

Abstract
Nonsteroidal anti-inf lammatory drugs (NSAIDs) are widely prescribed for the treatment of inflammatory diseases, but their use is frequently related to hypersensitivity reactions. This review outlines our current knowledge of NSAID hypersensitivity (NHS) with regard to its pathogenic, molecular, and genetic mechanisms, as well as diagnosis and treatment. The presentation of NHS varies from a local (skin and/or airways) reaction to systemic reactions, including anaphylaxis. At the molecular level, NHS reactions can be classified as cross-reactive (mediated by cyclooxygenase inhibition) or selective (specific activation of immunoglobulin E antibodies or T cells). Genetic polymorphisms and epigenetic factors have been shown to be closely associated with NHS, and may be useful as predictive markers. To diagnose NHS, inhalation or oral challenge tests are applied, with the exclusion of any cross-reactive NSAIDs. For patients diagnosed with NHS, absolute avoidance of NSAIDs/aspirin is essential, and pharmacological treatment, including biologics, is often used to control their respiratory and cutaneous symptoms. Finally, desensitization is recommended only for selected patients with NHS. However, further research is required to develop new diagnostic methods and more effective treatments against NHS.
Keywords: Anti-inflammatory agents, non-steroidal; Drug hypersensitivity; Etiology; Genetic predisposition to disease
INTRODUCTION
Nonsteroidal anti-inflammatory drugs (NSAIDs) are widely prescribed for the treatment of various inflammatory diseases. However, NSAIDs can cause drug hypersensitivity reactions, varying from local reactions in the skin and/or airways to systemic symptoms, including life-threatening anaphylaxis [1,2]. NSAID hypersensitivity (NHS) has been reported in 4.3% to 11% of patients with asthma [3] and in 27% to 35% of patients with chronic urticaria (CU) [4]. Moreover, NHS contributes to 13.3% of anaphylaxis cases in Korean adults [5]. While various terms (e.g., sensitivity, idiosyncrasy, or intolerance) have been used to describe unintended and unpredictable adverse drug reactions, the term “hypersensitivity” seems appropriate to describe the adverse reaction to NSAIDs, as its pathogenesis can involve both immunological and nonimmunological reactions [6,7].
NHS reactions may be classified as cross-reactive against several and chemically unrelated NSAIDs, or selective for a single compound or a group of chemically related compounds. Cross-reactive NHS reactions can manifest as three major clinical phenotypes: (1) NSAID-exacerbated respiratory disease (NERD), occurring in patients with underlying chronic upper and lower airway respiratory diseases; (2) NSAID-exacerbated cutaneous disease (NECD), occurring in patients with a history of CU; and (3) NSAID-induced urticaria/angioedema (NIUA), occurring in otherwise healthy subjects (without a history of CU) [2,8]. Anaphylaxis is commonly seen in NIUA. In addition, cross-reactive NHS may induce both respiratory and cutaneous symptoms in a subgroup of patients [9]. Selective NHS reactions manifest as two clinical phenotypes: (1) single NIUA/anaphylaxis reactions, which develop immediately after drug intake (immunoglobulin E [IgE]-mediated); and (2) NSAID-induced delayed hypersensitivity reactions, which develop more than 24 hours after drug intake (T cell-mediated) [2].
The pathogenesis of NHS reactions is thought to be related to various mechanisms, including: (1) a non-immunological mechanism, through the inhibition of enzymes involved in the arachidonic acid (AA) pathway; and (2) an immunological mechanism, involving the production of drug-specific IgE antibodies or T cell activation. Moreover, genetic polymorphisms and epigenetic factors have been implicated in the pathogenesis of, and susceptibility to, NHS. This review summarizes current knowledge regarding the pathogenic mechanisms, associated genetic and epigenetic factors, diagnostic methods, and treatments for patients with NHS.
PATHOGENIC MECHANISMS OF NHS
The proposed pathogenic mechanisms and clinical manifestations of NHS are shown in Table 1 and Fig. 1 [10]. NHS reactions can be subdivided as cross-reactive, in which the cyclooxygenase 1 (COX-1) pathway is inhibited, or selective, resulting from an immunological (allergic) reaction to the drugs and characterized by IgE-mediated (acute) or T cell-mediated (delayed) reactions [2,11-13]. In addition, novel mechanisms related to oxidative stress and platelet activation that could contribute to the pathogenesis of NHS have been reported [14-17].
Table 1.
Pathogenic mechanisms and clinical manifestations of NSAID hypersensitivity
| Type of reaction | Time of onseta | Clinical manifestation | Mechanism |
|---|---|---|---|
| Cross reactive | Acute | NERD | COX-1 inhibition |
| Oxidative stress | |||
| NECD | COX-1 inhibition | ||
| NIUA | COX-1 inhibition | ||
| Platelet activation | |||
| Selective | Acute | SNIUAA | IgE-mediated |
| Delayed | NIDHR | T cell-mediated |
NSAID, nonsteroidal anti-inf lammatory drug; NERD, NSAID-exacerbated respiratory disease; COX-1, cyclooxygenase 1; NECD, NSAID-exacerbated cutaneous disease; NIUA, NSAID-induced urticaria/angioedema; SNIUAA, single-NSAID-induced urticaria/anaphylaxis; IgE, immunoglobulin E; NIDHR, NSAID-induced delayed hypersensitivity reaction.
Acute onset, clinical symptoms occur immediately or a few hours after drug administration; delayed onset, clinical symptoms occur 24 hours or more after drug administration.
Figure 1.
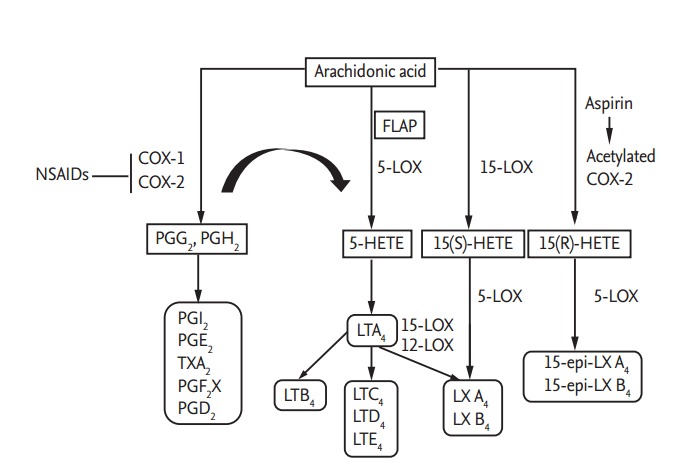
Arachidonic acid (AA) metabolism involved in the pathogenic mechanisms of nonsteroidal anti-inflammatory drug (NSAID) hypersensitivity. AA can be metabolized by either the cyclooxygenase (COX) pathway or 5-lipoxygenase (5-LOX) pathway [25]. Inhibition of COX enzymes by NSAIDs shifts the conversion of AA to the 5-LOX pathway, leading to decreased production of prostaglandins (PGs) and increased production of cysteinyl leukotrienes (LTs). Lipoxin (LX) A4/B4 are synthesized either from LTA4 or from 15S-hydroxyeicosatetraenoic acid (15(S)-HETE). In addition, aspirin acetylates COX-2, transforming AA to 15(R)-HETE and then to 15-epi-LX by 5-LOX [125]. In NSAID-exacerbated respiratory disease patients, the production of LX and 15-epi-LX, which have anti-inflammatory effects, could be decreased, possibly due to the excessive usage of AA for LT synthesis. FLAP, 5-lipoxygenase-activating protein.
Metabolism of arachidonic acid and the effects of NSAIDs
AA can be metabolized by either the COX pathway or the 5-lipoxygenase (5-LOX) pathway, with conversion to prostanoids or cysteinyl leukotrienes (CysLTs), respectively [18]. There are two isoforms of COX: COX-1 (expressed constitutively in most cells) and COX-2 (expressed either constitutively or in responses to inflammatory stimuli) [2]. The COX enzymes catalyze AA to prostaglandin (PG) G2 and then to PGH2, proceeding to the syntheses of PGE2, PGI2, PGD2, PGF2α, and thromboxane A2 (TXA2) by tissue-specific isomerase and synthase enzymes [18]. During prostanoid syntheses, COX-1 couples preferentially to thromboxane synthase (TBXAS), PGF synthase, and cytosolic PGE synthase (PGES) isozymes, while COX-2 shows preferential coupling with PGI synthase and microsomal PGES isozymes [19]. In the 5-LOX pathway, 5-LOX converts AA into LTA4, which is then converted to LTB4 (by LTA4 hydrolase) or catalyzed to LTC4 (by LTC4 synthase), followed by LTD4 and then E4 syntheses [20].
NSAIDs act by suppressing the activities of (1) COX isoenzymes, and (2) the enzymes responsible for prostanoid biosynthesis from AA [18,21,22]. Certain NSAIDs preferentially inhibit COX-1 and only partially inhibit COX-2 (e.g., aspirin, indomethacin, naproxen, and diclofenac); thereby, inhibiting production of protective PGs. Newer NSAIDs that inhibit COX-2 primarily (e.g., nimesulide, meloxicam) or specifically (e.g., celecoxib, rofecoxib) can suppress the inflammatory prostanoids, and only slightly decrease protective PG production [2].
COX-1 inhibition
By suppressing COX-1 activity, aspirin and some classical NSAIDs inhibit the synthesis of PGs, especially PGE2; thereby, increasing the production of CysLTs (LTA4, LTB4, LTC4, LTD4) [2,17,23,24]. PGEE2 has a protective effect against bronchoconstriction, releases inflammatory mediators from mast cells, and recruits immune cells to inflammatory sites [25,26]. On the other hand, CysLTs induce bronchoconstriction, airway inflammation, cell recruitment, and platelet activation [27-30]. In addition, aspirin can acetylate COX-2, leading to the generation of 15-hydroeicosatetraenoic acid (15-HETE), which is then transformed into lipoxin (LX) or 15-epimer-LX (15-epiLX) by 5-LOX. Both LX and 15-epi-LX exert anti-inflammatory effects by inhibiting neutrophil and eosinophil chemotaxis, as well as blocking LTC4-induced bronchoconstriction [23,31-33]. However, in NERD patients, the levels of LXA4 (downstream product of LXs) and 15-epi-LX in plasma and nasal lavage fluid were reported to decrease after lysine-aspirin nasal challenge [23,31]. A possible reason for the reduced LXA4 and 15-epi-LX levels in these patients is that the cellular AA resources are already overused for the synthesis of CysLTs [23].
IgE-mediated allergic reactions
NSAIDs can also elicit the production of specific IgE antibodies, which bind to their high-affinity receptors (FceRIα and FcεRII) on the surface of mast cells and basophils and provide multivalent binding sites for drug antigens. When the drug antigen is cross-linked with IgE bound to the receptors, the mast cells or basophils are stimulated to release preformed mediators (e.g., histamine) and produce new mediators [34]. Although a few studies showed high serum-specific IgE level production by NSAIDs, these results were not replicated and require further investigation [13,35-37].
Despite the conflicting results, one serum-specific IgE that has been confirmed to be involved in NHS is the IgE against thyroid peroxide (TPO), which was first discovered by Altrichter et al. [38] in 2011. More recently, high TPO-specific IgE levels were found in patients with NIUA and NECD compared to healthy controls [39]. Moreover, when TPO was added to basophils isolated from these NIUA and NECD patients, the expression of CD203c (a surface marker for basophil activation) increased [39]. These findings suggest a role of TPO-specific IgE in the pathogenesis of NECD/NIUA via activation of basophils or mast cells [39]. Furthermore, crosslinking of an allergen to the neighboring IgE molecules bound to FceRI can trigger calcium influx, resulting in basophil degranulation [40]. Moreover, in patients with CU and food-dependent, exercise-induced anaphylaxis, aspirin treatment was found to increase phosphorylation of the spleen tyrosine kinase (Syk) in IgE-sensitized basophils [16]. Finally, other NSAIDs (e.g., diclofenac, salicylate, and ketoprofen) were also found to indirectly enhance IgE-mediated histamine release from human basophils through activation of the Syk kinase pathway [40,41].
T cell-mediated mechanisms
NSAIDs can induce delayed-type reactions mediated by T cell activation [2,42]. In the context of maturation signals resulting from drug-related stress, disease, or trauma, dendritic cells (DCs) in the skin and mucous membrane recognize and transport drug antigen complexes to the regional lymph nodes [43]. In the lymph nodes, DCs introduce the drug antigens to naïve T lymphocytes and stimulate the production of antigen-specific T cells. Subsequently, drug antigen-specific T cells migrate to the target organs and secrete cytokines and cytotoxins upon re-exposure to the drugs [43]. Based on the main effector cells involved, T cell-mediated NHS mechanisms are classified into four subtypes: (1) IVa, involving type 1 helper (Th1) cells and monocytes and the production of interferon g, interleukin 1 (IL-1), and IL-2 cytokines; (2) IVb, involving Th2 cells and eosinophils and the production of IL-4, IL-5, and IL-13; (3) IVc, involving cytotoxic T cells and the production of perforin, granzyme B, and Fas ligand; and (4) IVd, involving CD4+ T cells, CD8+ T cells, and neutrophils, and the production of IL-8 and granulocyte-macrophage colony-stimulating factor [7,42].
Oxidative stress and apoptosis in NHS
Another mechanism underlying NHS involves oxidative stress and the induction of pro-inflammatory mediator production (cytokines and chemokines), which can aggravate airway inflammation, bronchospasm, and mucin secretion [14]. For example, Antczak et al. [44] showed that the levels of 8-isoprostanes, metabolized products from AA that are induced by reactive oxygen species, are increased in the exhaled breath condensate of patients with NERD [45], suggesting a role of oxidative stress in NHS. Moreover, aspirin can trigger apoptosis by inducing oxidative stress as well as by inhibiting expression of the anti-apoptotic protein, Beclin-2 (Bcl2) [17]. NSAIDs may reduce Bcl2 expression by blocking the IL6-IL6R-STAT3 signaling pathway. In turn, decreased Bcl2 expression initiates tumor necrosis factor (TNF)-related apoptosis-inducing ligand and TNF-a generation, which then elicit apoptosis [17].
Platelet activation in NHS
While platelet activation has long been known to be involved in the pathogenic mechanisms of asthma and urticaria [46,47], its role in NHS has not been studied in depth. However, Palikhe et al. [15] recently found a higher level of soluble P-selectin, which is a marker for activated platelets, in patients with NECD than in those with aspirin-tolerant CU or healthy controls. In addition, while aspirin treatment abolished the expression of soluble P-selectin and P2Y12 on platelets in aspirin-tolerant CU patients, no such inhibitory effect was observed in platelets from NECD patients [15]. These findings suggest that platelet activation contributes to the pathogenesis of NECD.
GENETIC AND EPIGENETIC FACTORS INVOLVED IN NHS
A family history of aspirin intolerance has been reported in 6% of patients, thereby suggesting a role of genetic factors in NHS pathogenesis [48]. However, most genome-wide association and case-control studies have focused on NERD, and only a limited number of studies have investigated genetic factors involved in NECD/NIUA. These studies concentrated on the genes related to the suspected pathomechanisms of the disease, including AA metabolism pathways, IgE, inflammatory mediators, and cytokines, the human leukocyte antigen (HLA) system, and other miscellaneous factors [8,49]. Genetic polymorphisms that have been found to be associated with NHS are shown in Table 2. In addition, epigenetic factors have recently been found to be associated with the pathogenic mechanisms of NHS, which are also summarized in this section.
Table 2.
Genetic polymorphisms involved in NSAID hypersensitivity
| Pathway/gene name | SNPs | Associated clinical phenotype | Population |
|---|---|---|---|
| Arachidonic acid | |||
| LTC4S | -444A>C | C allele was associated with NERD and increased urine LTE4 level after aspirin challenge. | Polish [50, 51] |
| CYSLTR1 | -634C>T, -475 A>C, -336 A>G | Those SNPs were associated with NERD and could affect gene transcriptional activity. | Korean [55-57] |
| CYSLTR2 | -189 T>C | ||
| COX-2 | -765 G>C | This SNP could affect gene transcriptional activity and contribute to PGD2 production in NERD patients. | Polish [60] |
| ALOX5 | -1708 G>A, 21 C>T, 270 G>A, 1728 G>A | -1708 A allele was associated with NECD/NIUA. | Korean [53, 61] |
| Haplotype (GCGA) was associated with NERD. | |||
| ALOX15 | -272 C>A | NECD/NIUA | Polish [62] |
| TBXA2R | -4684 C>T | This SNP affected gene transcriptional activity. C allele was associated with NERD, while TT genotype was associated with NIUA. | Korean [64, 65] |
| 795 T>C | This SNP was associated with NERD and % fall in FEV1 after aspirin challenge. | Korean [66] | |
| TBXAS1 | 141931T>A | This SNP was associated with NERD in a Korean, and NIUA in a Spanish population. | Korean [67] |
| Spain [68] | |||
| PTGER2 | -616 C>G, -166 G>A | NERD | Korean [65] |
| PTGER3 | -1709 T>A, 173288G>T, 195000A>G | NERD | Korean [65, 70] |
| PTGER4 | -1254 A>G | This SNP could affect gene transcriptional activity and was associated with NERD as well as NECD. | Korean [65, 69] |
| PTGIR | 1915 T>A | NERD | Korean [65] |
| Basophil/mast cell and eosinophil activation | |||
| FCER1G | -237 A>G | This SNP was associated with NERD and increased total IgE serum level in NERD patients. | Korean [72] |
| FCER1A | -344 T>A | This SNP was associated with NECD and increased total IgE serum level in NECD patients. | Korean [72, 73] |
| NERD patients carrying CC/CT genotype had a higher serum level of IgE to staphylococcal enterotoxin A than those with CC genotype. | |||
| HNMT | 939 A>G | This SNP was associated with NECD, could affect HNMT mRNA stability, protein expression, and enzymatic activity of HNMT. | Korean [75] |
| IL-4 | -589 T>C, -33 T>C | These SNPs were associated with NERD and could affect gene transcriptional activity. | Korean [78] |
| IL-13 | -1510 A>C, -1055 C>T | These SNPs were associated with rhinosinusitis in NERD patients. | Korean [79, 80] |
| 110 G>A (Arg 110Gln) | This SNP was associated with increased blood eosinophil count in NERD patients. | Korean [80] | |
| -1111 C>T | This SNP was associated with NERD. | Japanese [81] | |
| Other inflammatory mediators and cytokines | |||
| IL-5Rα | -5993 G>A | This SNP was associated with the production of serum-specific IgE against staphylococca enterotoxin A in NERD patients. | Korean [83] |
| TNF-α | -1031 T>C, -863 C>A | NECD/NIUA | Korean [84] |
| -308 G>A | This SNP was associated with chronic rhinosinusitis and nasal polyposis in NERD. | Hungarian [85] | |
| IL-10 | -819 T>C | NERD | Korean [87] |
| IL-18 | -607 A>C | -607 A>C and the haplotype (CG) were associated with NIUA. The (CG) haplotype increased transcriptional activity and neutrophil chemotaxis. | Korean [86] |
| -137 G>C | |||
| IL-17A | -737 C>T | NERD | Japan [81] |
| IL-17Rα | -1075 A>G, -947 A>G, -50 C>T | Major alleles of these SNPs were associated with NERD, higher IL-17RA mRNA and protein expression in CD14+ peripheral monocytes. | Korean [88] |
| HLA system | |||
| DPB1*0301 | - | This allele was associated with NERD and could interact with TNF-α polymorphisms to increase NERD susceptibility. | Polish [90] |
| Korean [91, 92] | |||
| DPB1 | Rs3128965 | This SNP was associated with NERD, increased airway hyper-responsiveness in aspirin challenge test, and increased secretion of 15-HETE from peripheral blood leukocytes. | Korean [92] |
| Exm537513 | NERD | Korean [94] | |
| DPB1*0401 | - | This allele had a protective role against NERD. | Polish [90] |
| German [93] | |||
| DQB1*0301, DQB1*011 | - | There alleles had a protective role against NERD. | Iran [95] |
| DQB1*0302, DRB1*04 | - | NERD | Iran [95] |
| DQB1*0609 [DRB1*1303 /DQB1*0 609/DPB1*0201] | - | NECD | Korean [96] |
| Cw4, Cw7 | - | These alleles may have a protective role against NECD. | Italian [97] |
| B44 | - | NECD | Italian [97] |
| Other pathways | |||
| ATF6B | Rs2228628, Rs8111 | These SNPs were associated with a decline in %FEV1 after aspirin challenge test. | Korean [98] |
| CYP2C19 | 681 G>A, 636 G>A | NERD | Japan [99] |
| DPP10 | Rs1704175 | This SNP was associated with NERD and the serum level of DPP10 in asthmatic patients. | Korean [100] |
| P2Y12 | 18 C>T | This SNP was associated with platelet P2Y12 expression in NERD patients. | Korean [101] |
| 742 T>A | This SNP was associated with the nasal secretion level of eosinophil cationic protein in NERD patients. | ||
NSAID, nonsteroidal anti-inflammatory drug; SNP, single nucleotide polymorphism; LTC4S, leukotriene C4 synthase; NERD, NSAID-exacerbated respiratory disease; LTE4, leukotriene E4; CYSLTR1, CysLT receptor 1 gene; CYSLTR2, CysLT receptor 2 gene; COX-2, cyclooxygenase 2; PGD2, prostaglandin D2; ALOX5, 5-lipoxygenase; NECD, NSAID-exacerbated cutaneous disease; NIUA, NSAID-induced urticaria/angioedema; TBXA2R, TXA2 receptor gene; FEV1, forced expiratory volume in 1 second; TBXAS1, TBXA1 synthase gene; PTGER, prostaglandin E receptor; PTGIR, prostagladin I receptor; FCER1G, Fc epsilon receptor 1 gamma; IgE, immunoglobulin E; FCER1A, Fc epsilon receptor 1 alpha; HNMT, histamine N-methyltransferase; IL-4, interleukin 4; TNF-α, tumor necrosis factor α; HLA, human leukocyte antigen; 15-HETE, 15-hydroeicosatetraenoic acid; ATF6B, activating transcription factor 6β gene; CYP, cytochrome P450; DPP10, dipeptidyl-peptidase 10 gene.
Genes involved in AA metabolism pathways
Early studies on NHS-associated genes focused on the gene encoding leukotriene C4 synthase (LTC4S). The C allele of LTC4S -444A>C (rs730012) was reported to be a risk factor for NERD, and was associated with an increase in urine LTE4 level after aspirin challenge test [50]. In addition, NIUA was found to aggregate in Polish families inheriting the LTC4S -444C allele [51]. However, these findings were not replicated in American, Japanese, or Korean populations [52-54]. Instead, an association of NERD with three single nucleotide polymorphisms (SNPs) in the CysLT receptor 1 gene (CYSLTR1 -634C>T, -475A>C, and -336A>G) and one SNP in CYSLTR2 (-189T>C) was identified in Korean populations. These SNPs were also found to affect gene transcriptional activity [55-57], consistent with the observed increases in CysLTR1 and CysLTR2 expression found in nasal mucosal inflammatory cells of NERD patients [58,59].
In Polish subjects with NERD, a promoter SNP in COX-2 (-765G>C) was found to affect transcriptional activity and contribute to the production of PGD2 [60], while four SNPs in the 5-LOX gene (ALOX5 -1708G>A, 21C>T, 270G>A, and 1728G>A) were associated with NERD in a Korean population [53]. The ALOX5 -1708A allele was also associated with NECD/NIUA in a Korean population [61]. In addition, a promoter SNP in ALOX15 (-272C>A) was found to be associated with NECD/NIUA in a Spanish population. However, associations of ALOX12 and ALOX15 polymorphisms with NHS were not observed in a Korean population [62,63].
The transcriptional function of the -4684C>T SNP in the TXA2 receptor gene (TBXA2R) was reported previously, and was associated with NIUA [64]. Consistently, the TBXA2R -4684C allele was shown to be associated with NERD, while the TBXA2R -4684TT genotype was associated with NIUA in a Korean population [64,65]. A nonsynonymous SNP of TBXA2R (795T>C) was also found to be associated with NERD, as well as with a drop in forced expiratory volume in 1 second (FEV1) after inhaled aspirin challenge [66]. Similarly, the SNP in intron 9 (rs6962291, 141931T>A) in the TBXA1 synthase gene (TBXAS1) was shown to be associated with NERD in a Korean population [67], as well as with NIUA in a Spanish population [68]. In a Korean population, NERD was significantly associated with genetic polymorphisms of the prostanoid receptors, prostaglandin E receptor 2 (PTGER2) (-616C>G, -166G>A), PTGER3 (-1709T>A), PTGER4 (-1254A>G), and PTGIR (1915T>A) [65]. Moreover, PTGER4 -1254A>G was found to affect transcriptional activity and was associated with NECD [69]. Another study indicated an association of NERD with rs7543182 (173288G>T) and rs959 (195000A>G) in PTGER3 [70].
Genes involved in mast cell/basophil and eosinophil activation
The high-affinity IgE receptor (FcεRI) plays a crucial role in the activation and degranulation of mast cells and basophils, resulting in the release of a variety of cytokines and leukotrienes [71]. Palikhe et al. [72] demonstrated an association of FCER1G -237A>G with NERD and increased total IgE serum levels in a Korean population with NERD. On the other hand, the C allele of FCER1A -344T>C was associated with NECD and increased total serum IgE level as well as atopy rate in NECD patients [73]. Although no association of FCER1A -344T>C with NERD was found, the FCER1A -344CT/TT genotype was associated with a higher serum level of IgE specific antibody to Staphylococcal enterotoxin A compared to the CC genotype [72]. Nevertheless, these findings could not be replicated in a recent study with a small population of Taiwanese CU patients [34].
The cellular level of histamine, a key mediator released from basophils/mast cells, is regulated by histamine N-methyltransferase (HNMT) [74]. Therefore, the HNMT 939A>G polymorphism may contribute to NECD by influencing HNMT mRNA stability, protein expression, and HNMT enzymatic activity [75,76]. In addition, IL-4 and IL-13 are important cytokines involved in mast cell, basophil, and/or eosinophil activation and function, and share a common receptor subunit (IL-4Rα) [77]. The IL-4 -589T>C and -33T>C polymorphisms could affect the promoter transcriptional activity, and were shown to be associated with NERD risk [78]. Although associations of IL-13polymorphisms (-1510A>C, -1055C>T, and 110G>A [Arg110Gln]) with NERD and NECD were not observed in a Korean population, higher frequencies of rhinosinusitis were noted in NERD patients carrying the IL-13 -1510A allele, as well as the -1055CC genotype, compared to non-carriers [79,80]. Moreover, the GG genotype of IL-13 110G>A was significantly associated with an increased blood eosinophil count in NERD patients [80]. In addition, in a Japanese population, the minor allele of IL-13 -1111C>T was reported to be associated with NERD [81]. Finally, IL-5 and its receptor IL-5R are necessary for the survival and activation of eosinophils [82], and Losol et al. [83] recently described an association between an IL-5Rα polymorphism (-5993G>A) and the production of serum-specific IgE antibodies against Staphylococcal enterotoxin A. However, no association of this IL-5Rα polymorphism with NERD susceptibility was found.
Other inflammatory mediators and cytokine-related genes
Among the five SNPs of the TNF-α gene investigated in our previous study (TNF-α -1031T>C, -863C>A, -857C>T, -308G>A, and -238G>A), only two (TNF-α -1031T>C and -863C>A) were significantly associated with NECD/NIUA risk [84]. Associations of chronic rhinosinusitis (CRS) and nasal polyps with TNF-α -308G>A in NERD patients were reported in a Hungarian population, although no association with NERD susceptibility was observed [85]. Kim et al. [86] demonstrated an association of NIUA with IL-18 -607A>C, as well as with the haplotype (CG) of IL-18 -607A>C and -137G>C, which could increase transcriptional activity and neutrophil chemotaxis. In addition, associations of NERD with other cytokine gene polymorphisms, including IL-10 (-819T>C), IL-17A (-737C>T), and IL-17Rα (-1075A>G, -947A>G, -50C>T) were also reported in some studies [81,87,88]. These findings suggest that NHS is associated with numerous genetic polymorphisms of inflammatory cytokines, the functions of which in the pathogenesis of NHS should be investigated further.
HLA genes
HLA molecules play a crucial role in T cell activation. Classical HLA class I molecules (HLA-A, HLA-B, and HLA-C) are responsible for presenting drug antigens to CD8+ T cells, while HLA class II molecules (HLA-DP, HLA-DQ, and HLA-DR) present antigens to CD4+ T cells [89]. Among them, HLA class II alleles are well-known genetic factors associated with NHS. The association of NERD with HLA-DPB1*0301 was first reported in a Polish population, and these findings were then replicated in a Korean population [90,91]. Interestingly, HLA-DPB1*0301 combined with certain TNF-α polymorphisms increases the susceptibility to NERD [91]. More recently, the A allele of rs3128965 in HLA-DPB1 was found to be associated with NERD, increased airway hyper-responsiveness after inhaled aspirin challenge test, and increased secretion of 15-HETE from peripheral blood leukocytes treated with AA [92]. In contrast, the HLA-DPB1*0401 allele was reported as a protective factor against NERD susceptibility in studies with Polish and German populations [90,93]. A novel exonic polymorphism (exm537513) in HLA-DPB1 associated with NERD was also recently identified [94]. In addition, the combination of exm537513 with six other exonic SNPs from other genes (exm83523, exm1884673, exm538564, exm2264237, exm396794, and exm791954) provides good predictive value for NERD (area under the curve of 0.75 with 34% sensitivity and 93% specificity) [94]. In addition, Esmaeilzadeh et al. [95] reported that HLA-DQB1*0302, HLA-DRB1*04, and their haplotype were risk factors of NERD, while HLA-DQB1*0301 and HLA-DRB1*011 were negatively associated with NERD.
NECD has also been found to be associated with HLA-DRB1*1302, HLA-DQB1*0609, and the haplotype of HLA-DRB1*1303/DQB1*0609/DPB1*0201 in a Korean population [96]. Furthermore, NECD has also been reported to be associated with HLA class I alleles. Pacor et al. [97] showed that HLA-Cw4 and HLA-Cw7 could have a protective role against NECD, while HLA-B44 was a risk factor. The study by Pacor et al. [97] indicates that HLA class I molecules are involved in the pathogenesis of NHS, and further investigations are required.
Genes related to other pathways
Several genetic risk factors for NERD related to other mechanisms have also been identified. For example, polymorphisms of the activating transcription factor 6β gene (ATF6B rs2228628 and rs8111) were associated with decline in FEV1 % after inhaled aspirin challenge test [98]. Moreover, two SNPs in cytochrome P450 CYP2C19 (681G>A and 636G>A) were found to be associated with NERD and the predicted FEV1 % value [99]. Kim et al. [100] recently reported an association of dipeptidyl-peptidase 10 gene (DPP10) rs1704175 (TT genotype) with NERD, and the TT genotype was associated with a higher serum DPP10 level compared to the CC/CT genotypes in asthmatic patients. The purinergic receptor gene polymorphism (P2RY12 742T>C) was associated with the nasal secretion level of an eosinophil cationic protein, while P2RY12 18C>T was associated with platelet P2Y12 expression level in NERD patients [101]. Nevertheless, the underlying mechanisms by which these genes contribute to NERD pathogenesis remain to be elucidated.
Epigenetic mechanisms
A recent study evaluated DNA methylation levels in nasal polyp tissues of patients with NERD and aspirin-tolerant asthma (ATA) [102]. Comparison of the NERD and ATA groups indicated increased methylation levels of 332 loci in 296 genes, while 158 loci in 141 genes had decreased methylation levels. In addition, four genes involved in AA metabolism (PGD synthase [PGDS], PTGES, ALOX5 activating protein [ALOX5AP], and leukotriene B4 receptor [LTB4R]) were found to have different DNA methylation levels between NERD and ATA patients. These findings highlight the value of nasal polyp profiles for suggesting potential susceptibility genes and pathways involved in the pathogenesis of NERD [102].
MANAGEMENT OF PATIENTS WITH NHS
Clinical manifestations
Symptoms of acute NHS include rhinitis/asthma, urticaria/angioedema, and anaphylaxis [82,103]. Patients with NERD have hypersensitivity in the upper (CRS, nasal polyps) and lower (asthma) airways, called Samter’s triad [104]. In cases of acute NERD, rhinorrhea and nasal congestion followed by dyspnea and chest tightness appear within 30 to 120 minutes after administration of NSAIDs. The chronic form of NERD, i.e., preexisting inflammation of the upper and lower airways that is exacerbated by ingestion of NSAIDs, usually appears in the third decade of life, and is characterized by a variety of symptoms, including nasal congestion, hyposmia, pansinusitis, laryngospasm, and bronchospasm [105,106]. The phenotype of the disease progresses to severe and corticosteroid-dependent asthma with NHS, and NERD can persist throughout life [107,108]. Cutaneous (such as urticaria) and gastric symptoms may accompany the respiratory symptoms in these patients.
In NECD, symptoms occur within 1 to 4 hours after ingestion of a culprit drug, whereas delayed reactions may occur up to 24 hours. Hives usually disappear within several hours but may persist for days, and some NECD patients may concurrently develop respiratory symptoms [4]. In 30% of CU patients, hypersensitivity reactions occur following ingestion of aspirin or NSAIDs [109]. While aggravation of underlying urticaria by ingestion of the culprit drug can occur in these CU patients, the symptoms may fluctuate depending on how well the underlying disease is controlled [110].
Anaphylaxis is another type of acute reaction in NHS, which is commonly combined with NIUA. Within a few minutes of ingestion or administration of the culprit drug, generalized urticaria and skin swelling may progress into anaphylaxis. Some patients develop both symptoms of NERD and CU [24]. Drugs notorious for causing anaphylaxis are pyrazolones, ibuprofen, diclofenac, aspirin, and paracetamol [24]. In delayed reactions, symptoms occur within days or even weeks after exposure, but may be induced earlier by reintroduction. Cutaneous reactions (such as fixed drug eruption, severe bullous cutaneous reaction, and maculopapular drug eruption) mostly occur, although rarely organ-specific symptoms (aseptic meningitis, pneumonitis, or nephritis) are reported [24,111].
Diagnosis
The initial step in the diagnosis of NHS is examination of clinical history. Carefully obtained information on symptom descriptions, symptom chronology (including order of incidence or previous exposure to the culprit drugs), and underlying comorbidities (including asthma, CRS, nasal polyps, or CU) are key for NHS diagnosis [6,112]. Adverse reactions to NSAIDs are those where the patient had an adverse drug reaction within 6 hours of NSAID intake, or if such a reaction was recorded by a physician, or if a reaction reoccurred to the same or a distinct NSAID at different times [109]. However, further diagnostic modalities should be applied, as clinical history is never enough to identify the culprit drugs or mechanisms of the reaction. Therefore, skin prick and intradermal tests are useful tools to confirm or exclude sensitization in acute reactions related to an IgE-dependent mechanism, while a patch test is used to identify a delayed reaction related to T cell-mediated responses [6]. However, the sensitivity and specificity of these tests remain unclear and, to date, no standardized tests have been established [6,24]. Moreover, as an in vitro study, the basophil activation test may provide useful information; however, it is not used widely due to its low sensitivity and difficulties in setting up the method.
While skin or in vitro tests are useful for confirming the culprit drugs, the oral challenge test remains the gold standard for NHS diagnosis [113]. Indeed, an l-lysine-aspirin challenge test and/or aspirin oral challenge test should be performed to confirm the diagnosis of potential NERD patients [106,113]. These oral challenge tests to NSAIDs show very high sensitivity and specificity, exceeding 90% [2]. For the aspirin oral challenge test, the European Academy of Allergy and Clinical Immunology/Global Allergy and Asthma European Network (EAACI/GA2 LEN) guidelines outline a 2-day protocol where four placebo capsules are administered on the first day, and then four increasing doses of aspirin (71, 117, 312, and 500 mg) are given within a 2-hour interval on the second day, followed by FEV1 evaluation every 30 minutes [113]. In Korea, the l-lysine-aspirin bronchoprovocation test is widely applied to confirm a diagnosis of NERD; however, to perform these bronchoprovocation tests, the patient should be in a stable condition and the tests should not be performed in a patient with FEV1 < 70%. Moreover, the bronchoprovocation test should be implemented under the guidance of physicians and technicians equipped with emergency resuscitative equipment [113,114]. The contraindications of challenge tests are acute severe exacerbations, a history of anaphylaxis, or severe cutaneous adverse reactions. It is important to note that although negative results to challenge test may be observed in highly suspicious cases, the diagnosis of NHS should not be excluded, especially in patients on long-term corticosteroid treatment or those with well-controlled underlying diseases.
Recently, Kowalski et al. [2] outlined a simplified, practical diagnostic algorithm for NHS, as follows: (1) the allergist should evaluate if the symptoms are predictable (intolerant) or unpredictable (hypersensitivity); (2) the onset time of the reactions should be assessed; and (3) the pattern of clinical symptoms and underlying chronic disease should be analyzed. In addition, identifying a history of tolerance or intolerance to other NSAIDs helps suggest appropriate NSAIDs for use in challenge tests to confirm or exclude cross-reactivity with other NSAIDs.
Treatment
Immediate NSAID cessation and strict avoidance of the culprit drug and any cross-reacting NSAIDs are of the utmost importance in both early and delayed NHS reactions. Moreover, written information, including lists of culprit and alternative medications, should be provided to NHS patients [7,112].
Most NERD patients have moderate to severe asthma, and medium to high doses of inhaled corticosteroid/long-acting β agonist and leukotriene modifiers should be maintained to control lower respiratory symptoms. The upper airway symptoms related to CRS and/or nasal polyps should be controlled to improve bronchial symptoms, either medically using intranasal corticosteroids, or surgically if necessary [115]. In cases of severe asthma with frequent asthma exacerbations, biologics, such as anti-IgE antibodies, may have beneficial effects for controlling upper and lower respiratory symptoms [116-118]. The suggested biologics for use in these patients include omalizumab, mepolizumab, and benralizumab [119-121]. For the treatment of CRS/nasal polyps, along with medical treatment (e.g., intranasal corticosteroids), consultation with an ear, nose, and throat specialist for surgical treatment, such as sinus endoscopy, is advised [122].
Systemic steroids and antihistamines may be used during acute, severe cutaneous reactions. However, in severe and delayed reactions (e.g., Stevens-Johnson syndrome [SJS] and toxic epidermal necrolysis [TEN]) withdrawal of the culprit drugs is the only proven treatment to decrease fatality. Although other treatments have been suggested for severe adverse reactions (including systemic corticosteroids, intravenous Igs, or immunosuppressive drugs [e.g., cyclosporin and infliximab]), these treatments remain controversial [24].
Finally, desensitization, which involves the induction of a temporary state of tolerance/unresponsiveness to the drug (mostly aspirin) responsible for NHS, may be an optional treatment for NERD and is expected to improve upper and lower respiratory symptoms [123,124]. Once desensitized, patients should take the drug regularly to maintain tolerance, as intolerance could recur after 2 to 5 days of cessation [124]. Oh et al. [125] reported two cases of NECD that were successfully controlled by aspirin desensitization. Unfortunately, protocols for drug desensitization have not been standardized to date; however, slower protocols tend to be more effective than rush protocols [114]. Despite these successes, the EAACI guidelines indicate that desensitization is absolutely contraindicated in cases of severe or threatening delayed drug hypersensitivity reactions (such as SJS, TEN, or drug reactions with eosinophilia and systemic symptoms) [114].
CONCLUSIONS
NHS is a heterogeneous disease with several clinical phenotypes involving various pathogenic mechanisms. In addition to COX pathway alterations, NHS is related to immune responses (including IgE production and T cell activation), oxidative stress, and platelet activation. While recent genetic studies have identified some predictive markers for NHS susceptibility and treatment response in different populations, these require further investigation. In future, an improved understanding of the functional and genetic/epigenetic pathogenic mechanisms of NHS will help in the development of new diagnostic methods and effective treatments.
Acknowledgments
This study was supported by a grant from the Korean Health Technology R&D Project, Ministry of Health and Welfare, Republic of Korea (H14C2628).
Footnotes
No potential conflict of interest relevant to this article was reported.
REFERENCES
- 1.Conaghan PG. A turbulent decade for NSAIDs: update on current concepts of classification, epidemiology, comparative efficacy, and toxicity. Rheumatol Int. 2012;32:1491–1502. doi: 10.1007/s00296-011-2263-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kowalski ML, Makowska JS. Seven steps to the diagnosis of NSAIDs hypersensitivity: how to apply a new classification in real practice? Allergy Asthma Immunol Res. 2015;7:312–320. doi: 10.4168/aair.2015.7.4.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kasper L, Sladek K, Duplaga M, et al. Prevalence of asthma with aspirin hypersensitivity in the adult population of Poland. Allergy. 2003;58:1064–1066. doi: 10.1034/j.1398-9995.2003.00267.x. [DOI] [PubMed] [Google Scholar]
- 4.Erbagci Z. Multiple NSAID intolerance in chronic idiopathic urticaria is correlated with delayed, pronounced and prolonged autoreactivity. J Dermatol. 2004;31:376–382. doi: 10.1111/j.1346-8138.2004.tb00688.x. [DOI] [PubMed] [Google Scholar]
- 5.Ye YM, Kim MK, Kang HR, et al. Predictors of the severity and serious outcomes of anaphylaxis in Korean adults: a multicenter retrospective case study. Allergy Asthma Immunol Res. 2015;7:22–29. doi: 10.4168/aair.2015.7.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Demoly P, Adkinson NF, Brockow K, et al. International Consensus on drug allergy. Allergy. 2014;69:420–437. doi: 10.1111/all.12350. [DOI] [PubMed] [Google Scholar]
- 7.Kowalski ML, Makowska JS, Blanca M, et al. Hypersensitivity to nonsteroidal anti-inflammatory drugs (NSAIDs). Classification, diagnosis and management: review of the EAACI/ENDA(#) and GA2LEN/HANNA*. Allergy. 2011;66:818–829. doi: 10.1111/j.1398-9995.2011.02557.x. [DOI] [PubMed] [Google Scholar]
- 8.Gomez F, Perkins JR, Garcia-Martin E, Canto G, Cornejo-Garcia JA. Genetic basis of hypersensitivity reactions to nonsteroidal anti-inflammatory drugs. Curr Opin Allergy Clin Immunol. 2015;15:285–293. doi: 10.1097/ACI.0000000000000178. [DOI] [PubMed] [Google Scholar]
- 9.Dona I, Blanca-Lopez N, Cornejo-Garcia JA, et al. Characteristics of subjects experiencing hypersensitivity to non-steroidal anti-inflammatory drugs: patterns of response. Clin Exp Allergy. 2011;41:86–95. doi: 10.1111/j.1365-2222.2010.03651.x. [DOI] [PubMed] [Google Scholar]
- 10.Kowal-Bielecka O, Kowal K, Distler O, Gay S. Mechanisms of disease: leukotrienes and lipoxins in scleroderma lung disease. Insights and potential therapeutic implications. Nat Clin Pract Rheumatol. 2007;3:43–51. doi: 10.1038/ncprheum0375. [DOI] [PubMed] [Google Scholar]
- 11.Hofmeier KS. Hypersensitivity reactions to modern antiplatelet and anticoagulant drugs. Allergo J Int. 2015;24:58–66. doi: 10.1007/s40629-015-0048-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blanca-Lopez N, Cornejo-Garcia JA, Plaza-Seron MC, et al. Hypersensitivity to nonsteroidal anti-inflammatory drugs in children and adolescents: cross-intolerance reactions. J Investig Allergol Clin Immunol. 2015;25:259–269. [PubMed] [Google Scholar]
- 13.Stone SF, Phillips EJ, Wiese MD, Heddle RJ, Brown SG. Immediate-type hypersensitivity drug reactions. Br J Clin Pharmacol. 2014;78:1–13. doi: 10.1111/bcp.12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cho YS, Moon HB. The role of oxidative stress in the pathogenesis of asthma. Allergy Asthma Immunol Res. 2010;2:183–187. doi: 10.4168/aair.2010.2.3.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Palikhe S, Palikhe NS, Kim SH, Yoo HS, Shin YS, Park HS. Elevated platelet activation in patients with chronic urticaria: a comparison between aspirin-intolerant and aspirin-tolerant groups. Ann Allergy Asthma Immunol. 2014;113:276–281. doi: 10.1016/j.anai.2014.06.011. [DOI] [PubMed] [Google Scholar]
- 16.Matsuo H, Yokooji T, Morita H, et al. Aspirin augments IgE-mediated histamine release from human peripheral basophils via Syk kinase activation. Allergol Int. 2013;62:503–511. doi: 10.2332/allergolint.13-OA-0536. [DOI] [PubMed] [Google Scholar]
- 17.Kacprzak D, Pawliczak R. Does aspirin-induced oxidative stress cause asthma exacerbation? Arch Med Sci. 2015;11:494–504. doi: 10.5114/aoms.2014.41960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bruno A, Tacconelli S, Patrignani P. Variability in the response to non-steroidal anti-inflammatory drugs: mechanisms and perspectives. Basic Clin Pharmacol Toxicol. 2014;114:56–63. doi: 10.1111/bcpt.12117. [DOI] [PubMed] [Google Scholar]
- 19.Reiss AB, Edelman SD. Recent insights into the role of prostanoids in atherosclerotic vascular disease. Curr Vasc Pharmacol. 2006;4:395–408. doi: 10.2174/157016106778521652. [DOI] [PubMed] [Google Scholar]
- 20.Hedi H, Norbert G. 5-Lipoxygenase pathway, dendritic cells, and adaptive immunity. J Biomed Biotechnol. 2004;2004:99–105. doi: 10.1155/S1110724304310041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meade EA, Smith WL, DeWitt DL. Differential inhibition of prostaglandin endoperoxide synthase (cyclooxygenase) isozymes by aspirin and other non-steroidal anti-inflammatory drugs. J Biol Chem. 1993;268:6610–6614. [PubMed] [Google Scholar]
- 22.Vane JR. The mode of action of aspirin and similar compounds. J Allergy Clin Immunol. 1976;58:691–712. doi: 10.1016/0091-6749(76)90181-0. [DOI] [PubMed] [Google Scholar]
- 23.Kupczyk M, Antczak A, Kuprys-Lipinska I, Kuna P. Lipoxin A4 generation is decreased in aspirin-sensitive patients in lysine-aspirin nasal challenge in vivo model. Allergy. 2009;64:1746–1752. doi: 10.1111/j.1398-9995.2009.02047.x. [DOI] [PubMed] [Google Scholar]
- 24.Kowalski ML, Asero R, Bavbek S, et al. Classification and practical approach to the diagnosis and management of hypersensitivity to nonsteroidal anti-inflammatory drugs. Allergy. 2013;68:1219–1232. doi: 10.1111/all.12260. [DOI] [PubMed] [Google Scholar]
- 25.Sastre B, del Pozo V. Role of PGE2 in asthma and non-asthmatic eosinophilic bronchitis. Mediators Inflamm. 2012;2012:645383. doi: 10.1155/2012/645383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ricciotti E, FitzGerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. 2011;31:986–1000. doi: 10.1161/ATVBAHA.110.207449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beller TC, Maekawa A, Friend DS, Austen KF, Kanaoka Y. Targeted gene disruption reveals the role of the cysteinyl leukotriene 2 receptor in increased vascular permeability and in bleomycin-induced pulmonary fibrosis in mice. J Biol Chem. 2004;279:46129–46134. doi: 10.1074/jbc.M407057200. [DOI] [PubMed] [Google Scholar]
- 28.Paruchuri S, Tashimo H, Feng C, et al. Leukotriene E4-induced pulmonary inflammation is mediated by the P2Y12 receptor. J Exp Med. 2009;206:2543–2555. doi: 10.1084/jem.20091240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gauvreau GM, Parameswaran KN, Watson RM, O’Byrne PM. Inhaled leukotriene E(4), but not leukotriene D(4), increased airway inflammatory cells in subjects with atopic asthma. Am J Respir Crit Care Med. 2001;164(8 Pt 1):1495–1500. doi: 10.1164/ajrccm.164.8.2102033. [DOI] [PubMed] [Google Scholar]
- 30.Cummings HE, Liu T, Feng C, et al. Cutting edge: leukotriene C4 activates mouse platelets in plasma exclusively through the type 2 cysteinyl leukotriene receptor. J Immunol. 2013;191:5807–5810. doi: 10.4049/jimmunol.1302187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sanak M, Levy BD, Clish CB, et al. Aspirin-tolerant asthmatics generate more lipoxins than aspirin-intolerant asthmatics. Eur Respir J. 2000;16:44–49. doi: 10.1034/j.1399-3003.2000.16a08.x. [DOI] [PubMed] [Google Scholar]
- 32.Fierro IM, Colgan SP, Bernasconi G, et al. Lipoxin A4 and aspirin-triggered 15-epi-lipoxin A4 inhibit human neutrophil migration: comparisons between synthetic 15 epimers in chemotaxis and transmigration with microvessel endothelial cells and epithelial cells. J Immunol. 2003;170:2688–2694. doi: 10.4049/jimmunol.170.5.2688. [DOI] [PubMed] [Google Scholar]
- 33.Bonnans C, Levy BD. Lipid mediators as agonists for the resolution of acute lung inflammation and injury. Am J Respir Cell Mol Biol. 2007;36:201–205. doi: 10.1165/rcmb.2006-0269TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hsieh CW, Lee JW, Liao EC, Tsai JJ. A disease marker for aspirin-induced chronic urticaria. Int J Mol Sci. 2014;15:12591–12603. doi: 10.3390/ijms150712591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blanca M, Perez E, Garcia JJ, et al. Angioedema and IgE antibodies to aspirin: a case report. Ann Allergy. 1989;62:295–298. [PubMed] [Google Scholar]
- 36.Bolze S, Bromet N, Gay-Feutry C, Massiere F, Boulieu R, Hulot T. Development of an in vitro screening model for the biosynthesis of acyl glucuronide metabolites and the assessment of their reactivity toward human serum albumin. Drug Metab Dispos. 2002;30:404–413. doi: 10.1124/dmd.30.4.404. [DOI] [PubMed] [Google Scholar]
- 37.Berkes EA. Anaphylactic and anaphylactoid reactions to aspirin and other NSAIDs. Clin Rev Allergy Immunol. 2003;24:137–148. doi: 10.1385/CRIAI:24:2:137. [DOI] [PubMed] [Google Scholar]
- 38.Altrichter S, Peter HJ, Pisarevskaja D, Metz M, Martus P, Maurer M. IgE mediated autoallergy against thyroid peroxidase: a novel pathomechanism of chronic spontaneous urticaria? PLoS One. 2011;6: doi: 10.1371/journal.pone.0014794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shin YS, Suh DH, Yang EM, Ye YM, Park HS. Serum specific IgE to thyroid peroxidase activates basophils in aspirin intolerant urticaria. J Korean Med Sci. 2015;30:705–709. doi: 10.3346/jkms.2015.30.6.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gibbs BF, Rathling A, Zillikens D, Huber M, Haas H. Initial Fc epsilon RI-mediated signal strength plays a key role in regulating basophil signaling and deactivation. J Allergy Clin Immunol. 2006;118:1060–1067. doi: 10.1016/j.jaci.2006.07.022. [DOI] [PubMed] [Google Scholar]
- 41.Wojnar RJ, Hearn T, Starkweather S. Augmentation of allergic histamine release from human leukocytes by non-steroidal anti-inflammatory-analgesic agents. J Allergy Clin Immunol. 1980;66:37–45. doi: 10.1016/0091-6749(80)90136-0. [DOI] [PubMed] [Google Scholar]
- 42.Hyman MH. Delayed drug hypersensitivity reactions. Ann Intern Med. 2004;140:W35. doi: 10.7326/0003-4819-140-9-200405040-00028-w1. [DOI] [PubMed] [Google Scholar]
- 43.Gallo PM, Gallucci S. The dendritic cell response to classic, emerging, and homeostatic danger signals. Implications for autoimmunity. Front Immunol. 2013;4:138. doi: 10.3389/fimmu.2013.00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Antczak A, Montuschi P, Kharitonov S, Gorski P, Barnes PJ. Increased exhaled cysteinyl-leukotrienes and 8-isoprostane in aspirin-induced asthma. Am J Respir Crit Care Med. 2002;166:301–306. doi: 10.1164/rccm.2101021. [DOI] [PubMed] [Google Scholar]
- 45.Dworski R, Murray JJ, Roberts LJ, 2nd, et al. Allergen-induced synthesis of F(2)-isoprostanes in atopic asthmatics. Evidence for oxidant stress. Am J Respir Crit Care Med. 1999;160:1947–1951. doi: 10.1164/ajrccm.160.6.9903064. [DOI] [PubMed] [Google Scholar]
- 46.Idzko M, Pitchford S, Page C. Role of platelets in allergic airway inflammation. J Allergy Clin Immunol. 2015;135:1416–1423. doi: 10.1016/j.jaci.2015.04.028. [DOI] [PubMed] [Google Scholar]
- 47.Chandrashekar L, Rajappa M, Sundar I, et al. Platelet activation in chronic urticaria and its correlation with disease severity. Platelets. 2014;25:162–165. doi: 10.3109/09537104.2013.786822. [DOI] [PubMed] [Google Scholar]
- 48.Szczeklik A, Nizankowska E, Duplaga M. Natural history of aspirin-induced asthma. AIANE Investigators. European Network on Aspirin-Induced Asthma. Eur Respir J. 2000;16:432–436. doi: 10.1034/j.1399-3003.2000.016003432.x. [DOI] [PubMed] [Google Scholar]
- 49.Losol P, Yoo HS, Park HS. Molecular genetic mechanisms of chronic urticaria. Allergy Asthma Immunol Res. 2014;6:13–21. doi: 10.4168/aair.2014.6.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sanak M, Pierzchalska M, Bazan-Socha S, Szczeklik A. Enhanced expression of the leukotriene C(4) synthase due to overactive transcription of an allelic variant associated with aspirin-intolerant asthma. Am J Respir Cell Mol Biol. 2000;23:290–296. doi: 10.1165/ajrcmb.23.3.4051. [DOI] [PubMed] [Google Scholar]
- 51.Mastalerz L, Setkowicz M, Sanak M, Rybarczyk H, Szczeklik A. Familial aggregation of aspirin-induced urticaria and leukotriene C synthase allelic variant. Br J Dermatol. 2006;154:256–260. doi: 10.1111/j.1365-2133.2005.06851.x. [DOI] [PubMed] [Google Scholar]
- 52.Van Sambeek R, Stevenson DD, Baldasaro M, et al. 5’ Flanking region polymorphism of the gene encoding leukotriene C4 synthase does not correlate with the aspirin-intolerant asthma phenotype in the United States. J Allergy Clin Immunol. 2000;106(1 Pt 1):72–76. doi: 10.1067/mai.2000.107603. [DOI] [PubMed] [Google Scholar]
- 53.Choi JH, Park HS, Oh HB, et al. Leukotriene-related gene polymorphisms in ASA-intolerant asthma: an association with a haplotype of 5-lipoxygenase. Hum Genet. 2004;114:337–344. doi: 10.1007/s00439-004-1082-1. [DOI] [PubMed] [Google Scholar]
- 54.Kawagishi Y, Mita H, Taniguchi M, et al. Leukotriene C4 synthase promoter polymorphism in Japanese patients with aspirin-induced asthma. J Allergy Clin Immunol. 2002;109:936–942. doi: 10.1067/mai.2002.124466. [DOI] [PubMed] [Google Scholar]
- 55.Kim SH, Oh JM, Kim YS, et al. Cysteinyl leukotriene receptor 1 promoter polymorphism is associated with aspirin-intolerant asthma in males. Clin Exp Allergy. 2006;36:433–439. doi: 10.1111/j.1365-2222.2006.02457.x. [DOI] [PubMed] [Google Scholar]
- 56.Kim SH, Ye YM, Hur GY, et al. CysLTR1 promoter polymorphism and requirement for leukotriene receptor antagonist in aspirin-intolerant asthma patients. Pharmacogenomics. 2007;8:1143–1150. doi: 10.2217/14622416.8.9.1143. [DOI] [PubMed] [Google Scholar]
- 57.Park JS, Chang HS, Park CS, et al. Association analysis of cysteinyl-leukotriene receptor 2 (CYSLTR2) polymorphisms with aspirin intolerance in asthmatics. Pharmacogenet Genomics. 2005;15:483–492. doi: 10.1097/01.fpc.0000166456.84905.a0. [DOI] [PubMed] [Google Scholar]
- 58.Sousa AR, Parikh A, Scadding G, Corrigan CJ, Lee TH. Leukotriene-receptor expression on nasal mucosal inflammatory cells in aspirin-sensitive rhinosinusitis. N Engl J Med. 2002;347:1493–1499. doi: 10.1056/NEJMoa013508. [DOI] [PubMed] [Google Scholar]
- 59.Adamusiak AM, Stasikowska-Kanicka O, Lewandowska-Polak A, et al. Expression of arachidonate metabolism enzymes and receptors in nasal polyps of aspirin-hypersensitive asthmatics. Int Arch Allergy Immunol. 2012;157:354–362. doi: 10.1159/000329744. [DOI] [PubMed] [Google Scholar]
- 60.Szczeklik W, Sanak M, Szczeklik A. Functional effects and gender association of COX-2 gene polymorphism G-765C in bronchial asthma. J Allergy Clin Immunol. 2004;114:248–253. doi: 10.1016/j.jaci.2004.05.030. [DOI] [PubMed] [Google Scholar]
- 61.Kim SH, Choi JH, Holloway JW, et al. Leukotriene-related gene polymorphisms in patients with aspirin-intolerant urticaria and aspirin-intolerant asthma: differing contributions of ALOX5 polymorphism in Korean population. J Korean Med Sci. 2005;20:926–931. doi: 10.3346/jkms.2005.20.6.926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cornejo-Garcia JA, Jagemann LR, Blanca-Lopez N, et al. Genetic variants of the arachidonic acid pathway in non-steroidal anti-inflammatory drug-induced acute urticaria. Clin Exp Allergy. 2012;42:1772–1781. doi: 10.1111/j.1365-2222.2012.04078.x. [DOI] [PubMed] [Google Scholar]
- 63.Lee HY, Kim SH, Ye YM, Choi GS, Park HS. Lack of association of ALOX12 and ALOX15 polymorphisms with aspirin-exacerbated respiratory disease in Korean patients. Ann Allergy Asthma Immunol. 2009;103:84–86. doi: 10.1016/S1081-1206(10)60152-4. [DOI] [PubMed] [Google Scholar]
- 64.Palikhe NS, Kim SH, Lee HY, Kim JH, Ye YM, Park HS. Association of thromboxane A2 receptor (TBXA2R) gene polymorphism in patients with aspirin-intolerant acute urticaria. Clin Exp Allergy. 2011;41:179–185. doi: 10.1111/j.1365-2222.2010.03642.x. [DOI] [PubMed] [Google Scholar]
- 65.Kim SH, Kim YK, Park HW, et al. Association between polymorphisms in prostanoid receptor genes and aspirin-intolerant asthma. Pharmacogenet Genomics. 2007;17:295–304. doi: 10.1097/01.fpc.0000239977.61841.fe. [DOI] [PubMed] [Google Scholar]
- 66.Kim SH, Choi JH, Park HS, et al. Association of thromboxane A2 receptor gene polymorphism with the phenotype of acetyl salicylic acid-intolerant asthma. Clin Exp Allergy. 2005;35:585–590. doi: 10.1111/j.1365-2222.2005.02220.x. [DOI] [PubMed] [Google Scholar]
- 67.Oh SH, Kim YH, Park SM, et al. Association analysis of thromboxane A synthase 1 gene polymorphisms with aspirin intolerance in asthmatic patients. Pharmacogenomics. 2011;12:351–363. doi: 10.2217/pgs.10.181. [DOI] [PubMed] [Google Scholar]
- 68.Vidal C, Porras-Hurtado L, Cruz R, et al. Association of thromboxane A1 synthase (TBXAS1) gene polymorphism with acute urticaria induced by nonsteroidal anti-inflammatory drugs. J Allergy Clin Immunol. 2013;132:989–991. doi: 10.1016/j.jaci.2013.04.045. [DOI] [PubMed] [Google Scholar]
- 69.Palikhe NS, Sin HJ, Kim SH, et al. Genetic variability of prostaglandin E2 receptor subtype EP4 gene in aspirin-intolerant chronic urticaria. J Hum Genet. 2012;57:494–499. doi: 10.1038/jhg.2012.55. [DOI] [PubMed] [Google Scholar]
- 70.Park BL, Park SM, Park JS, et al. Association of PTGER gene family polymorphisms with aspirin intolerant asthma in Korean asthmatics. BMB Rep. 2010;43:445–449. doi: 10.5483/bmbrep.2010.43.6.445. [DOI] [PubMed] [Google Scholar]
- 71.Kawakami T, Kashiwakura J, Kawakami Y. Histamine-releasing factor and immunoglobulins in asthma and allergy. Allergy Asthma Immunol Res. 2014;6:6–12. doi: 10.4168/aair.2014.6.1.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Palikhe NS, Kim SH, Cho BY, Ye YM, Hur GY, Park HS. Association of three sets of high-affinity IgE receptor (FcepsilonR1) polymorphisms with aspirin-intolerant asthma. Respir Med. 2008;102:1132–1139. doi: 10.1016/j.rmed.2008.03.017. [DOI] [PubMed] [Google Scholar]
- 73.Bae JS, Kim SH, Ye YM, et al. Significant association of FcepsilonRIalpha promoter polymorphisms with aspirin-intolerant chronic urticaria. J Allergy Clin Immunol. 2007;119:449–456. doi: 10.1016/j.jaci.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 74.Yamauchi K, Sekizawa K, Suzuki H, et al. Structure and function of human histamine N-methyltransferase: critical enzyme in histamine metabolism in airway. Am J Physiol. 1994;267(3 Pt 1):L342–L349. doi: 10.1152/ajplung.1994.267.3.L342. [DOI] [PubMed] [Google Scholar]
- 75.Kim SH, Kang YM, Kim SH, et al. Histamine N-methyltransferase 939A>G polymorphism affects mRNA stability in patients with acetylsalicylic acid-intolerant chronic urticaria. Allergy. 2009;64:213–221. doi: 10.1111/j.1398-9995.2008.01795.x. [DOI] [PubMed] [Google Scholar]
- 76.Pope SM, Brandt EB, Mishra A, et al. IL-13 induces eosinophil recruitment into the lung by an IL-5- and eotaxin-dependent mechanism. J Allergy Clin Immunol. 2001;108:594–601. doi: 10.1067/mai.2001.118600. [DOI] [PubMed] [Google Scholar]
- 77.McLeod JJ, Baker B, Ryan JJ. Mast cell production and response to IL-4 and IL-13. Cytokine. 2015;75:57–61. doi: 10.1016/j.cyto.2015.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kim BS, Park SM, Uhm TG, et al. Effect of single nucleotide polymorphisms within the interleukin-4 promoter on aspirin intolerance in asthmatics and interleukin-4 promoter activity. Pharmacogenet Genomics. 2010;20:748–758. doi: 10.1097/FPC.0b013e3283402155. [DOI] [PubMed] [Google Scholar]
- 79.Palikhe NS, Kim SH, Choi GS, Ye YM, Park HS. No evidence of association between interleukin-13 gene polymorphism in aspirin intolerant chronic urticaria. Allergy Asthma Immunol Res. 2009;1:36–40. doi: 10.4168/aair.2009.1.1.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Palikhe NS, Kim SH, Cho BY, et al. IL-13 gene polymorphisms are associated with rhinosinusitis and eosinophilic inflammation in aspirin intolerant asthma. Allergy Asthma Immunol Res. 2010;2:134–140. doi: 10.4168/aair.2010.2.2.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kohyama K, Abe S, Kodaira K, et al. IL-13 and IL-17A gene polymorphisms in Japanese patients with aspirin-exacerbated respiratory disease. Ann Allergy Asthma Immunol. 2011;107:510–516. doi: 10.1016/j.anai.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 82.Choi JH, Kim MA, Park HS. An update on the pathogenesis of the upper airways in aspirin-exacerbated respiratory disease. Curr Opin Allergy Clin Immunol. 2014;14:1–6. doi: 10.1097/ACI.0000000000000021. [DOI] [PubMed] [Google Scholar]
- 83.Losol P, Kim SH, Shin YS, Ye YM, Park HS. A genetic effect of IL-5 receptor α polymorphism in patients with aspirin-exacerbated respiratory disease. Exp Mol Med. 2013;45: doi: 10.1038/emm.2013.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Choi JH, Kim SH, Cho BY, et al. Association of TNF-alpha promoter polymorphisms with aspirin-induced urticaria. J Clin Pharm Ther. 2009;34:231–238. doi: 10.1111/j.1365-2710.2008.00979.x. [DOI] [PubMed] [Google Scholar]
- 85.Szabo K, Kiricsi A, Revesz M, et al. The -308 G>A SNP of TNFA is a factor predisposing to chronic rhinosinusitis associated with nasal polyposis in aspirin-sensitive Hungarian individuals: conclusions of a genetic study with multiple stratifications. Int Immunol. 2013;25:383–388. doi: 10.1093/intimm/dxs162. [DOI] [PubMed] [Google Scholar]
- 86.Kim SH, Son JK, Yang EM, Kim JE, Park HS. A functional promoter polymorphism of the human IL18 gene is associated with aspirin-induced urticaria. Br J Dermatol. 2011;165:976–984. doi: 10.1111/j.1365-2133.2011.10467.x. [DOI] [PubMed] [Google Scholar]
- 87.Palikhe NS, Kim SH, Jin HJ, Nam YH, Park HS. Association of interleukin 10 promoter polymorphism at -819 T>C with aspirin-induced urticaria in a Korean population. Ann Allergy Asthma Immunol. 2011;107:544–546. doi: 10.1016/j.anai.2011.08.014. [DOI] [PubMed] [Google Scholar]
- 88.Park JS, Park BL, Kim MO, et al. Association of single nucleotide polymorphisms on Interleukin 17 receptor A (IL17RA) gene with aspirin hypersensitivity in asthmatics. Hum Immunol. 2013;74:598–606. doi: 10.1016/j.humimm.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 89.Bronietzki AW, Schuster M, Schmitz I. Autophagy in T-cell development, activation and differentiation. Immunol Cell Biol. 2015;93:25–34. doi: 10.1038/icb.2014.81. [DOI] [PubMed] [Google Scholar]
- 90.Dekker JW, Nizankowska E, Schmitz-Schumann M, et al. Aspirin-induced asthma and HLA-DRB1 and HLA-DPB1 genotypes. Clin Exp Allergy. 1997;27:574–577. [PubMed] [Google Scholar]
- 91.Choi JH, Lee KW, Oh HB, et al. HLA association in aspirin-intolerant asthma: DPB1*0301 as a strong marker in a Korean population. J Allergy Clin Immunol. 2004;113:562–564. doi: 10.1016/j.jaci.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 92.Kim SH, Ye YM, Lee SK, et al. Association of TNF-alpha genetic polymorphism with HLA DPB1*0301. Clin Exp Allergy. 2006;36:1247–1253. doi: 10.1111/j.1365-2222.2006.02567.x. [DOI] [PubMed] [Google Scholar]
- 93.Lympany PA, Welsh KI, Christie PE, Schmitz-Schumann M, Kemeny DM, Lee TH. An analysis with sequence-specific oligonucleotide probes of the association between aspirin-induced asthma and antigens of the HLA system. J Allergy Clin Immunol. 1993;92(1 Pt 1):114–123. doi: 10.1016/0091-6749(93)90045-h. [DOI] [PubMed] [Google Scholar]
- 94.Shin SW, Park BL, Chang H, et al. Exonic variants associated with development of aspirin exacerbated respiratory diseases. PLoS One. 2014;9: doi: 10.1371/journal.pone.0111887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Esmaeilzadeh H, Nabavi M, Amirzargar AA, et al. HLA-DRB and HLA-DQ genetic variability in patients with aspirin-exacerbated respiratory disease. Am J Rhinol Allergy. 2015;29: doi: 10.2500/ajra.2015.29.4154. [DOI] [PubMed] [Google Scholar]
- 96.Kim SH, Choi JH, Lee KW, et al. The human leucocyte antigen-DRB1*1302-DQB1*0609-DPB1*0201 haplotype may be a strong genetic marker for aspirin-induced urticaria. Clin Exp Allergy. 2005;35:339–344. doi: 10.1111/j.1365-2222.2004.02197.x. [DOI] [PubMed] [Google Scholar]
- 97.Pacor ML, Di Lorenzo G, Mansueto P, et al. Relationship between human leucocyte antigen class I and class II and chronic idiopathic urticarial associated with aspirin and/or NSAIDs hypersensitivity. Mediators Inflamm. 2006;2006:62489. doi: 10.1155/MI/2006/62489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Park TJ, Kim JH, Pasaje CF, et al. Polymorphisms of ATF6B are potentially associated with fev1 decline by aspirin provocation in asthmatics. Allergy Asthma Immunol Res. 2014;6:142–148. doi: 10.4168/aair.2014.6.2.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kohyama K, Abe S, Kodaira K, et al. Polymorphisms of the CYP2C19 gene in Japanese patients with aspirin-exacerbated respiratory disease. J Allergy Clin Immunol. 2011;128:1117–1120. doi: 10.1016/j.jaci.2011.07.013. [DOI] [PubMed] [Google Scholar]
- 100.Kim SH, Choi H, Yoon MG, Ye YM, Park HS. Dipeptidyl-peptidase 10 as a genetic biomarker for the aspirin-exacerbated respiratory disease phenotype. Ann Allergy Asthma Immunol. 2015;114:208–213. doi: 10.1016/j.anai.2014.12.003. [DOI] [PubMed] [Google Scholar]
- 101.Losol P, Palikhe NS, Lee JW, et al. Association of P2RY12 polymorphisms with eosinophil and platelet activation in patients with aspirin-exacerbated respiratory disease. Ann Allergy Asthma Immunol. 2015;114:423–424. doi: 10.1016/j.anai.2015.02.009. [DOI] [PubMed] [Google Scholar]
- 102.Cheong HS, Park SM, Kim MO, et al. Genome-wide methylation profile of nasal polyps: relation to aspirin hypersensitivity in asthmatics. Allergy. 2011;66:637–644. doi: 10.1111/j.1398-9995.2010.02514.x. [DOI] [PubMed] [Google Scholar]
- 103.Choi JH, Kim JH, Park HS. Upper airways in aspirin-exacerbated respiratory disease. Curr Opin Allergy Clin Immunol. 2015;15:21–26. doi: 10.1097/ACI.0000000000000122. [DOI] [PubMed] [Google Scholar]
- 104.Samter M, Beers RF., Jr Concerning the nature of intolerance to aspirin. J Allergy. 1967;40:281–293. doi: 10.1016/0021-8707(67)90076-7. [DOI] [PubMed] [Google Scholar]
- 105.Graefe H, Roebke C, Schafer D, Meyer JE. Aspirin sensitivity and chronic rhinosinusitis with polyps: a fatal combination. J Allergy (Cairo) 2012;2012:817910. doi: 10.1155/2012/817910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lee RU, Stevenson DD. Aspirin-exacerbated respiratory disease: evaluation and management. Allergy Asthma Immunol Res. 2011;3:3–10. doi: 10.4168/aair.2011.3.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jenkins C, Costello J, Hodge L. Systematic review of prevalence of aspirin induced asthma and its implications for clinical practice. BMJ. 2004;328:434. doi: 10.1136/bmj.328.7437.434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Baenkler HW. Salicylate intolerance: pathophysiology, clinical spectrum, diagnosis and treatment. Dtsch Arztebl Int. 2008;105:137–142. doi: 10.3238/arztebl.2008.0137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Asero R, Bavbek S, Blanca M, et al. Clinical management of patients with a history of urticaria/angioedema induced by multiple NSAIDs: an expert panel review. Int Arch Allergy Immunol. 2013;160:126–133. doi: 10.1159/000342424. [DOI] [PubMed] [Google Scholar]
- 110.Parejo PA, Garcia-Agundez JA, Cornejo-Garcia JA, et al. Association study of functional polymorphisms in genes involved in histamine homeostasis and multiple NSAID: triggered urticaria and/or angioedema and anaphylaxis in patients without pre-existing chronic urticaria (MNSAID-UA) J Allergy Clin Immunol. 2013;131(2 Suppl):AB169. [Google Scholar]
- 111.Johansson SG, Bieber T, Dahl R, et al. Revised nomenclature for allergy for global use: report of the Nomenclature Review Committee of the World Allergy Organization, October 2003. J Allergy Clin Immunol. 2004;113:832–836. doi: 10.1016/j.jaci.2003.12.591. [DOI] [PubMed] [Google Scholar]
- 112.Simons FE, Ardusso LR, Bilo MB, et al. 2012 Update: World Allergy Organization Guidelines for the assessment and management of anaphylaxis. Curr Opin Allergy Clin Immunol. 2012;12:389–399. doi: 10.1097/ACI.0b013e328355b7e4. [DOI] [PubMed] [Google Scholar]
- 113.Nizankowska-Mogilnicka E, Bochenek G, Mastalerz L, et al. EAACI/GA2LEN guideline: aspirin provocation tests for diagnosis of aspirin hypersensitivity. Allergy. 2007;62:1111–1118. doi: 10.1111/j.1398-9995.2007.01409.x. [DOI] [PubMed] [Google Scholar]
- 114.Scherer K, Brockow K, Aberer W, et al. Desensitization in delayed drug hypersensitivity reactions: an EAACI position paper of the Drug Allergy Interest Group. Allergy. 2013;68:844–852. doi: 10.1111/all.12161. [DOI] [PubMed] [Google Scholar]
- 115.Mullol J, Picado C. Rhinosinusitis and nasal polyps in aspirin-exacerbated respiratory disease. Immunol Allergy Clin North Am. 2013;33:163–176. doi: 10.1016/j.iac.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 116.Pinto JM, Mehta N, DiTineo M, Wang J, Baroody FM, Naclerio RM. A randomized, double-blind, placebo-controlled trial of anti-IgE for chronic rhinosinusitis. Rhinology. 2010;48:318–324. doi: 10.4193/Rhino09.144. [DOI] [PubMed] [Google Scholar]
- 117.Vennera Mdel C, Picado C, Mullol J, Alobid I, Bernal-Sprekelsen M. Efficacy of omalizumab in the treatment of nasal polyps. Thorax. 2011;66:824–825. doi: 10.1136/thx.2010.152835. [DOI] [PubMed] [Google Scholar]
- 118.Mesonjesi E, Ziu EP, Priftanji A, Qama D. Omalizumab for severe allergic asthma: a life changing drug? Clin Transl Allergy. 2015;5(Suppl 2):P18. [Google Scholar]
- 119.Brightling CE, Chanez P, Leigh R, et al. Efficacy and safety of tralokinumab in patients with severe uncontrolled asthma: a randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Respir Med. 2015;3:692–701. doi: 10.1016/S2213-2600(15)00197-6. [DOI] [PubMed] [Google Scholar]
- 120.Darveaux J, Busse WW. Biologics in asthma: the next step toward personalized treatment. J Allergy Clin Immunol Pract. 2015;3:152–160. doi: 10.1016/j.jaip.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Fainardi V, Pisi G, Chetta A. Mepolizumab in the treatment of severe eosinophilic asthma. Immunotherapy. 2016;8:27–34. doi: 10.2217/imt.15.102. [DOI] [PubMed] [Google Scholar]
- 122.Riechelmann H, Europaischen Akademie fur Allergie und Klinische Immunologie (EAACI) und der European Rhinologic Society (ERS) Chronic rhinosinusitis: EPOS 2012 part I. Laryngorhinootologie. 2013;92:193–201. doi: 10.1055/s-0033-1333704. [DOI] [PubMed] [Google Scholar]
- 123.Cernadas JR, Brockow K, Romano A, et al. General considerations on rapid desensitization for drug hypersensitivity: a consensus statement. Allergy. 2010;65:1357–1366. doi: 10.1111/j.1398-9995.2010.02441.x. [DOI] [PubMed] [Google Scholar]
- 124.White AA, Stevenson DD. Aspirin-exacerbated respiratory disease: update on pathogenesis and desensitization. Semin Respir Crit Care Med. 2012;33:588–594. doi: 10.1055/s-0032-1325618. [DOI] [PubMed] [Google Scholar]
- 125.Oh MS, Seong GM, Lee HS, Lim GC, Lee J. Two cases of aspirin sensitive, intractable chronic urticaria successfully controlled by aspirin desensitization. Korean J Asthma Allergy Clin Immunol. 2012;32:51–55. [Google Scholar]