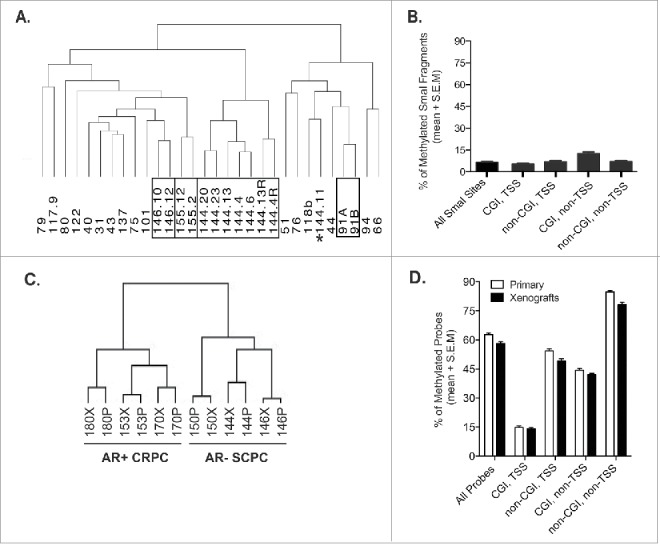
A. Unsupervised hierarchical clustering (Ward linkage, Euclidean distance) using all the MCAM M values for the 16,621 SmaI sites after LOWESS normalization. Black boxes group single patient xenograft sublines, whereas the asterisks indicates a xenograft subline that does not cluster with sublines from the same patient donor. B. Frequency of methylated SmaI fragments, normalized by the fraction of the gene, across all samples according to their relationship to CGIs and to the promoter region (±1 kb of TSS) of known Reference Sequence genes. C. Unsupervised hierarchical clustering (Ward linkage, Euclidean distance) using all β-values from the Illumina 450K array of six xenografts and their corresponding patient donor. D. Frequency of methylated probes, normalized by the fraction of the gene, total and by compartment, for primary patient samples and xenografts evaluated on Illumina 450K arrays.
To validate the MCAM results, we selected 19 sequences contained within 17 promoter-associated (±1 kb from the closest TSS) CGIs for pyrosequencing. Using 10% as the cutoff for hypermethylated sequences and a normalized log2 ratio of Cy5/Cy3 tumor:normal signal ≥1.3, we obtained a sensitivity of 89.8%, a specificity of 67.0%, a positive predictive value of 71.0%, and a negative predictive value of 87.9% (Supplementary Fig. S2), consistent with our previous experience with the MCAM method.17
In subsequent analyses, we excluded 1,800 SmaI sites to account for nonspecific hybridization of contaminant mouse DNA in the xenograft DNA samples and for tissue-specific hypermethylation of PrEC (normal prostate cells), compared with normal blood (1,317 from mouse DNA, 553 from PrEC DNA, and 70 from both), leaving a total of 14,821 SmaI fragments for analysis. We averaged the log2 ratio values of technical and biologic replicates of the same tumors and, with the 1.3 cutoff, found that the frequency of hypermethylated SmaI fragments ranged from 2.2% to 12.7% (median, 6.6%) per xenograft. However, 80% of the studied SmaI fragments were unmethylated across all samples, and correlated hypermethylation was rare; only 527 of 14,821 SmaI fragments (3.6%) were hypermethylated in 50% or more of the tumors (Fig. 1B). We classified the 14,821 SmaI fragments according to their relationship to CGIs and to the promoter region (±1 kb of TSS) of known Reference Sequence genes. We observed that the compartment consisting of non-promoter CGIs was generally 2–3 times more hypermethylated than any other compartment and that promoters constituted the least-hypermethylated compartment (Fig. 1B). Generally, there was good agreement among the compartments: xenograft lines with the lowest frequencies of methylated promoter CGIs, for example, also had very low frequencies of hypermethylation in other compartments (data not shown).
Additionally, we examined the relationship between the xenografts and the corresponding patient tumor DNA methylation profiles by using six xenograft-patient tumor pairs—three AR− models meeting SCPC criteria (MDA 144-13, MDA 146-10, and MDA 150) and three AR+ADENO models (MDA 153, MDA 170, and MDA 180). For this analysis, we expanded the CpG site coverage to include CGI shores 22 and intergenic and intragenic regions using Illumina 450K arrays. Of these six models, MDA 144-4 and MDA 146-10 had also been analyzed by MCAM. Unsupervised hierarchical clustering using all β-values segregated the AR−SCPC and AR+ADENO samples into two ggroups and showed high correlation between the DNA methylation profiles of the xenografts and the corresponding patient tumors (Fig. 1C). The frequency of hypermethylated probes (β-value above 0.2), total and by compartment, were similar between the xenografts and the corresponding patient tumors, suggesting that methylation patterns carry over from the patient to the xenograft models (Fig. 1D).
Differences in DNA methylation profiles between AR− and AR+ prostate tumors
To determine whether the MCAM DNA methylation profiles of AR−SCPC and AR+ADENO prostate tumors were distinct, we excluded samples with squamous cell morphology (MDA 100), sarcomatoid morphology (MDA 62), and the xenografts for which morphology was unknown (MDA 46 and MDA 102). This left 11 AR−SCPC, 3 AR−LCNEC (previously found to be biologically similar to AR−SCPC),7 and one mixed AR−SCPC and AR+ADENO xenograft samples (n = 15) derived from five patient tumors. There were an additional 5 AR− samples that did not meet SCPC criteria (AR−ADENO) and 11 AR+ADENO xenograft samples derived from 15 patient tumors. We averaged the MCAM probe M values per patient and converted them to categorical values (log2 R/G ≥ 1.3 = methylated; log2 R/G < 1.3 = unmethylated). Unsupervised hierarchical clustering using probes with at least two methylated samples (resulting in 3,031 probes) segregated the patients into three groups: group A contained nine of the 11 AR+ADENO samples and group B was composed entirely of AR− samples. Group C was composed of outliers that had lower than average methylation but was composed mostly of AR− tumors (Fig. 2A).
Figure 2.
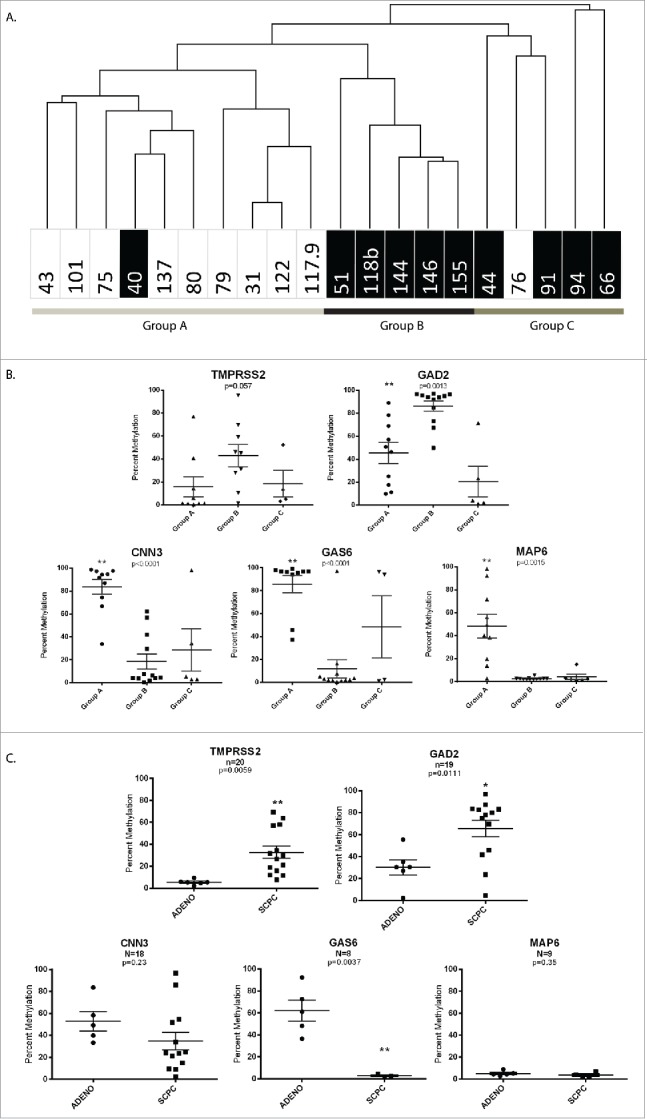
A. Unsupervised hierarchical clustering using MCAM probes with at least two methylated samples (resulting in 3,031 probes) segregated the tumors into three groups. The black boxes indicate AR− xenografts. B. Differentially methylated sequences identified by MCAM using the Marker Selection tool (GENE-E software) validated by pyrosequencing the differentially methylated sequences identified by MCAM. C. The methylation levels of the validated genes in the patient DNA samples. Statistical significance based on Student t-test is indicated by asterisk marks. (*) represents P< 0.05 and (**) represents P < 0 .005.
We used pyrosequencing to validate the differentially methylated sequences identified by MCAM. Using the Marker Selection tool (GENE-E software), we generated a list of the top 100 differentially methylated probes between group A and group B (group C was excluded because of its below-average methylation status). Of these differentially methylated sequences detected by MCAM, we selected four genes (GAS6, GAD2, MAP6, and CNN3) for validation on the basis of several criteria: a P-value less than 0.01, only probes located in the CGIs, probes with a frequency of 70% in one group and less than 30% in the other, gene function, and confirmation that the gene was methylated in the AR−SCPC patient samples analyzed by the Illumina 450K array. Additionally, we chose to validate TMPRSS2 solely on the basis of results from the Illumina arrays because of gene function and previous reports of methylation status in AR–negative prostate cancer cell lines 23 We then used pyrosequencing to quantify the methylation levels of these genes in the xenograft samples (Fig. 2B). Finally, we examined the methylation levels of the validated genes in the patient DNA samples. Three of the five genes, (TMPRSS2, GAD2, and GAS6) were differentially methylated (P<0.05) between SCPC and ADENO patient samples (Fig. 2C).
Promoter DNA methylation not a cause of AR silencing in AR−SCPC xenografts
Previously, we had found that both AR protein and mRNA transcripts were absent in our AR−SCPC models, and array comparative genomic hybridization experiments did not reveal AR deletions.8 Struck by the low levels of methylation observed in the AR promoter CpG island of the AR−SCPC xenografts (Fig.3A-B), we used bisulfite pyrosequencing to confirm this observation, measuring the methylation of the AR promoter–associated CGI in DNA extracted from 11 AR+ and 19 AR− PDX, a xenograft-derived cell line (MDA 144-13), as well as prostate cancer cell lines LNCaP, PC3, NCI-H660, and DU145. Of the cell lines, only PC3 and DU145 cells had more than 15% methylation, confirming the findings that others have reported.24 All but four of the xenografts had less than 15% methylation at the AR promoter, regardless of AR expression status (Fig. 3C). Therefore, we concluded that DNA methylation of the AR promoter is infrequent in CRPC.
Figure 3.
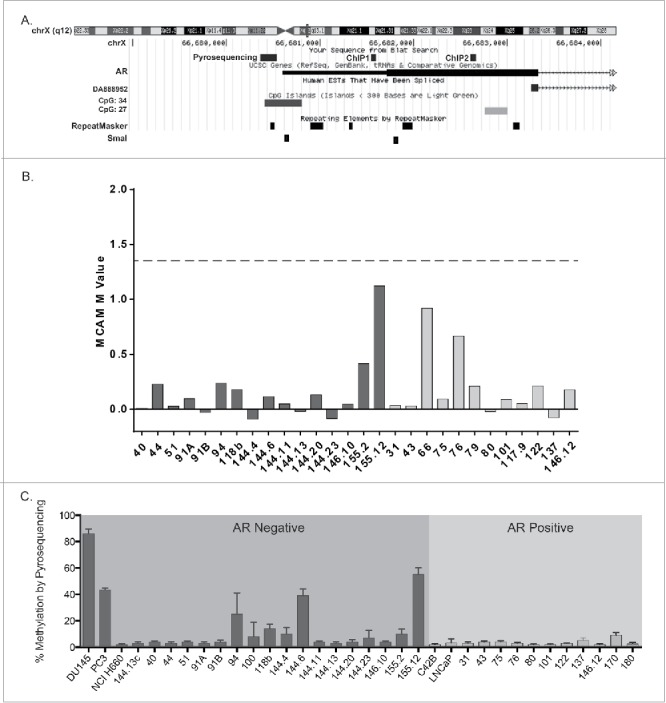
A. Map showing the sequence location of the pyrosequencing and ChIP q-PCR primers. B. MCAM M-values at the AR promoter in patient derived xenografts (AR-negative, dark gray; AR-positive, light gray). C. Frequency of AR promoter-associated methylation in prostate cancer patient derived xenografts and cell lines (AR-negative black, AR-positive gray) using bisulfite pyrosequencing.
H3K27me3 enrichment at the AR promoter in AR-SCPC xenografts and AR expression in AR-SCPC cells treated with DZNep
We then examined the chromatin markings on the AR promoter using ChIP-qPCR in the previously described AR−SCPC/LCNEC (MDA 144-13, MDA 144-4, MDA 155-2, and MDA 146-10) and AR+ADENO (MDA 170-4 and MDA 180-30 xenografts 7,8 as well as in three established prostate cancer cell lines [one AR–positive (LNCaP) and two AR–negative (PC3 and DU145)]. We evaluated both active (H3K4me3 and H3K9ac) and repressive (H3K9me2 and H3K27me3) histone modifications, using the marking of the constitutively expressed gene β-actin and human β-globin, a repressed gene in prostate tissue, as controls for these experiments. As expected, the only samples with AR marking by H3K4me3 and/or H3K9ac were the two AR–positive xenografts MDA PCa 170.4 and MDA PCa 180.30 and AR–positive LNCaP cells. Marking by repressive histone modifications varied: H3K27me3 was enriched in all but one of the AR–negative samples, and H3K9me2 was also found in two of the four xenografts (Fig. 4).
Figure 4.

AR promoter histone modification using ChIP-PCR for repressive marks (H3K27me3 and H3K9me2) in gray and active histone marks (H3K4me3 and H3K9ac) in black were assayed using primers covering two regions of the AR exon 1 as shown in Fig. 3A. Shown here is the average for both regions. HBB and ACTB were used as repressive and active mark controls respectively. The y-axis indicates fold enrichment compared to total histone H3 using the CT method.
H3K27me3 gene silencing involves polycomb group protein EZH2, which correlates with the aggressiveness of prostate cancer and is known to be overexpressed in several cancers.25 We therefore investigated the global protein expression of H3K27me3 and EZH2 in our xenograft samples and cell lines. Using Western blot analysis, we observed similar levels of H3K27me3 and EZH2 protein in AR–negative xenografts, cell lines and AR–positive xenografts (Fig. 5A).
Figure 5.
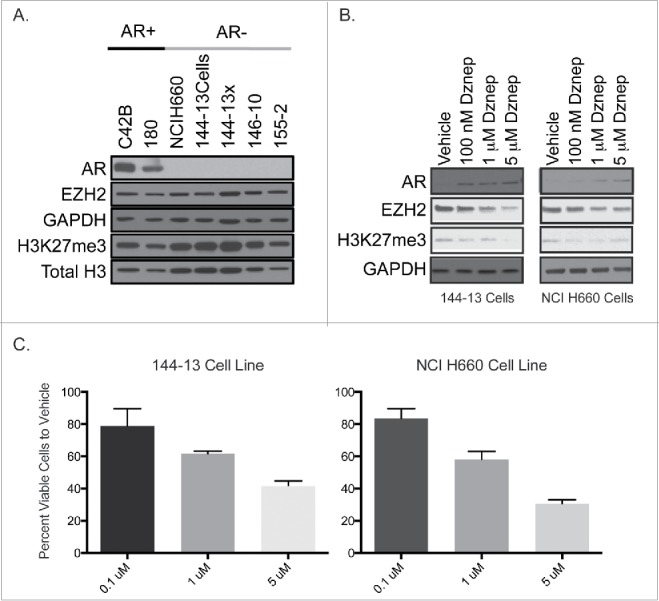
A. Global levels of AR, EZH2 and H3K27me3 by protein gel blot analysis in AR+ cell line C42B and xenograft 180 and AR- SCPC cell line NCI H660 and xenografts (144-13, 146-10, 155-2). B. Western blot analysis of the AR, EZH2, and H3K27me3 protein following 72 hDZNep treatment in 144-13 and NCIH660 cell lines. C. Percent of viable cells after 72 h of DZNep treatment as compared to vehicle.
Treatment of the NCI-H660 and MDA 144-13 cell lines with DZNep, a known S-adenosylhomocysteine hydrolase inhibitor that leads to the indirect inhibition of methyltransferase activity by blocking S-adenosyl-methionine–dependent reactions,26 resulted in AR expression (Fig. 5B) and dose-dependent cell growth inhibition (Fig. 5C), which were associated with a decrease in the levels of EZH2 and H3K27me3.
Discussion
To our knowledge, we are the first to compare AR−SCPC and AR+ADENO DNA methylation profiles, to identify candidate methylation markers for distinguishing these subtypes, and to show that AR silencing in AR−SCPC can be reversed with DZNep. Our findings also support the value of patient-derived xenografts in studying the biology of CRPC.
One limitation of our study was that the segregation of xenografts by the MCAM methylation profiles was not perfect. Misclassification could be due to the possibility that, despite different morphology, some of the AR+ADENO samples shared biology with the AR−SCPC samples. Our previous studies have shown that tumors of various morphologies share clinical and molecular features (such as loss of RB1 and p53 mutations) with the AR−SCPC 27,28 indicating that the underlying biology of clinically relevant subsets of CRPC is not necessarily reflected in a specific morphology. A second possibility is that AR−SCPC, the recognizable extreme of this biological spectrum, is still a heterogeneous group, and that this heterogeneity could be reflected in different DNA methylomes. A third possibility is that MCAM could not capture the differentially methylated regions because this method is designed to detect methylation predominantly at promoter CGIs. Although fewer samples were analyzed by Illumina arrays than by MCAM, the Illumina arrays provided a cleaner segregation, possibly because of the greater depth of analysis and the contribution of non-promoter non-CGI promoter methylation to distinguishing between phenotypes. Additional analyses with more samples are needed to confirm the hypothesis that DNA methylation profiles can distinguish clinically relevant subsets of CRPC.
Nonetheless, we identified and validated candidate DNA methylation markers, TMPRSS2 and GAD2, that could be used to detect emerging AR−SCPC. Since developing such markers for clinical use is our ultimate goal, our future research will analyze serum samples from men with AR−SCPC using reduced representation bisulfite sequencing, which should help identify not only differentially methylated regions but also markers that are most likely to be detected in peripheral circulation.
The expression of AR in AR−SCPC models suggests that this aggressive phenotype can be reversed with epigenetic drugs. Previous reports have shown a correlation between decreased H3K27me3 after DZNep treatment and increased gene expression.29,30 However, DZNep is not specific for EZH2, so our future studies will use GSK126,31 an S-adenosyl-methionine–competitive, small molecule inhibitor of EZH2 methyltransferase activity, to confirm that AR expression in AR−SCPC cells is due to depletion of H3K27me3 at the AR promoter after EZH2 inhibition.
The question is whether inducing AR expression in AR−SCPC will be clinically beneficial. AR is considered the principal oncogenic driver of most CRPC. However, one study proposed that AR also has tumor suppressor functions.32 Indeed, other researchers found that the downstream effects of AR signaling are context dependent: the transcriptional profiles attributed to AR signaling are distinct in castration-sensitive prostate cancer and CRPC.33 Additional research is needed to determine what, if any, downstream AR signaling is restored by epigenetic therapies in AR−SCPC and whether AR signaling is associated with tumor growth inhibition (and presumably differentiation), as anticipated by our DZNep results. Depending on the transcriptional program that might be activated, combinations of epigenetic drugs with AR agonists or antagonists could be effective therapies.
In summary, our results support the hypothesis that AR−SCPC and AR+ADENO have distinct DNA methylation profiles that may serve as classifying markers for patients with prostate cancer. We also showed that H3K27me3 enrichment at the AR promoter is associated with AR silencing in AR−SCPC, which appears to be reversible when EZH2 is inhibited, suggesting the AR−SCPC phenotype is potentially reversible through epigenetic manipulation.
Supplementary Material
Biographies
*Marcos R. H. Estécio contributed equally to this work and are co-first authors.
Brittany Kleb: Performed the majority of the experiments, data analyses, figure generation and manuscript preparation.
Marcos R. H. Estécio: Provided technical supervision and expertise, and performed data analyses and interpretation; also contributed to writing of manuscript and generation of the figures.
Jiexin Zhang: Contributed to the bioinformatics and statistical analyses.
Vassiliki Tzelepi: Evaluated the morphology and immunohistochemical staining of the samples.
Woonbok Chung: Performed MCAM in the PrEC cell line.
Jaroslav Jelinek: Provided primers and assistance for mouse DNA contamination assay, as well as assistance with data analyses and interpretation.
Jean-Pierre Issa: Provided expertise and assistance with data analyses and interpretation.
Nora M. Navone: Provided PDX samples.
Salahaldin Tahir: Performed western blots.
Sankar Maity: Provided expertise and assistance with data analyses and interpretation.
Ana Aparicio: Designed and supervised experiments, performed data analyses and interpretation, and supervised manuscript preparation and figure generation
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors wish to acknowledge the sources of funding that made this work possible including the NIH/NCI (under award numbers P30CA016672 (Bioinformatics Shared Resource) and P50CA140388) and the Joan Stanford Alexander Family Fund.
References
- 1.Ryan CJ, Molina A, Li J, Kheoh T, Small EJ, Haqq CM, Grant RP, de Bono JS, Scher HI. Serum androgens as prognostic biomarkers in castration-resistant prostate cancer: results from an analysis of a randomized phase III trial. J Clin Oncol 2013; 31(22): p:2791-8; PMID:23816964; http://dx.doi.org/23337756 10.1200/JCO.2012.45.4595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rathkopf D, Scher HI. Androgen receptor antagonists in castration-resistant prostate cancer. Cancer J 2013; 19(1): p:43-9; PMID:23337756; http://dx.doi.org/ 10.1097/PPO.0b013e318282635a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tanaka M, Suzuki Y, Takaoka K, Suzuki N, Murakami S, Matsuzaki O, Shimazaki J. Progression of prostate cancer to neuroendocrine cell tumor. Int J Urol 2001; 8(8): p:431-6; discussion 437; PMID:11555007; http://dx.doi.org/ 10.1046/j.1442-2042.2001.00347.x [DOI] [PubMed] [Google Scholar]
- 4.Moore SR, Reinberg Y, Zhang G, Small cell carcinoma of prostate: effectiveness of hormonal versus chemotherapy. Urology 1992; 39(5):p:411-6; PMID:1315995; http://dx.doi.org/12118020 10.1016/0090-4295(92)90235-O [DOI] [PubMed] [Google Scholar]
- 5.Papandreou CN, Daliani DD, Thall PF, Tu SM, Wang X, Reyes A, Troncoso P, Logothetis CJ. Results of a phase II study with doxorubicin, etoposide, and cisplatin in patients with fully characterized small-cell carcinoma of the prostate. J Clin Oncol 2002; 20(14): p:3072-80; PMID:12118020; http://dx.doi.org/ 10.1200/JCO.2002.12.065 [DOI] [PubMed] [Google Scholar]
- 6.Ro JY, Têtu B, Ayala AG, Ordóñez NG. Small cell carcinoma of the prostate. II. Immunohistochemical and electron microscopic studies of 18 cases. Cancer 1987; 59(5):p:977-82; PMID:2434204; http://dx.doi.org/22156612 10.1002/1097-0142(19870301)59:5<977::AID-CNCR2820590521>3.0.CO;2-G [DOI] [PubMed] [Google Scholar]
- 7.Aparicio A, Tzelepi V, Araujo JC, Guo CC, Liang S, Troncoso P, Logothetis CJ, Navone NM, Maity SN. Neuroendocrine prostate cancer xenografts with large-cell and small-cell features derived from a single patient's tumor: morphological, immunohistochemical, and gene expression profiles. Prostate 2011; 71(8): p:846-56; PMID:21456067; http://dx.doi.org/22156612 10.1002/pros.21301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tzelepi V, Zhang J, Lu JF, Kleb B, Wu G, Wan X, Hoang A, Efstathiou E, Sircar K, Navone NM, et al.. Modeling a lethal prostate cancer variant with small-cell carcinoma features. Clin Cancer Res 2012; 18(3): p:666-77; PMID:22156612; http://dx.doi.org/ 10.1158/1078-0432.CCR-11-1867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clegg N, Ferguson C, True LD, Arnold H, Moorman A, Quinn JE, Vessella RL, Nelson PS. Molecular characterization of prostatic small-cell neuroendocrine carcinoma. Prostate 2003; 55(1): p:55-64; PMID:12640661; http://dx.doi.org/ 10.1002/pros.10217 [DOI] [PubMed] [Google Scholar]
- 10.Lapuk AV, Wu C, Wyatt AW, McPherson A, McConeghy BJ, Brahmbhatt S, Mo F, Zoubeidi A, Anderson S, Bell RH, et al.. From sequence to molecular pathology, and a mechanism driving the neuroendocrine phenotype in prostate cancer. J Pathol 2012; 227(3): p:286-97; PMID:22553170; http://dx.doi.org/ 10.1002/path.4047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, Zhang X, Bernstein BE, Nusbaum C, Jaffe DB, et al.. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 2008; 454(7205): p:766-70; PMID:18600261; http://dx.doi.org/10.1038/nature07107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Navone NM, Olive M, Troncoso P. Isolation and culture of prostate cancer cell lines. Methods Mol Med, 2004; 88: p:121-32; PMID:14634223; http://dx.doi.org/10.1385/1-59259-406-9:121 [DOI] [PubMed] [Google Scholar]
- 13.Li ZG, Mathew P, Yang J, Starbuck MW, Zurita AJ, Liu J, Sikes C, Multani AS, Efstathiou E, Lopez A, et al.. Androgen receptor-negative human prostate cancer cells induce osteogenesis in mice through FGF9-mediated mechanisms. J Clin Invest 2008; 118(8): p:2697-710; PMID:18618013; http://dx.doi.org/ 10.1172/JCI33637C1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tang W, David FB, Wilson MM, Barwick BG, Leyland-Jones BR, Bouzyk MM. DNA extraction from formalin-fixed, paraffin-embedded tissue. Cold Spring Harb Protoc 2009; 2009(2): p:pdb prot5138; PMID:20147068; http://dx.doi.org/20818844 10.1101/pdb.prot5138 [DOI] [PubMed] [Google Scholar]
- 15.Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, O'Dwyer PJ, Lee RJ, Grippo JF, Nolop K, et al.. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med, 2010; 363(9): p:809-19; PMID:20818844; http://dx.doi.org/ 10.1056/NEJMoa1002011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Toyota M, Ho C, Ohe-Toyota M, Baylin SB, Issa JP. Inactivation of CACNA1G, a T-type calcium channel gene, by aberrant methylation of its 5′ CpG island in human tumors. Cancer Res 1999; 59(18): p:4535-41; PMID:10493502 [PubMed] [Google Scholar]
- 17.Estecio MR, Yan PS, Ibrahim AE, Tellez CS, Shen L, Huang TH, Issa JP. High-throughput methylation profiling by MCA coupled to CpG island microarray. Genome Res 2007; 17(10): p:1529-36; PMID:17785535; http://dx.doi.org/ 10.1101/gr.6417007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang YH, Dudoit S, Luu P, et al. Normalization for cDNA microarray data: a robust composite method addressing single and multiple slide systematic variation. Nucleic acids research 2002; 30(4):e15; PMID:11842121; http://dx.doi.org/10.1093/nar/30.4.e15 12866414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Colella S, Shen L, Baggerly KA, Issa JP, Krahe R. Sensitive and quantitative universal Pyrosequencing methylation analysis of CpG sites. Biotechniques, 2003; 35(1): p:146-50; PMID:12866414 [DOI] [PubMed] [Google Scholar]
- 20.Shen L, Toyota M, Kondo Y, Lin E, Zhang L, Guo Y, Hernandez NS, Chen X, Ahmed S, Konishi K, et al.. Integrated genetic and epigenetic analysis identifies three different subclasses of colon cancer. Proc Natl Acad Sci U S A 2007; 104(47): p:18654-9; PMID:18003927; http://dx.doi.org/ 10.1073/pnas.0704652104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li ZG, Yang J, Vazquez ES, Rose D, Vakar-Lopez F, Mathew P, Lopez A, Logothetis CJ, Lin SH, Navone NM. Low-density lipoprotein receptor-related protein 5 (LRP5) mediates the prostate cancer-induced formation of new bone. Oncogene 2008; 27(5): p:596-603; PMID:17700537; http://dx.doi.org/19151715 10.1038/sj.onc.1210694 [DOI] [PubMed] [Google Scholar]
- 22.Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, Cui H, Gabo K, Rongione M, Webster M, et al.. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet 2009; 41(2): p:178-86; PMID:19151715; http://dx.doi.org/ 10.1038/ng.298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chu M, Chang Y, Li P, Guo Y, Zhang K, Gao W. Androgen receptor is negatively correlated with the methylation-mediated transcriptional repression of miR-375 in human prostate cancer cells. Oncol Rep 2014; 31(1): p:34-40; PMID:24173286; http://dx.doi.org/10.1038/sj.onc.1210694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yegnasubramanian S, Kowalski J, Gonzalgo ML, Zahurak M, Piantadosi S, Walsh PC, Bova GS, De Marzo AM, Isaacs WB, Nelson WG. Hypermethylation of CpG islands in primary and metastatic human prostate cancer. Cancer Res 2004; 64(6): p:1975-86; PMID:15026333; http://dx.doi.org/ 10.1158/0008-5472.CAN-03-3972 [DOI] [PubMed] [Google Scholar]
- 25.Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, Ghosh D, Pienta KJ, Sewalt RG, Otte AP, et al., The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002; 419(6907): p:624-9; PMID:12374981; http://dx.doi.org/ 10.1038/nature01075 [DOI] [PubMed] [Google Scholar]
- 26.Jones PA, Liang G. Rethinking how DNA methylation patterns are maintained. Nat Rev Genet 2009; 10(11): p:805-11; PMID:19789556; http://dx.doi.org/ 10.1038/nrg2651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aparicio AM, Harzstark AL, Corn PG, Wen S, Araujo JC, Tu SM, Pagliaro LC, Kim J, Millikan RE, Ryan C, et al., Platinum-based chemotherapy for variant castrate-resistant prostate cancer. Clin Cancer Res 2013; 19(13): p:3621-30; PMID:23649003; http://dx.doi.org/ 10.1158/1078-0432.CCR-12-3791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aparicio AM, Shen L, Tapia EL, et al. Combined Tumor Suppressor Defects Characterize Clinically Defined Aggressive Variant Prostate Cancers. Clinical cancer research 2015; PMID:26546618; http://dx.doi.org/10.1158/1078-0432.CCR-15-1259 17437993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tan J, Yang X, Zhuang L, Jiang X, Chen W, Lee PL, Karuturi RK, Tan PB, Liu ET, Yu Q. Pharmacologic disruption of Polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev 2007; 21(9): p:1050-63; PMID:17437993; http://dx.doi.org/ 10.1101/gad.1524107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miranda TB, Jones PA. DNA methylation: the nuts and bolts of repression. J Cell Physiol 2007; 213(2): p:384-90; PMID:17708532; http://dx.doi.org/17369840 10.1002/jcp.21224 [DOI] [PubMed] [Google Scholar]
- 31.McCabe NP, De S, Vasanji A, Brainard J, Byzova TV. Prostate cancer specific integrin alphavbeta3 modulates bone metastatic growth and tissue remodeling. Oncogene 2007; 26(42): p:6238-43; PMID:17369840; http://dx.doi.org/ 10.1038/sj.onc.1210429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Niu Y, Altuwaijri S, Lai KP, Wu CT, Ricke WA, Messing EM, Yao J, Yeh S, Chang C. Androgen receptor is a tumor suppressor and proliferator in prostate cancer. Proc Natl Acad Sci U S A 2008; 105(34): p:12182-7; PMID:18723679; http://dx.doi.org/ 10.1073/pnas.0804700105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Q, Li W, Zhang Y, Yuan X, Xu K, Yu J, Chen Z, Beroukhim R, Wang H, Lupien M, et al., Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell 2009; 138(2): p:245-56; PMID:19632176; http://dx.doi.org/ 10.1016/j.cell.2009.04.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.