

Abstract
NSF (N-ethylmaleimide Sensitive Factor), first discovered in 1988, is a key factor for eukaryotic trafficking, including protein and hormone secretion and neurotransmitter release. It is a member of the AAA+ family (ATPases Associated with diverse cellular Activities). NSF disassembles SNARE (Soluble N-ethylmaleimide sensitive factor Attachment protein REceptors) complexes in conjunction with SNAP (Soluble N-ethylmaleimide sensitive factor Attachment Protein) adaptor proteins. Structural studies of NSF and its complex with SNARE and SNAP (known as 20S supercomplex) started about twenty years ago. Crystal structures of individual N and D2 domains of NSF and low-resolution electron microscopy structures of full-length NSF and 20S supercomplex have been reported over the years. Nevertheless, the molecular architecture of the 20S supercomplex and the molecular mechanism of NSF-mediated SNARE complex disassembly remained unclear until recently. Here we review recent atomic-resolution or near-atomic resolution structures of NSF and of the 20S supercomplex, and recent insights into the molecular mechanism and energy requirements of NSF. We also compare NSF with other known AAA+ family members.
Keywords: eukaryotic trafficking, SNAREs, NSF, ATPase, protein disassembly
Graphical Abstract
1. Introduction
NSF was the first protein found to play a key role in eukaryotic trafficking [1,2]. It is a member of the AAA+ family consisting of two ATPase rings (known as Type II AAA+). In concert with the adaptor protein, SNAP, NSF disassembles the SNARE complex into individual proteins upon ATP hydrolysis [3–5]. Although earlier work suggested that disruption of the SNARE complex by ATP hydrolysis drives fusion [3], subsequent work clarified that it is actually the SNARE complex formation that drives fusion [4,5]. By disassembling post-fusion SNARE complexes, NSF is essential for maintaining pools of fusion-ready individual SNARE proteins that mediate membrane fusion in a variety of cellular processes, including neurotransmitter release, protein transport, and hormone secretion [6–9]. Most eukaryotic organisms encode only one NSF gene. The known exception is fly in which two homologues of NSF have been reported [10,11]. By contrast, there are three homologues of SNAP proteins in mammals [12,13], and dozens of different SNARE complexes in a typical eukaryotic cell depending on the specific compartment [14]. Nevertheless, NSF is responsible for recycling all the SNARE complexes. In addition to this recycling function, NSF may also play a role in establishing fusogenic SNARE complexes, for example, in conjunction with Munc18 and Munc13 [15]. Prior to ATP hydrolysis, NSF, SNAP, and SNARE complex form the so-called 20S supercomplex [3]. Other functions of NSF have also been suggested [16–21].
Phylogenetically, NSF belongs to the “classic clade” of AAA+ proteins [22]. The closest homologue of NSF is VCP/p97 (Valosin-Containing Protein, also known as Cdc48p in yeast), which is thought to be involved in translocation of ubiquitylated substrates from the endoplasmic reticulum to the cytoplasm for proteasome degradation, a process known as ERAD (Endoplasmic-Reticulum-Associated protein Degradation) [23,24]. NSF also shares close homology to prokaryotic AAA+ ATPases ClpA/B/C, which also contain tandem AAA+ domains and act as chaperones that clear protein aggregates upon ATP hydrolysis. NSF can be regarded as a special and dedicated chaperone for the SNARE complex.
NSF is a homomeric hexamer with a total molecular weight of ~500 kDa. Each protomer consists of an N-terminal domain (termed N) and two ATPase domains (termed D1 and D2, respectively) (Fig. 1). The D1 domains are probably responsible for the majority of the ATPase activity of NSF whereas the D2 domains are primarily responsible for hexamerization [25]. The N domains are involved in adaptor and substrate binding [26]. Previously, the crystal structures of the D2 and N domains have been determined [27–30], with the former regarded as a prototype of an AAA+ ATPase domain. The N domain consists of two subdomains termed NA and NB (Fig. 1 top). D2 domain also consists of two subdomains termed α/β and α, which is a characteristic feature of all AAA+ domains (Fig. 1 bottom).
Fig. 1. Domain architecture of NSF.

Crystal structures of the N domain (PDB accession code: 1qcs), and the D2 domain (PDB accession code: 1nsf) are shown. The structure of the D1 domain is from the cryo-EM structure of NSF in the ATP state (PDB accession code: 3j94). The structures are shown in scale, with subdomains marked. D1 and D2 domains are aligned in a similar orientation with all the conserved secondary structure elements of the AAA+ domain (defined in [22]) labeled. Compared to the D2 domain, the D1 domain has a characteristic bent α2 helix.
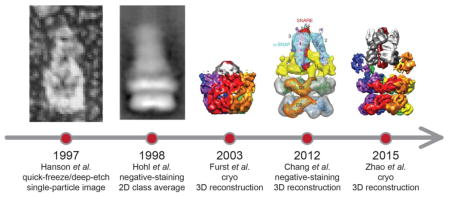
Low-resolution (> 11 Å) structural studies of full-length NSF and the 20S supercomplex have been carried out using quick-freeze/deep-etch (Fig. 2a), negative-staining electron microscopy (Fig. 2b), and cryo-electron microscopy (Figs. 2c,d) (cryo-EM) [5,31–34]. However, these earlier studies did not allow tracing of the unknown D1 domain, and they did not reveal clearly interpretable density for SNAP proteins and the SNARE complex. Here we review recent advances in obtaining atomic-resolution or near-atomic resolution structures of NSF and the 20S supercomplex [35] (Fig. 2e), along with a subsequent cryo-EM study [36], and progress in obtaining the dynamics of SNARE complex disassembly [37–40]. We also compare NSF with other known AAA+ family members, in particular, VCP/p97.
Fig. 2. A history of EM studies on 20S supercomplex.

Representative images or reconstructions are shown on the time axis. They are adapted from Figure 6 of Hanson et al., 1997 (panel a), Figure 2 of Hohl et al., 1998 (panel b), and Figure 6 of Chang et al, 2012 (panel d), respectively. In addition, the maps of Furst et al., 2003 (panel c) and Zhao et al., 2015 (EMD-6206, state I, panel e) are recolored in the same scheme as in Figures 3 and 4.
2. Structures of full-length NSF and 20S supercomplex
The first structural study of full-length NSF and 20S supercomplex using cryo-EM was published 12 years ago at 11 Å resolution [31] (Fig. 2c). In retrospect, due to suboptimal sample quality and limitations in EM technology at that time, the reconstruction did not reveal detailed structural information, and there was no clear density for the NSF N domains, the SNARE complex, and αSNAP proteins. Although it was possible to dock the crystal structure of the D2 ring into the EM density map, the “best” fit of the D2 ring was actually upside down, and the orientation of the D1 domain was uncertain. This is an important lesson that fitting low-resolution EM maps that do not show clear secondary structure details can be misleading.
Many years later, improved cryo-EM maps of full-length NSF in different nucleotide states were published [34], which revealed the correct orientation of both D1 and D2 rings of NSF (Fig. 2d). The improvement in EM map quality was achieved by using more homogenous sample obtained by monomerizing NSF with apyrase, followed by reconstitution of the hexamer in the desired nucleotide state. The density maps of NSF were symmetrized but nonetheless suggested large conformational changes of the N domains upon ATP hydrolysis. Yet, the resolution (at best, 9.2 Å) of the density maps of NSF was still insufficient to allow de novo model building of the unknown D1 domain, and the authors resorted to docking a homology model of the D1 domain, along with the crystal structures of the D2 and N domains, into the EM density maps. A low-resolution (~15 Å) 3D reconstruction of the 20S supercomplex was also obtained from negative stained EM micrographs with C3 symmetry superimposed (Fig. 2d). However, this reconstruction did not reveal clear density for NSF N domains and the SNARE complex. Features that were interpreted as αSNAP proteins were arranged in C3 symmetry. Moreover, the symmetry averaging probably produced an incorrect number of SNAP molecules and lacked the resolution to discern the correctness of the fitted model (Fig. 2d) (see also the simulated effects of averaging presented in [35] that showed that averaging can deteriorate map quality and resolution if the underlying structure is asymmetric).
The limitations and lack of resolution of these earlier studies were alleviated by high-resolution cryo-EM structures of full-length NSF in both ATP- and ADP-bound states as well as near atomic-resolution structures the full-length 20S supercomplex (Fig. 3) [35]. The structure of ATP-bound NSF was determined to a resolution of 4.2 Å, which allowed building a de novo model of the D1 domain. Compared to the D2 domain, the D1 domain has two unique bent helices, α0 and α2, and a longer α7 helix (Fig. 1 middle). It also contains a pore loop with aminoacid sequence YVG that is not present in the D2 domain (Fig. 1 middle). The quality and resolution of the EM data enabled data analysis and reconstruction without imposing any symmetry. Two key factors that led to these higher resolution structure determinations were: (1) further improved sample homogeniety and (2) advancement in cryo-EM technology over the past three years (reviewed in [41,42]). The higher sample homogeneity was achieved by a new protocol for NSF purification and reconstitution in order to ensure a specific nucleotide-bound state [35]. Different from the method used by Chang et al. [34], this approach resulted in monomerization of NSF and complete removal of nucleotide by repeated size-exclusion runs in phosphate buffer. The reassembly of NSF was subsequently carried out in a buffer containing specific nucleotide and EDTA instead of Mg2+ to prevent hydrolysis.
Fig. 3. EM structure of the 20S supercomplex (state I) at 7.6 Å resolution in the presence of AMPPNP.

Structures of individual proteins domains had been previously determined by X-ray crystallography or cryo-EM at higher resolution as indicated by references in the figure, and were fit to the EM density map of the 20S supercomplex.
The most unexpected feature of the cryo-EM structures of NSF and the 20S supercomplex is their pronounced asymmetry (Fig. 4a) [35]. The unprecedented resolution that was achieved for both full-length NSF and 20S supercomplex structures yielded new mechanistic insights as reviewed in more details in the following sections.
Fig. 4. Cryo-EM structures of ATP- and ADP-bound full-length NSF.

The cryo-EM structures are derived from PDB entries 3j94 and 3j95 with the N domains docked into the density of unsharpened maps of ATP- and ADP-bound NSF respectively as rigid bodies, using the crystal structure of the N domain (PDB accession code: 1qcs). (a), Side views. The six chains are colored red, orange, yellow, green, blue, and purple counter-clockwise as viewed from the top. The red chain is always the one with the lowest α2 helix in the D1 domain. (b), Top views of the N domains. The D1 rings are colored white. (c), Top views of the D1 domains. (d), Top views of the D2 domains. Conformational changes upon ATP hydrolysis are highlighted in the ATP-bound structures on the left in panels (b–d). All structures are shown in scale.
2.1. Cryo-EM structures of full-length NSF in ATP- and ADP-bound states
ATP-bound NSF consists of a planar and six-fold symmetric D2 ring (Fig. 4d left) [35]. The D1 ring is reminiscent of a right-handed “split washer”, with each chain stepping up about 5 A except chain F (purple), which bridges back to Chain A (red) (Fig. 4c left). The N domains are rather flexible and asymmetrically organized on top of the D1 ring (Fig. 4b left). In comparison, ADP-bound NSF consists of a D2 ring that exhibits slight deviations from six-fold symmetry (Fig. 4d right). The D1 ring is more expanded with a large opening between Chains A and F, resembling an open “flat washer” (Fig. 4c right). Two out of six N domains are flipped down along the sides of the ATPase rings (Fig. 4b right).
The cryo-EM NSF structures [35] suggest large conformational changes initiated from ATP hydrolysis in the D1 ring, which then lead to changes in the N domains and the D2 ring. This is in agreement with previous biochemical data suggesting that the D1 domains have higher ATPase activity and are responsible for the majority of ATP consumption of NSF [25]. Superposition of all D1 domains suggests that the translation of α7 helix in the α subdomain may result in coupling ATP hydrolysis to conformational changes [35]. The conformational change of the D1 ring from a closed “split washer” to an open “flat washer” leads to a small gap in the D2 ring and flip of two of the N domains along the sides of the ATPase rings (Fig. 4b–d). However, it is still unknown if the small gap in the D2 ring has any functional consequence. The opening of the D1 ring and the flip of the N domains are probably coupled to the mechanical forces applied through αSNAP to disassemble the SNARE complex.
2.2. Cryo-EM structures of the 20S supercomplex
The tower-like 20S supercomplex cryo-EM structures are striking examples of broken symmetry: the assembly is six-fold symmetric at the bottom (D2 ring), and pseudo four-fold symmetric at the top (SNARE complex and αSNAP proteins) (Fig. 5) [35]. The D1 and D2 rings are in a similar state as that of the cryo-EM structure of ATP-bound NSF [35]. However, the N domains are much better resolved compared to the NSF-only structures. The N domains interact only with the αSNAP proteins, but not with the substrate SNARE complex. The transition of the symmetry is accomplished by two binding modalities of αSNAP proteins: one αSNAP can bind to either one N domain or two N domains [35].
Fig. 5. Cryo-EM structures of the 20S supercomplex.

NSF is colored the same as in Fig. 2. αSNAP is colored white and black. SNARE complex is colored dark red. (a) 20S supercomplex with truncated neuronal SNARE complex (state I, PDB accession code: 3j96). (b) V7-20S supercomplex that involves the Vamp-7 SNARE complex. The particular construct of the SNARE complex is shown at the bottom right of each panel. The dark red shades correspond to the actual segments included in the structure.
The stoichiometry of αSNAP in the 20S supercomplex is variable and depends on the particular SNARE complex. In the 7.6 Å resolution cryo-EM structure of the 20S supercomplex, four αSNAPs bind to the neuronal SNARE complex [35]. This study used a truncated neuronal SNARE complex without the N-terminal domain of syntaxin-1A [43]. Likewise, four bound αSNAPs were found in a subsequent lower-resolution (13 Å) cryo-EM reconstruction of the 20S supercomplex [36] using a more complete fragment of the neuronal SNARE complex (including the syntaxin-1A N-terminal Habc domain), confirming that the αSNAP stoichiometry is independent of the particular construct of syntaxin-1A used for the SNARE complex.
For a different SNARE complex involving VAMP-7, syntaxin-1A, and SNAP-25, only two αSNAPs were bound in the 20S supercomplex (referred to as V7-20S supercomplex) (Fig. 5b) [35]. The reason for the different αSNAP stoichiometry is not fully understood, but it may be related to the N-terminal longin domain of VAMP-7 that was included in the complex (Fig. 5 insets). Despite different stoichiometry, the αSNAP proteins bound to the SNARE complex in a similar fashion as in the neuronal 20S supercomplex—a right-handed wrap around the left-handed coiled coil (see further discussions below). The stoichiometries of both the 20S and the V7-20S supercomplexes were verified in solution by using composition-gradient light scattering experiments [35].
Using 3D classification algorithm for single-particle cryo-EM [44,45], the mode of interaction between six N domains and four αSNAPs (from the 20S supercomplex with the truncated neuronal SNARE complex) was classified into three different states, out of 9 possible asymmetric configurations (Fig. 6a) [35]. These states represent snapshots of a working NSF, and suggest rotational motions of the N domains prior to the flip along the sides of the ATPase rings caused by ATP hydrolysis. Such motions may play a role in NSF-mediated SNARE complex disassembly by unwinding and loosening the SNARE complex before pulling apart individual components. The opposite twist observed for the αSNAP proteins (right handed) and the SNARE complex helical bundle (left-handed) supports this idea. Further studies on the 20S supercomplex will be needed to test this hypothesis. Note that in the subsequent cryo-EM study [36], the number of particles was substantially smaller (12,500 compared to 116,000 in the study by [35]), resulting in lower resolution and the apparently inability to observe substates in the 3D reconstructions.
Fig. 6. Different states of the 20S supercomplex.

Cartoon representations of the top views are shown. (a) There are nine asymmetric configurations with four SNAP proteins. (b) The actual pattern observed for the V7-20S supercomplex that involves the Vamp-7 SNARE complex. There are two SNAP proteins, and four resolved N domains in the structure (see Fig. 3b). The black circles indicate the configurations that have been observed [35].
Electrostatic interactions dominate the binding interfaces between the N domains and the αSNAPs, and between the αSNAPs and the SNARE complex [35]. Four positively charged residues of the N domain (R10, R67, K68, K104) interact with either one of the two groups of negatively charged residues at the C-terminus of αSNAP (D217, E249, E252, E253 as one group; D290, E291, E292, D293 as the other group). The importance of the four residues on the N domains is in agreement with a previous mutagenesis study [46]. Note that there is a mismatch between the six N domains and eight (in the case of four αSNAPs) or four (in the case of two αSNAPs) potential binding sites on αSNAP. A potential switch between the binding sites may lead to the rotational motions as mentioned above.
Positively charged residues of αSNAP (K122, K163, K203, R239) interact with the negatively charged surface areas of the SNARE complex. In particular, the two residues close to the ionic layer of the SNARE complex (K122, K163) are critical for the disassembly reaction. Earlier mutagenesis studies had suggested a critical role of these two residues [47]. Due to the resolution limit (7.6 Å) of the cryo-EM structure, specific side chain conformations are not well resolved. Nevertheless, these observations provide an explanation of the promiscuity that one NSF can disassemble many different species of SNARE complex along with just a few SNAP homologues in vivo: Electrostatic interactions are not as specific as hydrogen bonding interactions and only require conserved electrostatic charge patterns for recognition. Indeed, all SNARE complexes and SNAP proteins with known structures have conserved electrostatic charge patterns [37,48,49]. Therefore, one NSF may use its N domains to recognize multiple SNAP species, and one SNAP protein may bind to multiple species of the SNARE complex.
3. Mechanism of NSF mediated SNARE complex disassembly
The estimated number of ATP molecules required for SNARE complex disassembly ranges from 10 to 50 [38,39]. A recent study demonstrated that a set of initially bound ATP molecules are sufficient for complete SNARE complex disassembly, i.e., replacement with fresh ATP molecules is not required for the disassembly reaction [40]. They achieved this measurement by preassembling surface-immobilized single 20S supercomplexes under non-hydrolyzing conditions, followed by removing free ATP molecules in the solution, and then monitoring disassembly by disappearance of fluorescent dye labeled synaptobrevin, a component of the SNARE complex. Since the maximum number of initially bound ATP molecules is twelve, this sets an upper limit on the number of ATP hydrolysis events required for SNARE complex disassembly. This observation is in agreement with the model (i.e., single ATP turn-over) that was proposed based on the cryo-EM structures and biochemical assays by [35]. However, we note that extra ATP may be consumed for any failed disassembly events. Therefore, such failed disassembly events may explain the apparent differences between previous experiments and the single molecule experiment by [40] since the failed events would be counted towards ATP consumption in an ensemble experiment.
After a membrane fusion event, SNAP proteins recognize and bind to the resulting cis-SNARE complex. For the neuronal SNARE complex, previous studies suggested 1:1 or 1:3 stoichiometries for SNAP/SNARE binding [50]. These earlier studies deduced the stoichiometry by cross-linking the complex or measuring the light scattering, which may be sensitive to the experimental conditions. Recently, a 3:1 stoichiometry was also reported by kinetic analysis, and synergistic effects were observed for an engineered trimeric αSNAP [39]. It is therefore likely that the 3:1 stoichiometry represents the number of SNAP proteins that are functionally required by NSF for one neuronal SNARE complex disassembly, which may be different than the number of SNAP proteins bound to the neuronal SNARE complex in the initial ATP-bound state (four as observed in the cryo-EM structures of the 20S supercomplex involving the neuronal SNARE complex [35,36]). The structures also suggest that more than four bound SNAP proteins would be unlikely due to steric hindrance. ATP-loaded NSF binds to the SNAP/SNARE complex to form a 20S supercomplex. In the cell, the 20S supercomplex is probably a transient entity since ATPase activity is greatly enhanced once NSF binds to SNAP and SNARE complex [38].
The 20S supercomplex resembles a loaded spring and the disassembly occurs upon ATP hydrolysis [35,40]. The overall dynamics of the disassembly reaction has been studied in solution by ensemble methods [37–39], and by single-molecule fluorescence spectroscopy and magnetic tweezers [40]. Upon ATP hydrolysis, there is a latency period of tens of seconds with accumulating tension in the SNARE complex, followed by a release of tension within 20 milliseconds that leads to the disassembly of the SNARE complex [40]. In this work it was also speculated that the binding of the SNAP molecules alone may partially disassemble the SNARE complex based on single-molecule FRET measurements [40]. However, partial disassembly of the SNARE complex by SNAP molecules has not been observed in any of the cryo-EM structures of the 20S supercomplex [35,36]. Moreover, SNAP is likely to associate with membranes in vivo (consistent with the location of the membrane-binding loops for the αSNAP proteins observed in the cryo-EM structures), and this membrane association makes SNARE complex disassembly more efficient [51], arguing against partial unfolding of the SNARE complex by SNAP molecules alone.
There may be multiple intermediate steps that lead to disassembly, for example, unwinding of the SNARE complex as evidenced by the presence of four 20S supercomplex states [35]. Since single-round ATP turnover is sufficient for disassembly, some of these intermediate steps could be associated with certain subsets of hydrolysis states of the 20S supercomplex. After the disassembly of the SNARE complex, NSF is recharged with ATP for the next round of SNARE complex binding and disassembly, along with closure of the D1 ring, and reverse movement of the N domains.
The kinetics of the disassembly is conserved across different SNARE complexes, i.e., independent of the N terminal domains of the SNARE complex [37], suggesting a conserved mechanism to disassemble all the species of SNARE complexes. The disassembly rate decreased when using an “extended” SNARE complex (essentially fusing two SNARE complexes together) [38]. This decrease in disassembly rate was initially interpreted as a processive mechanism occurring during the translocation of the substrate through the center of both NSF ATPase rings. However, the cryo-EM structures of the 20S complex [35] argues against this translocation model since an opening of the D2 ATPase ring is not observed. Thus, the effect may be related to impaired formation of an effective SNAP-SNARE subcomplex to initiate the disassembly (see further discussion of the pore loop below).
4. More questions about NSF
Although significant progress has been made in elucidating the mechanism of NSF, there are many remaining questions. For example, the relation between SNAP homologues and their stoichiometry in 20S supercomplex is unknown, and different SNAP homologues may participate in the formation of one 20S supercomplex. It is also unknown if different SNAP homologues target different SNARE complexes, although the surface charge distributions of both SNAPs and SNAREs is highly conserved [47–49].
The details of the SNAP/SNARE interaction that directly leads to SNARE complex disassembly are unknown, e.g., on which part(s) of the SNARE complex do SNAPs apply force? As mentioned above, basic residues on the surface of the SNAP proteins are critical for disassembly, especially the two lysine residues close to the ionic layer of the SNARE complex [35]. Therefore, one would expect that the ionic layer may be critical for force transmission between SNAPs to the SNARE complex, although there are contradictory results in the literature about the importance of the ionic layer [52,53]. Other mechanisms are also possible. For example, a conserved hydrophobic patch connecting helices 9 and 10 of αSNAP may form a “chock” protruding into the groove of the SNARE four-helix bundle, akin a “sliding” mechanism [36], although this particular study used a lower resolution cryo-EM reconstruction along with masking and averaging of the SNARE/αSNAP subcomplex to reveal this detail. Thus, due to the limited resolution and static nature of the current single-particle cryo-EM reconstructions of the 20S supercomplex, these and other possible models remain to be verified.
Another interesting question concerns the mode of interactions between the N domains and the SNAPs. Assuming four SNAPs are bound, in theory there would be nine possible asymmetric configurations (Fig. 6a). However, only three of them were observed in the cryo-EM reconstructions of the 20S supercomplex by [35]. Multiple patterns of configurations are also possible in the case of only two SNAPs interaction with the VAMP-7 containing 20S supercomplex, but only one was observed in the cryo-EM reconstruction (Fig. 6b). The limited number of states may be due to the EM data, i.e., there were not enough good particles to allow classification into more states. Moreover, it is unclear why only two N domains are flipped along the sides of the ATPase rings as observed in the ADP-bound NSF structure (Fig. 4).
Although the N domains of NSF do not directly interact with the SNARE complex in the cryo-EM structures, it is possible that NSF may interact with the SNARE complex through the pore loops of the D1 domains. However, the local densities of the cryo-EM maps were not well-resolved enough to confirm such interactions [35]. Pore loops are present in many AAA+ members that can translocate peptides through their central pore for degradation [54], and such a mechanism has also been proposed for NSF [38,55]. However, since the D2 ring of NSF does not contain such pore loops, and there is no opening of the D2 ring in the ADP- and ATP-bound states (Fig. 4d), a translocation through both rings is unlikely. Nevertheless, the pore loops may interact with individual SNARE proteins and act as anchor points for the N domains and SNAPs to apply a torque or shear force to the SNARE complex [35]. Moreover, it is possible that the parts of the SNARE complex that are unfolded “snorkel” through openings between the NSF N-domains and the D1 ring.
In addition to its role as a SNARE-complex disassembly machine, NSF also interacts with other cellular proteins besides the SNARE complexes [18–21]. However, the possible function and mechanisms involving these interactions are unclear.
5. Comparison of NSF with other AAA+ family members
AAA+ constitute a large family of ATPases found in both prokaryotes and eukaryotes. They are involved in many essential cellular processes and have remarkable functional heterogeneity [22]. A common feature of all AAA+ ATPases is converting chemical energy from ATP hydrolysis to mechanical forces that perform a wide range of tasks, such as DNA unwinding, protein unfolding, and protein complex disassembly [55]. The closest related member to NSF is VCP/p97 [22,56] which also contains three domains, termed N, D1, and D2. Interestingly, the activity of the ATPase domains D1 and D2 are opposite to those of NSF: D2 domain is the primary domain hydrolyzing ATP, whereas D1 domain has minimal ATPase activity in vitro [56]. The reason for the opposite ATPase activity is not fully understood – it may be related to the different functional roles of NSF and VCP/p97. The major function of VCP/p97 is to extract ubiquitylated proteins from ER (Endoplasmic Reticulum) membranes to cytosol for proteasome degradation [57], but the molecular mechanism remains to be elucidated. Crystal structures of full-length VCP/p97 in different nucleotide-bound states are available [58–60], as well as the structure of the complex between the N/D1 domain of VCP/p97 and one of its adaptor proteins, p47 [61,62]. In contrast to previous models of NSF [56,60], it is now known that the ATPase rings are arranged in the same direction. Thus, structurally, NSF and VCP/p97 are more similar than previously thought. Comparing the ATPase domains D1 and D2 of NSF (cryo-EM structures) and VCP/p97 (ADP-bound crystal structure), the two rings of the latter are more compact (Fig. 7a), possibly due to the crystal packing. Although the D1 and D2 rings are in the same orientation, there is a relative rotation between the two rings in the NSF structure. By contrast, the two rings are stacked right on top of each other without rotation in VCP/p97 (Fig. 7a). The difference in overall appearance also manifests itself in the relative orientation of each AAA+ domain, which can be visualized by the positions of individual α2 helices (Fig. 7b). The α2 helices of the D2 ring of NSF are nearly parallel to the plane of the ATPase ring in both ATP- and ADP-bound states as compared to that of VCP/p97, which are more tilted. The α2 helices of the D1 ring of NSF are rather tilted and are arranged in a “split washer” fashion in the ATP-bound state and in an “open washer” fashion in ADP-bound state, whereas in the D1 ring of VCP/p97, all α2 helices are parallel to the plane of the ATPase ring. For individual AAA+ domains, both D1 and D2 of VCP/p97 superimpose better to the D1 domain of NSF with a smaller root mean square deviation (RMSD) for the main chain atoms (Fig. 7c and e). The major difference is that NSF D1 domain has a characteristic bent α2 helix (Fig. 7e and f). Structurally, the NSF D2 domain is more similar to the VCP/p97 D1 domain, and the NSF D1 domain is more similar to the VCP/p97 D2 domain (Fig. 7c–f), which correlates with their opposite ATPase activities.
Fig. 7. Comparison of NSF and VCP/p97 structures.

(a) Overall structures of ATP-bound NSF (PDB accession code 3j94), ADP-bound NSF (PDB accession code 3j95), and ADP-bound VCP/p97 (PDB accession code 3cf3). Only ATPase rings are shown. The structures have been aligned and are on the same scale. (b) Same view as in (a) but only showing the α2 helices. Dashed lines are in the same positions as the top two lines in (a) to help visualization. (c)-(f) Superposition of AAA+ domains of NSF and VCP/p97. Root mean square deviations (RMSD) of the main chains are provided. α2 helices are highlighted in dashed boxes. The following PDB models are used: NSF D1 (accession code 3j94), NSF D2 (accession code 1nsf), VCP/p97 D1 (accession code 1e32), and VCP/p97 D2 (accession code 3cf0).
There is no structure available of full-length VCP/p97 with its adaptor proteins and substrates. NSF undergoes large conformational changes upon ATP hydrolysis as revealed by cryo-EM structures [35]. By contrast, VCP/p97 undergoes very small conformational changes judging from the crystal structures [60], but larger changes were seen based on SAXS experiments [63]. Cryo-EM has also been used to study the conformational changes of VCP/p97 upon ATP hydrolysis [64,65], however, due to the limited resolution and the application of symmetry, these earlier studies provided limited information. It is possible that VCP/p97 also undergoes large conformational changes upon ATP hydrolysis, which was not revealed by the crystal structures due to lattice restraints.
Crystal structures of other closely related Type II AAA+ members ClpA, ClpB and ClpC are available [66–68]. They do not show the asymmetric features observed for NSF. However, we note that ClpA and ClpB were not determined in a native oligomeric state, and six-fold symmetry was assumed in obtaining models of the assembly. In comparison, the well-characterized prokaryotic unfoldase ClpX, a Type I AAA+ consisting of a single ATPase ring, shows asymmetric arrangements in the crystal structures [69]. A similar argument as the VCP/p97 case can be applied here: It is likely that these Type II AAA+ also undergo large conformational changes upon ATP hydrolysis, which have yet been captured by the currently available structural studies. Besides NSF and ClpX, homomeric but asymmetric AAA+ ATPase ring has been observed for transcriptional activator NtrC1 [70,71], and more recently, the peroxisomal Pex1/Pex6 complex [72–74]. The relation between asymmetry and function has not been fully understood in these two cases. Nevertheless, this kind of asymmetry may be more prevalent than generally thought and may play key roles in the function of homomeric AAA+.
6. Concluding remarks
Recent progress in understanding the structure and molecular mechanism of NSF has made it one of the best characterized Type II AAA+ ATPases. In general, Type II AAA+ ATPases are difficult to study due to the complexity introduced by their double ATPase rings. The resulting larger molecule may make crystallization difficult since slight differences in a total of twelve ATPase domains may prevent crystal lattice formation. Most of Type II AAA+ ATPases also have a broad spectrum of substrates that are not as well characterized as that for NSF, and reconstituted systems to study the mechanism are often not yet available. Nevertheless, with the advancement of cryo-EM technologies, large protein structures can be determined to atomic resolutions without forming crystals, which will certainly benefit studying Type II AAA+ ATPases. In closing, there is an important caveat for structure determination of Type II AAA+ ATPases: imposing symmetry should be avoided during cryo-EM reconstruction whenever possible [35]. Although similar cautionary notes have been published previously [75,76], many previous EM studies of NSF and 20S supercomplex had imposed either C3 or C6 point group symmetry, obscuring asymmetric features that later turned out to be critical.
Supplementary Material
Highlights.
Recent progress in understanding the structure and molecular mechanism of NSF has made it one of the best characterized Type II AAA+ ATPases.
Cryo-electron microscopy is a powerful method for structural characterization of Type II AAA+ ATPases. However, imposing symmetry should be avoided during cryo-EM reconstruction whenever possible.
Although significant progress has been made in elucidating the mechanism of NSF, there are still many remaining questions.
Acknowledgments
We thank Arthur Lyubimov and William Weis for discussions and critical reading the manuscript, and the National Institutes of Health for support (R37MH63105 to A.T.B).
Abbreviations
- NSF
N-ethylmaleimide Sensitive Factor
- SNARE
Soluble N-ethylmaleimide sensitive factor Attachment protein REceptors
- SNAP
Soluble N-ethylmaleimide sensitive factor Attachment Protein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Block MR, Glick BS, Wilcox CA, Wieland FT, Rothman JE. Purification of an N-ethylmaleimide-sensitive protein catalyzing vesicular transport. Proc Natl Acad Sci U S A. 1988;85:7852–6. doi: 10.1073/pnas.85.21.7852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Malhotra V, Orci L, Glick BS, Block MR, Rothman JE. Role of an N-ethylmaleimide-sensitive transport component in promoting fusion of transport vesicles with cisternae of the Golgi stack. Cell. 1988;54:221–7. doi: 10.1016/0092-8674(88)90554-5. [DOI] [PubMed] [Google Scholar]
- 3.Söllner T, Bennett MK, Whiteheart SW, Scheller RH, Rothman JE. A protein assembly-disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell. 1993;75:409–18. doi: 10.1016/0092-8674(93)90376-2. [DOI] [PubMed] [Google Scholar]
- 4.Mayer A, Wickner W, Haas A. Sec18p (NSF)-driven release of Sec17p (alpha-SNAP) can precede docking and fusion of yeast vacuoles. Cell. 1996;85:83–94. doi: 10.1016/s0092-8674(00)81084-3. [DOI] [PubMed] [Google Scholar]
- 5.Hanson P, Roth R, Morisaki H, Jahn R, Heuser J. Structure and conformational changes in NSF and its membrane receptor complexes visualized by quick-freeze/deep-etch electron microscopy. Cell. 1997;90:523–35. doi: 10.1016/s0092-8674(00)80512-7. [DOI] [PubMed] [Google Scholar]
- 6.Wickner W, Schekman R. Membrane fusion. Nat Struct Mol Biol. 2008;15:658–64. doi: 10.1038/nsmb.1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Südhof TC. Neurotransmitter release: the last millisecond in the life of a synaptic vesicle. Neuron. 2013;80:675–90. doi: 10.1016/j.neuron.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sutton RB, Fasshauer D, Jahn R, Brunger AT. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2. 4 A resolution. Nature. 1998;395:347–53. doi: 10.1038/26412. [DOI] [PubMed] [Google Scholar]
- 9.Weber T, Zemelman BV, McNew JA, Westermann B, Gmachl M, Parlati F, et al. SNAREpins: minimal machinery for membrane fusion. Cell. 1998;92:759–72. doi: 10.1016/s0092-8674(00)81404-x. [DOI] [PubMed] [Google Scholar]
- 10.Boulianne GL, Trimble WS. Identification of a second homolog of N-ethylmaleimide-sensitive fusion protein that is expressed in the nervous system and secretory tissues of Drosophila. Proc Natl Acad Sci U S A. 1995;92:7095–9. doi: 10.1073/pnas.92.15.7095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pallanck L, Ordway RW, Ramaswami M, Chi WY, Krishnan KS, Ganetzky B. Distinct roles for N-ethylmaleimide-sensitive fusion protein (NSF) suggested by the identification of a second Drosophila NSF homolog. J Biol Chem. 1995;270:18742–4. doi: 10.1074/jbc.270.32.18742. [DOI] [PubMed] [Google Scholar]
- 12.Whiteheart SW, Griff IC, Brunner M, Clary DO, Mayer T, Buhrow SA, et al. SNAP family of NSF attachment proteins includes a brain-specific isoform. Nature. 1993;362:353–5. doi: 10.1038/362353a0. [DOI] [PubMed] [Google Scholar]
- 13.Stenbeck G. Soluble NSF-attachment proteins. Int J Biochem Cell Biol. 1998;30:573–7. doi: 10.1016/s1357-2725(97)00064-2. [DOI] [PubMed] [Google Scholar]
- 14.Jahn R, Scheller RH. SNAREs--engines for membrane fusion. Nat Rev Mol Cell Biol. 2006;7:631–43. doi: 10.1038/nrm2002. [DOI] [PubMed] [Google Scholar]
- 15.Ma C, Su L, Seven AB, Xu Y, Rizo J. Reconstitution of the vital functions of Munc18 and Munc13 in neurotransmitter release. Science. 2013;339:421–5. doi: 10.1126/science.1230473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martin HGS, Henley JM, Meyer G. Novel putative targets of N-ethylmaleimide sensitive fusion protein (NSF) and alpha/beta soluble NSF attachment proteins (SNAPs) include the Pak-binding nucleotide exchange factor betaPIX. J Cell Biochem. 2006;99:1203–15. doi: 10.1002/jcb.20998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao C, Slevin JT, Whiteheart SW. Cellular functions of NSF: not just SNAPs and SNAREs. FEBS Lett. 2007;581:2140–9. doi: 10.1016/j.febslet.2007.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gokhale A, Mullin AP, Zlatic SA, Easley CA, Merritt ME, Raj N, et al. The N-ethylmaleimide-sensitive factor and dysbindin interact to modulate synaptic plasticity. J Neurosci. 2015;35:7643–53. doi: 10.1523/JNEUROSCI.4724-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li T, Tan Y, Li Q, Chen H, Lv H, Xie W, et al. The Neurexin-NSF interaction regulates short-term synaptic depression. J Biol Chem. 2015 doi: 10.1074/jbc.M115.644583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nishimune A, Isaac JT, Molnar E, Noel J, Nash SR, Tagaya M, et al. NSF Binding to GluR2 Regulates Synaptic Transmission. Neuron. 1998;21:87–97. doi: 10.1016/S0896-6273(00)80517-6. [DOI] [PubMed] [Google Scholar]
- 21.Song I, Kamboj S, Xia J, Dong H, Liao D, Huganir RL. Interaction of the N-Ethylmaleimide–Sensitive Factor with AMPA Receptors. Neuron. 1998;21:393–400. doi: 10.1016/S0896-6273(00)80548-6. [DOI] [PubMed] [Google Scholar]
- 22.Erzberger JP, Berger JM. Evolutionary relationships and structural mechanisms of AAA+ proteins. Annu Rev Biophys Biomol Struct. 2006;35:93–114. doi: 10.1146/annurev.biophys.35.040405.101933. [DOI] [PubMed] [Google Scholar]
- 23.Brodsky JL. Cleaning up: ER-associated degradation to the rescue. Cell. 2012;151:1163–7. doi: 10.1016/j.cell.2012.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bagola K, Mehnert M, Jarosch E, Sommer T. Protein dislocation from the ER. Biochim Biophys Acta. 2011;1808:925–36. doi: 10.1016/j.bbamem.2010.06.025. [DOI] [PubMed] [Google Scholar]
- 25.Matveeva E, He P, Whiteheart S. N-Ethylmaleimide-sensitive fusion protein contains high and low affinity ATP-binding sites that are functionally distinct. J Biol Chem. 1997;272:26413–8. doi: 10.1074/jbc.272.42.26413. [DOI] [PubMed] [Google Scholar]
- 26.Whiteheart S, Schraw T, Matveeva E. N-ethylmaleimide sensitive factor (NSF) structure and function. Int Rev Cytol. 2001;207:71–112. doi: 10.1016/s0074-7696(01)07003-6. [DOI] [PubMed] [Google Scholar]
- 27.Yu R, Jahn R, Brunger A. NSF N-terminal domain crystal structure: models of NSF function. Mol Cell. 1999;4:97–107. doi: 10.1016/s1097-2765(00)80191-4. [DOI] [PubMed] [Google Scholar]
- 28.Lenzen C, Steinmann D, Whiteheart S, Weis W. Crystal Structure of the Hexamerization Domain of N-ethylmaleimide–Sensitive Fusion Protein. Cell. 1998;94:525–36. doi: 10.1016/s0092-8674(00)81593-7. [DOI] [PubMed] [Google Scholar]
- 29.May AP, Misura KM, Whiteheart SW, Weis WI. Crystal structure of the amino-terminal domain of N-ethylmaleimide-sensitive fusion protein. Nat Cell Biol. 1999;1:175–82. doi: 10.1038/11097. [DOI] [PubMed] [Google Scholar]
- 30.Yu RC, Hanson PI, Jahn R, Brünger AT. Structure of the ATP-dependent oligomerization domain of N-ethylmaleimide sensitive factor complexed with ATP. Nat Struct Biol. 1998;5:803–11. doi: 10.1038/1843. [DOI] [PubMed] [Google Scholar]
- 31.Furst J, Sutton RB, Chen J, Brunger AT, Grigorieff N. Electron cryomicroscopy structure of N-ethyl maleimide sensitive factor at 11 A resolution. EMBO J. 2003;22:4365–74. doi: 10.1093/emboj/cdg420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moeller A, Zhao C, Fried MG, Wilson-Kubalek EM, Carragher B, Whiteheart SW. Nucleotide-dependent conformational changes in the N-Ethylmaleimide Sensitive Factor (NSF) and their potential role in SNARE complex disassembly. J Struct Biol. 2012;177:335–43. doi: 10.1016/j.jsb.2011.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hohl TM, Parlati F, Wimmer C, Rothman JE, Söllner TH, Engelhardt H. Arrangement of subunits in 20 S particles consisting of NSF, SNAPs, and SNARE complexes. Mol Cell. 1998;2:539–48. doi: 10.1016/s1097-2765(00)80153-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chang L-F, Chen S, Liu C-C, Pan X, Jiang J, Bai X-C, et al. Structural characterization of full-length NSF and 20S particles. Nat Struct Mol Biol. 2012;19:268–75. doi: 10.1038/nsmb.2237. [DOI] [PubMed] [Google Scholar]
- 35.Zhao M, Wu S, Zhou Q, Vivona S, Cipriano DJ, Cheng Y, et al. Mechanistic insights into the recycling machine of the SNARE complex. Nature. 2015;518:61–7. doi: 10.1038/nature14148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou Q, Huang X, Sun S, Li X, Wang H-W, Sui S-F. Cryo-EM structure of SNAP-SNARE assembly in 20S particle. Cell Res. 2015;25:551–60. doi: 10.1038/cr.2015.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vivona S, Cipriano DJ, O’Leary S, Li YH, Fenn TD, Brunger AT. Disassembly of all SNARE complexes by N-ethylmaleimide-sensitive factor (NSF) is initiated by a conserved 1:1 interaction between α-soluble NSF attachment protein (SNAP) and SNARE complex. J Biol Chem. 2013;288:24984–91. doi: 10.1074/jbc.M113.489807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cipriano DJ, Jung J, Vivona S, Fenn TD, Brunger AT, Bryant Z. Processive ATP-driven Substrate Disassembly by the N-Ethylmaleimide-sensitive Factor (NSF) Molecular Machine. J Biol Chem. 2013;288:23436–45. doi: 10.1074/jbc.M113.476705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shah N, Colbert KN, Enos MD, Herschlag D, Weis WI. Three αSNAP and 10 ATP Molecules Are Used In SNARE Complex Disassembly by N-ethylmaleimide Sensitive Factor (NSF) J Biol Chem. 2014 doi: 10.1074/jbc.M114.620849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ryu J-K, Min D, Rah S-H, Kim SJ, Park Y, Kim H, et al. Spring-loaded unraveling of a single SNARE complex by NSF in one round of ATP turnover. Science. 2015;347:1485–9. doi: 10.1126/science.aaa5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cheng Y, Grigorieff N, Penczek PA, Walz T. A Primer to Single-Particle Cryo-Electron Microscopy. Cell. 2015;161:438–49. doi: 10.1016/j.cell.2015.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cheng Y. Single-Particle Cryo-EM at Crystallographic Resolution. Cell. 2015;161:450–7. doi: 10.1016/j.cell.2015.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ernst JA, Brunger AT. High resolution structure, stability, and synaptotagmin binding of a truncated neuronal SNARE complex. J Biol Chem. 2003;278:8630–6. doi: 10.1074/jbc.M211889200. [DOI] [PubMed] [Google Scholar]
- 44.Lyumkis D, Brilot AF, Theobald DL, Grigorieff N. Likelihood-based classification of cryo-EM images using FREALIGN. J Struct Biol. 2013;183:377–88. doi: 10.1016/j.jsb.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Scheres SHW. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol. 2012;180:519–30. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhao C, Matveeva EA, Ren Q, Whiteheart SW. Dissecting the N-ethylmaleimide-sensitive factor: required elements of the N and D1 domains. J Biol Chem. 2010;285:761–72. doi: 10.1074/jbc.M109.056739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marz KE, Lauer JM, Hanson PI. Defining the SNARE complex binding surface of alpha-SNAP: implications for SNARE complex disassembly. J Biol Chem. 2003;278:27000–8. doi: 10.1074/jbc.M302003200. [DOI] [PubMed] [Google Scholar]
- 48.Diao J, Liu R, Rong Y, Zhao M, Zhang J, Lai Y, et al. ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature. 2015;520:563–6. doi: 10.1038/nature14147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rice L, Brunger A. Crystal structure of the vesicular transport protein Sec17: implications for SNAP function in SNARE complex disassembly. Mol Cell. 1999;4:85–95. doi: 10.1016/s1097-2765(00)80190-2. [DOI] [PubMed] [Google Scholar]
- 50.Wimmer C, Hohl TM, Hughes CA, Müller SA, Söllner TH, Engel A, et al. Molecular mass, stoichiometry, and assembly of 20 S particles. J Biol Chem. 2001;276:29091–7. doi: 10.1074/jbc.M011292200. [DOI] [PubMed] [Google Scholar]
- 51.Winter U, Chen X, Fasshauer D. A conserved membrane attachment site in alpha-SNAP facilitates N-ethylmaleimide-sensitive factor (NSF)-driven SNARE complex disassembly. J Biol Chem. 2009;284:31817–26. doi: 10.1074/jbc.M109.045286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Scales SJ, Yoo BY, Scheller RH. The ionic layer is required for efficient dissociation of the SNARE complex by -SNAP and NSF. Proc Natl Acad Sci. 2001;98:14262–7. doi: 10.1073/pnas.251547598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lauer JM, Dalal S, Marz KE, Nonet ML, Hanson PI. SNARE complex zero layer residues are not critical for N-ethylmaleimide-sensitive factor-mediated disassembly. J Biol Chem. 2006;281:14823–32. doi: 10.1074/jbc.M512706200. [DOI] [PubMed] [Google Scholar]
- 54.Sauer RT, Baker TA. AAA+ proteases: ATP-fueled machines of protein destruction. Annu Rev Biochem. 2011;80:587–612. doi: 10.1146/annurev-biochem-060408-172623. [DOI] [PubMed] [Google Scholar]
- 55.Hanson PI, Whiteheart SW. AAA+ proteins: have engine, will work. Nat Rev Mol Cell Biol. 2005;6:519–29. doi: 10.1038/nrm1684. [DOI] [PubMed] [Google Scholar]
- 56.Brunger A, DeLaBarre B. NSF and p97/VCP: similar at first, different at last. FEBS Lett. 2003;555:126–33. doi: 10.1016/s0014-5793(03)01107-4. [DOI] [PubMed] [Google Scholar]
- 57.Yamanaka K, Sasagawa Y, Ogura T. Recent advances in p97/VCP/Cdc48 cellular functions. Biochim Biophys Acta. 2012;1823:130–7. doi: 10.1016/j.bbamcr.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 58.Huyton T, Pye VE, Briggs LC, Flynn TC, Beuron F, Kondo H, et al. The crystal structure of murine p97/VCP at 3. 6A. J Struct Biol. 2003;144:337–48. doi: 10.1016/j.jsb.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 59.Davies JM, Brunger AT, Weis WI. Improved structures of full-length p97, an AAA ATPase: implications for mechanisms of nucleotide-dependent conformational change. Structure. 2008;16:715–26. doi: 10.1016/j.str.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 60.DeLaBarre B, Brunger AT. Complete structure of p97/valosin-containing protein reveals communication between nucleotide domains. Nat Struct Biol. 2003;10:856–63. doi: 10.1038/nsb972. [DOI] [PubMed] [Google Scholar]
- 61.Dreveny I, Kondo H, Uchiyama K, Shaw A, Zhang X, Freemont PS. Structural basis of the interaction between the AAA ATPase p97/VCP and its adaptor protein p47. EMBO J. 2004;23:1030–9. doi: 10.1038/sj.emboj.7600139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang X, Shaw A, Bates P, Newman R. Structure of the AAA ATPase p97. Mol Cell. 2000;6:1473–84. doi: 10.1016/s1097-2765(00)00143-x. [DOI] [PubMed] [Google Scholar]
- 63.Davies JM, Tsuruta H, May AP, Weis WI. Conformational changes of p97 during nucleotide hydrolysis determined by small-angle X-Ray scattering. Structure. 2005;13:183–95. doi: 10.1016/j.str.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 64.Rouiller I, DeLaBarre B, May AP, Weis WI, Brunger AT, Milligan RA, et al. Conformational changes of the multifunction p97 AAA ATPase during its ATPase cycle. Nat Struct Biol. 2002;9:950–7. doi: 10.1038/nsb872. [DOI] [PubMed] [Google Scholar]
- 65.Rouiller I, Butel VM, Latterich M, Milligan RA, Wilson-Kubalek EM. A Major Conformational Change in p97 AAA ATPase upon ATP Binding. Mol Cell. 2000;6:1485–90. doi: 10.1016/S1097-2765(00)00144-1. [DOI] [PubMed] [Google Scholar]
- 66.Guo F, Maurizi MR, Esser L, Xia D. Crystal structure of ClpA, an Hsp100 chaperone and regulator of ClpAP protease. J Biol Chem. 2002;277:46743–52. doi: 10.1074/jbc.M207796200. [DOI] [PubMed] [Google Scholar]
- 67.Wang F, Mei Z, Qi Y, Yan C, Hu Q, Wang J, et al. Structure and mechanism of the hexameric MecA-ClpC molecular machine. Nature. 2011;471:331–5. doi: 10.1038/nature09780. [DOI] [PubMed] [Google Scholar]
- 68.Lee S, Sowa M, Watanabe Y, Sigler P. The structure of ClpB: a molecular chaperone that rescues proteins from an aggregated state. Cell. 2003;115:229–40. doi: 10.1016/s0092-8674(03)00807-9. [DOI] [PubMed] [Google Scholar]
- 69.Glynn SE, Martin A, Nager AR, Baker TA, Sauer RT. Structures of asymmetric ClpX hexamers reveal nucleotide-dependent motions in a AAA+ protein-unfolding machine. Cell. 2009;139:744–56. doi: 10.1016/j.cell.2009.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sysoeva TA, Chowdhury S, Guo L, Nixon BT. Nucleotide-induced asymmetry within ATPase activator ring drives σ54-RNAP interaction and ATP hydrolysis. Genes Dev. 2013;27:2500–11. doi: 10.1101/gad.229385.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dey S, Biswas M, Sen U, Dasgupta J. Unique ATPase site architecture triggers cis-mediated synchronized ATP binding in heptameric AAA+ ATPase domain of flagellar regulatory protein FlrC. J Biol Chem. 2015 doi: 10.1074/jbc.M114.611434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ciniawsky S, Grimm I, Saffian D, Girzalsky W, Erdmann R, Wendler P. Molecular snapshots of the Pex1/6 AAA+ complex in action. Nat Commun. 2015;6:7331. doi: 10.1038/ncomms8331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Blok NB, Tan D, Wang RY-R, Penczek PA, Baker D, DiMaio F, et al. Unique double-ring structure of the peroxisomal Pex1/Pex6 ATPase complex revealed by cryo-electron microscopy. Proc Natl Acad Sci. 2015:201500257. doi: 10.1073/pnas.1500257112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gardner BM, Chowdhury S, Lander GC, Martin A. The Pex1/Pex6 complex is a heterohexameric AAA+ motor with alternating and highly coordinated subunits. J Mol Biol. 2015 doi: 10.1016/j.jmb.2015.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Arias-Palomo E, O’Shea VL, Hood IV, Berger JM. The bacterial DnaC helicase loader is a DnaB ring breaker. Cell. 2013;153:438–48. doi: 10.1016/j.cell.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lyubimov AY, Costa A, Bleichert F, Botchan MR, Berger JM. ATP-dependent conformational dynamics underlie the functional asymmetry of the replicative helicase from a minimalist eukaryote. Proc Natl Acad Sci U S A. 2012;109:11999–2004. doi: 10.1073/pnas.1209406109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.