

Abstract
Whereas age increases microglial inflammatory activities and impairs their ability to effectively regulate their immune response, it is unclear at what age these exaggerated responses begin. We tested the hypotheses that augmented microglial responses to inflammatory challenge are present as early as middle age and that repeated stimulation of primed microglia in vivo would reveal microglial senescence. Microglial gene expression was investigated in a mouse model of repeated systemic inflammation induced by intraperitoneal injection of bacterial lipopolysaccharide (LPS). Following LPS, microglia from middle-aged mice (9–10 mo) displayed larger increases in Tnfα, Il-6, and Il-1β gene expression compared with young adults (2 mo). Similar results were observed in the spleens of middle-aged mice, indicating that exaggeration of both central and peripheral immune responses are already evident at early middle age. Interestingly, despite greater proinflammatory responses to the first LPS challenge in the aged mice, there were no age-dependent differences in either microglia or spleen following a subsequent LPS dose, suggesting that animals at this age retain the ability to effectively control their immune response following repeated challenge. The exacerbated microglial immune response to systemic inflammation at early middle age suggests that the CNS may be vulnerable to age-dependent alterations earlier than previously appreciated.
Keywords: mouse, M1/M2 mRNA, lipopolysaccharide, flow cytometry, spleen, gene expression
neuroinflammation, contributed to by microglia, is a hallmark of many neurodegenerative disorders, stroke, and traumatic injuries (2, 5, 50, 55). Chronic or poorly controlled neuroinflammation is detrimental and accelerates neurodegeneration (43). Although microglia, central nervous system (CNS) resident innate immune cells, have many beneficial activities, mounting evidence suggests that with aging, microglia undergo morphological and physiological changes that can lead to “parainflammation” (reviewed in Refs. 22, 38, 54, 56). This proinflammatory environment may increase susceptibility to neurodegenerative disorders and/or worsen ongoing pathology in the aging CNS. Indeed, age is the most significant risk factor for many neurodegenerative disorders, including Alzheimer's disease and Parkinson's disease.
Age-related morphological changes in microglia, typically in 18–20 mo old mice, are characterized by decreased ramification and perinuclear cytoplasm hypertrophy that are suggestive of increased proinflammatory activation. Elevated expression of several proinflammatory genes also support notion that microglia may be transitioning to an activated state with aging (14, 20, 30). A recent study revealed that microglia in the aged CNS have significantly reduced IL-4/IL-13 signaling that hinders their polarization toward the anti-inflammatory M2 phenotype (27). In addition to age-induced alterations in microglial proinflammatory characteristics, their ability to provide support and protection to neurons also deteriorates with age (48). Similar microglial transformations and parainflammation found in animal models have also been observed in the aged human hippocampus, superior frontal gyrus, and postcentral gyrus (15). This postmortem evidence of parainflammation is strengthened by in vivo observations of increased microglial activation in healthy older human subjects (mean age 53 yr) as assessed by positron emission tomography (PET) using R-[11C]PK11195, a ligand that preferentially binds to activated microglia in the CNS (45).
Interestingly, the biological onset of neurodegenerative disorders usually precedes clinical symptoms by several years to several decades (24), indicating that middle age may be an especially sensitive or critical period for the development of these diseases. Thus, changes in microglia during the middle-age period, and its associated parainflammation, may be important factors in the etiology of neurodegenerative disorders. Whereas age-related alterations in microglia are well documented in animal models of aging (>18–20 mo old mice and rats), it is not clear at what age these changes begin to occur. Thus, in this study, we tested the hypotheses that: 1) microglial responses to an initial inflammatory challenge will be elevated in middle-aged mice (9–10 mo) compared with young adult mice (2 mo), and 2) microglia from middle-aged mice will display properties of senescence involving stronger proinflammatory responses to a subsequent systemic inflammatory challenge. While microglial responses to repeated inflammatory challenge have been previously tested in vitro, they have not to our knowledge been tested in vivo, or in aged animals. To test these hypotheses, we used a model of repeated systemic inflammation induced by intraperitoneal (ip) injection of bacterial lipopolysaccharide (LPS).
METHODS
Animals.
Male ICR/CD1 mice were purchased from Charles River (Wilmington, MA) and housed in Association for Assessment and Accreditation of Laboratory Animal Care-accredited facilities under standard 12 h light/dark cycles with water and food available ad libitum. To study the effect of age, we used 2 mo old (young) and 9–10 mo old (middle-aged) adult mice (19). All experiments were conducted under protocols approved by the University of Wisconsin Institutional Animal Care and Use Committee. All efforts were taken to minimize the number of animals used for experimentation while allowing the formation of statistically relevant observations.
LPS treatment.
LPS (Escherichia coli 011:B4, Sigma Chemical) was administered by intraperitoneal (ip) injection at a dose of 5 mg/kg body wt. Control animals received ip injection of 100 μl PBS. Mice were killed 3 or 24 h after LPS or PBS administration, and transcardially perfused with ice-cold PBS followed by spleen and brain dissection. Another group of mice received a second dose of LPS (5 mg/kg) 24 h after the first injection, followed by spleen and brain harvesting 3 h later.
Microglial isolation.
Microglia were isolated from brains as we have described in detail previously (36). In brief: after perfusion with PBS, brains (including brain stem and cerebellum) were dissected, weighed, and enzymatically digested using Trypsin supplemented with DNase I for 20 min. Myelin was removed by centrifugation in 30% Percoll in HBSS. Cell pellets were resuspended in IMAG buffer (PBS supplemented with 0.5% BSA and 2 mM EDTA) with PE-anti-CD11b antibodies (Miltenyi Biotech) and incubated for 10 min at 4°C. After being washed, cells were incubated with anti-PE magnetic beads for 15 min. CD11b+ cells were separated in the magnetic field using MS columns (Miltenyi Biotech). Columns in the magnetic field were washed five times with IMAG buffer. After removing columns from the magnetic field, we eluted microglia with 1 ml of IMAG buffer. Cells were resuspended in TriReagent and frozen at −80°C until further use.
RNA extraction and quantitative RT-PCR.
Total RNA was extracted from isolated microglial cells or spleen tissue using Tri-Reagent (Sigma) according to the manufacturer's protocol. RNA was reverse-transcribed to cDNA using MMLV reverse transcriptase (Invitrogen, Grand Island, NY). This was followed by quantitative PCR using SYBR Green solution (Applied Biosystems). Primer sequences are provided in Table 1. Gene expression was normalized to 18S levels and relative gene expression was determined by the ΔΔCT method (28). Gene expression was considered undetectable if the CT values were >35 cycles. Data are expressed as fold change relative to the treatment indicated in each figure.
Table 1.
Primer sequences
| Gene | Forward Primer (5′→3′) | Reverse Primer (5′→3′) |
|---|---|---|
| Arginase | AGCCAATGAAGAGCTGGCTGGT | AACTGCCAGACTGTGGTCTCCA |
| Ifnγ | TGGCATAGATGTGGAAGAAAAGAG | TGCAGGATTTTCATGTCACCAT |
| iNos | TGACGCTCGGAACTGTAGCAC | TGATGGCCGACCTGATGTT |
| Il-1β | TCAAAGTGCCAGTGAACCCC | GGTCACAGCCAGTCCTCTTAC |
| Il-10 | GCCTTATCGGAAATGATCCA | TCTCACCCAGGGAATTCAAA |
| Tlr2 | CGAGTGGTGCAAGTACGAACTG | TGGTGTTCATTATCTTGCGCAG |
| Tlr4 | GAGGCAGCAGGTGGAATTGTAT | TTCGAGGCTTTTCCATCCAA |
| Tnfα | TGTAGCCCACGTCGTAGCAA | AGGTACAACCCATCGGCTGG |
| Ym1 | AAGCTCTCCAGAAGCAATCCT | TCAGAAGAATTGCCAGACCTGT |
Flow cytometry analysis.
After transcardial perfusion with ice-cold PBS, brains were dissected and homogenized in a glass-Teflon homogenizer and filtered through 70 μm cell strainer. Cells were resuspended in IMAG buffer and stained with PE-conjugated CD11b (Miltenyi) and FITC-conjugated CD45 antibodies (BD Pharmingen) for 10 min at 4°C. This CD45 antibody (30-F11 clone) reacts with all CD45 isoforms arising due to alternative splicing of exons 4, 5, and 6 (designated A, B, and C) and both alloantigens of CD45. After being washed, cells were fixed with 1.6% paraformaldehyde and analyzed by flow cytometry using a BD FACSCalibur. Cells were first gated based on forward-scatter and side-scatter parameters to exclude doublets and cell debris. Cells were then gated on CD11b+ immunoreactivity. CD45 expression was examined on CD11b+ gated cells. Data (n = 6–7) were analyzed with FlowJo software v.10. (TreeStar).
Statistical analysis.
Experiments were performed independently at least two times with three or four animals/group in each experiment. Results are expressed as means ± SE. Since there was no difference in basal gene expression detected at 3 and 24 h after PBS injection, both groups are combined in the “baseline” data shown on the graphs. Data were analyzed by one-way ANOVA followed by the Holm-Sidak or Fisher least significant difference post hoc test using SigmaStat software (Systat, San Jose, CA).
RESULTS
Microglial cell number and basal gene expression does not differ between young and middle-aged naïve mice.
The yield of isolated microglia quantified by trypan blue exclusion from the healthy brain was similar in young (1,589 ± 363 cells/mg tissue) and middle-aged (1,475 ± 420 cells/mg tissue) mice (Fig. 1A). Microglia expressed low levels of CD45 as determined by flow cytometry, and there were no age-related differences (Fig. 1B). We also evaluated basal gene expression of typical M1 and M2 markers. There were no age-dependent differences noted in basal gene expression (0 h, first arrow) of the M1 markers inducible nitric oxide synthase (iNos; Fig. 2A), tumor necrosis factor alpha (Tnfα; Fig. 2B), interleukin (Il)-6 (Fig. 2C), or Il-1β (Fig. 2D). Also there were no differences in basal microglial gene expression (0 h, first arrow) of the M2 markers Il-10 (Fig. 3A), arginase-1 (Fig. 3B), or Ym1 (Fig. 3C) between young and middle-aged animals. Baseline gene expression of Toll-like receptor (Tlr) 2 (Fig. 4A) and Tlr4 (Fig. 4B) were also similar between both ages.
Fig. 1.

Microglial number and expression of CD11b and CD45 after intraperitoneal lipopolysaccharide (LPS) injection. Young adult and middle-aged mice were injected with PBS or LPS (5 mg/kg ip) at times 0 and 24 h (arrows). Microglia were immunomagnetically isolated at 0, 3, and 24 h after the 1st LPS injection and 3 h after the 2nd PBS or LPS injection (24+3 h). The yield of isolated microglia are shown (A). Cells were also analyzed by flow cytometry for cell surface expression of CD11b and CD45 (B). Representative CD45 fluorescence intensity histograms (C) and CD45 mean fluorescent intensity quantification (D) are shown. 1 symbol, P < 0.05; 2 symbols, P < 0.01; 3 symbols, P < 0.001; *time point vs. baseline (0 h) in young mice; +time point vs. baseline (0 h) in middle-aged mice; #young adult vs. middle-aged mice at the indicated time point; $time point vs. all other time points in young adults; ‡time point vs. all other time points in middle-aged adults.
Fig. 2.

Microglial M1 proinflammatory gene mRNA levels after intraperitoneal LPS injection. Young adult and middle-aged mice were injected with PBS or LPS (5 mg/kg ip) at times 0 and 24 h (arrows). Microglia were immunomagnetically isolated at 0, 3, and 24 h after the 1st LPS injection and 3 h after the 2nd PBS or LPS injection (24+3 h). qRT-PCR was used to evaluate the expression of iNos (A), Tnfα (B), Il-6 (C), and Il-1β (D). 1 symbol, P < 0.05; 2 symbols, P < 0.01; 3 symbols, P < 0.001; *time point vs. baseline (0 h) in young adult mice; +time point vs. baseline (0 h) in middle-aged mice; #young adult vs. middle-aged mice at the indicated time point.
Fig. 3.
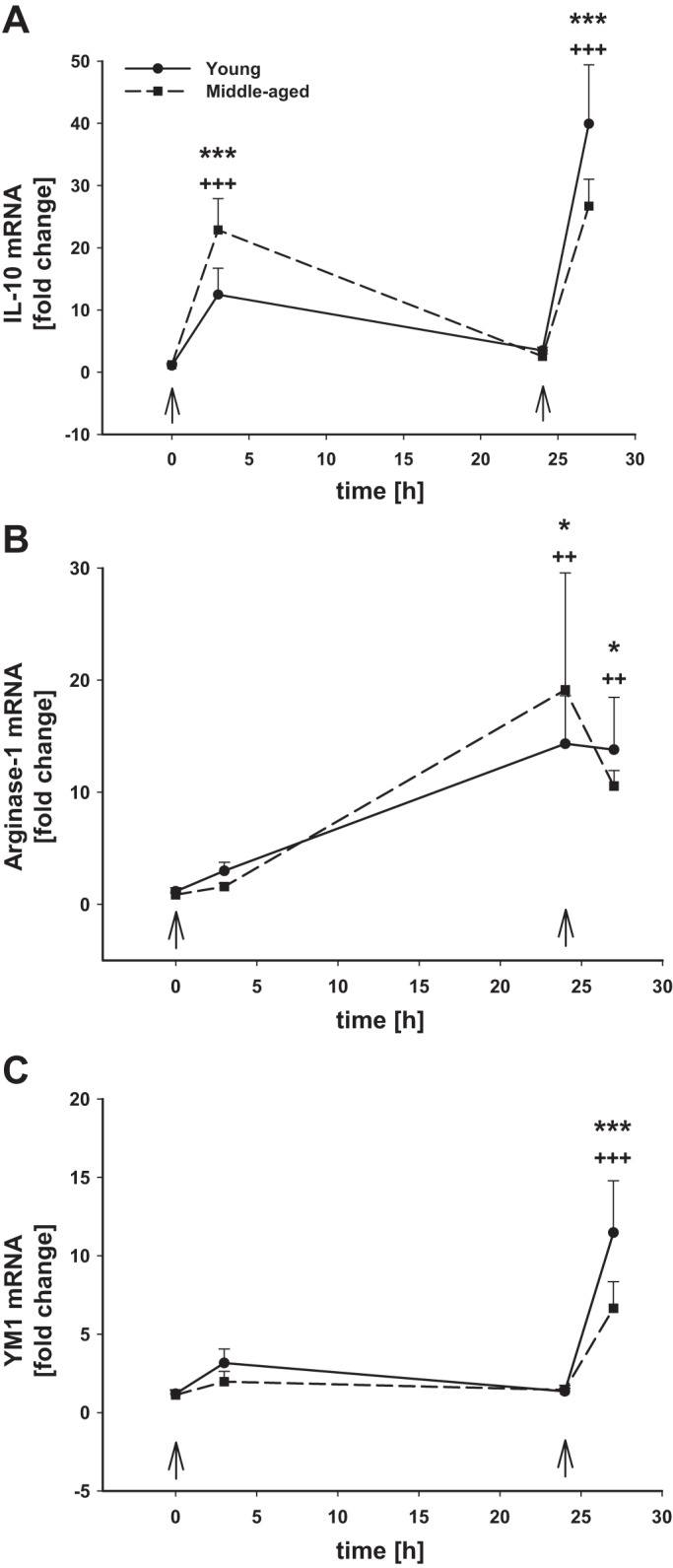
Microglial M2 anti-inflammatory gene mRNA levels after intraperitoneal LPS injection. Young adult and middle-aged mice were injected with PBS or LPS (5 mg/kg ip) at times 0 and 24 h (arrows). Microglia were immunomagnetically isolated at 0, 3, and 24 h after the 1st LPS injection and 3 h after the 2nd PBS or LPS injection (24+3 h). qRT-PCR was used to evaluate the expression of Il-10 (A), Arginase-1 (B), and Ym1 (C). 1 symbol, P < 0.05; 2 symbols, P < 0.01; 3 symbols, P < 0.001; *time point vs. baseline (0 h) in young adult mice; +time point vs. baseline (0 h) in middle-aged mice.
Fig. 4.
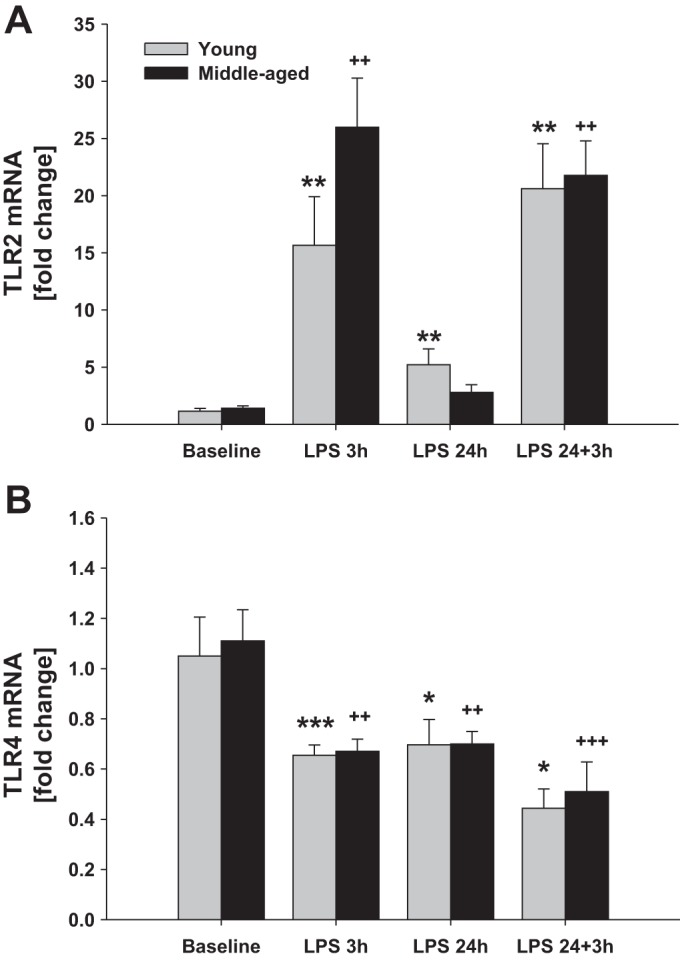
Microglial Tlr2 and Tlr4 mRNA levels after intraperitoneal LPS injection. Young adult and middle-aged mice were injected with either PBS (baseline) or LPS (5 mg/kg ip) at times 0 and 24 h. Microglia were immunomagnetically isolated at 3 and 24 h after the 1st LPS injection and 3 h after the 2nd LPS injection (24+3 h). qRT-PCR was used to evaluate the expression of Tlr2 (A) and Tlr4 (B). 1 symbol, P < 0.05; 2 symbols, P < 0.01; 3 symbols, P < 0.001; *time point vs. baseline (0 h) in young mice; +time point vs. baseline (0 h) in middle-aged mice.
Microglia from middle-aged mice exhibit stronger inflammatory responses to systemic administration of LPS.
Systemic inflammation was induced by intraperitoneal injection of LPS (5 mg/kg body wt). Microglia were examined at 3 and 24 h after injection. In young mice, the total number of microglia in the brain increased by 27% (P < 0.01) following 3 h after LPS injection and was further increased to 36% by 24 h after LPS (Fig. 1A). In middle-aged mice, microglial cell number was not statistically increased at either 3 or 24 h following LPS exposure (Fig. 1A), although there was a slight trend 17 and 9% toward an increase, respectively, at each time point. There were no differences in CD45 protein levels analyzed by flow cytometry in microglia from young and middle-aged animals, and their response to LPS was also similar (Fig. 1, B–D). The lack of significant changes in young adult microglial CD45 levels at 3 or 24 h after LPS injection suggests that macrophage influx is unlikely to be responsible for the increase in CD11b+ cell number observed in the brain at these time points.
In both young and aging mice, LPS-induced systemic inflammation resulted in significant upregulation of all M1 proinflammatory genes tested in microglia 3 h after LPS administration (Fig. 2). While there were no age-dependent differences in the magnitude of iNos upregulation (Fig. 2A), there were differences in the mRNA levels of the cytokines Tnfα (Fig. 2B), Il-6 (Fig. 2C), and Il-1β (Fig. 2D) that were significantly higher in microglia from the older animals. By 24 h post-LPS injection, the expression of all proinflammatory genes had returned close to baseline levels, although Tnfα and Il-1β still remained significantly elevated. The expression of the anti-inflammatory cytokine Il-10 followed a similar expression pattern to the proinflammatory cytokines with highest levels being observed 3 h after LPS injection; however, there was no significant difference in Il-10 mRNA levels between young and middle-aged mice (P = 0.073, n = 6–7) (Fig. 3A). By 24 h after LPS injection, Il-10 mRNA levels had returned to baseline levels in microglia from both ages. The expression of other markers of the M2 phenotype arginase-1 (Fig. 3B) and Ym1 (Fig. 3C) was not significantly changed at 3 h after LPS in microglia from mice of either age. Interestingly, however, 24 h after LPS injection when Il-10 mRNA levels were significantly downregulated from their peak at 3 h postinjection, the expression of arginase-1 was significantly upregulated by 14 and 22 times in young and aging mice, respectively, whereas Ym1 expression was not affected.
We also analyzed the expression of Tlr2 and Tlr4 in microglia from young and middle-aged mice following systemic LPS administration. Tlr2 expression did not differ by age at baseline (P = 0.438); it was upregulated baseline 15 and 5 times after 3 and 24 h of LPS, respectively, in young mice and 25 and 1.7 times in middle-aged animals (Fig. 4A). On the contrary, LPS downregulated microglial Tlr4 mRNA levels in mice of both ages relative to expression at baseline (Fig. 4B); basal Tlr4 levels did not differ by age (P = 0.787). We found that the expression of Tlr4 was decreased by 40% after 3 h of LPS injection in mice of both ages, an effect that was maintained for at least for 24 h after LPS administration.
Microglial responses to repeated systemic LPS administration are similar in young and older mice.
We hypothesized that, since the cytokine response to the first dose of LPS was greater in middle-aged microglia, repeated stimulation of these primed microglia may reveal further differences in responsiveness between young and middle-aged mice. Thus, a second dose of LPS was delivered intraperitoneally 24 h after the first injection when the expression of the proinflammatory genes measured here had almost returned to baseline levels. Microglial responses to this second LPS challenge were evaluated 3 h later. Contrary to the responses to the first LPS dose, we did not observe an increase in microglial number following the second injection of LPS in mice of either age (Fig. 1A). CD45 levels on CD11b+ cells as detected by flow cytometry were increased (Fig. 1, B–D), although there was no observable population of distinctly CD45high cells that would suggest macrophage infiltration. Rather, our data suggest an upregulation of cell surface CD45 levels by resident microglia. The M1 inflammatory gene response of microglia to the second LPS challenge was at least as strong as it was to the first LPS injection (Fig. 2, A–D), but this time we did not observe statistically significant differences with age, with the exception of Tnfα gene expression (Fig. 2B), which was higher in middle-aged microglia. Arginase-1 mRNA levels were high in both ages following the first LPS injection, and they remained upregulated after the second stimulation (Fig. 3B). Although the expression of Ym1 was unaffected by the first LPS injection, its mRNA levels were significantly increased after the second dose of LPS (Fig. 3C), and no age-dependent differences were observed.
Tlr2 (Fig. 4A) and Tlr4 (Fig. 4B) expression was not different following the second LPS challenge (LPS 24+3 h), and there was no difference observed with age. While Tlr2 mRNA levels at 24 h were either returning to or were already at baseline both ages, the second LPS challenge induced an increase in Tlr2 expression in microglia from both ages that was not different from the first LPS dose. Likewise, Tlr4 expression remained below baseline at 24 h, and the subsequent challenge with LPS did not reduce Tlr4 levels further; responses were similar in both ages.
Systemic inflammation induced by ip LPS is more severe in older mice.
Many peripheral immune cell types can make cytokine(s) that promote neuroinflammation following peripheral administration of an inflammatory stimulus such as LPS. Because many of these cell types are found in the spleen, we analyzed the expression of inflammatory cytokines in splenocytes as an indicator of the general peripheral immune response to reflect the contributions of as many individual peripheral immune cell populations as possible. Whereas basal proinflammatory gene expression in the spleen was similar in mice of both ages, with regard to Ifnγ (Fig. 5A), Il-1β (Fig. 5B), and Tnfα (Fig. 5C) mRNA levels, there were differences in the magnitude of the responses between young and aging mice. The first dose of LPS induced a strong upregulation of Ifnγ (Fig. 5A) in spleens of mice from both ages, but the response in the older mice was significantly higher than in the younger mice (70 vs. 25 times, respectively) 3 h after LPS injection. The aging animals also had significantly higher mRNA levels of Il-1β compared with the young adults (Fig. 5B), but there were no age-dependent differences in the expression of Tnfα (Fig. 5C). By 24 h post-LPS injection, all proinflammatory genes had significantly returned back to baseline levels in both ages. Interestingly, the second LPS injection did not induce expression of Ifnγ or Tnfα (Fig. 5, A and C) in the spleen like the first dose, but Il-1β mRNA was significantly upregulated in both young and middle-aged mice by ∼2-fold (Fig. 5B).
Fig. 5.
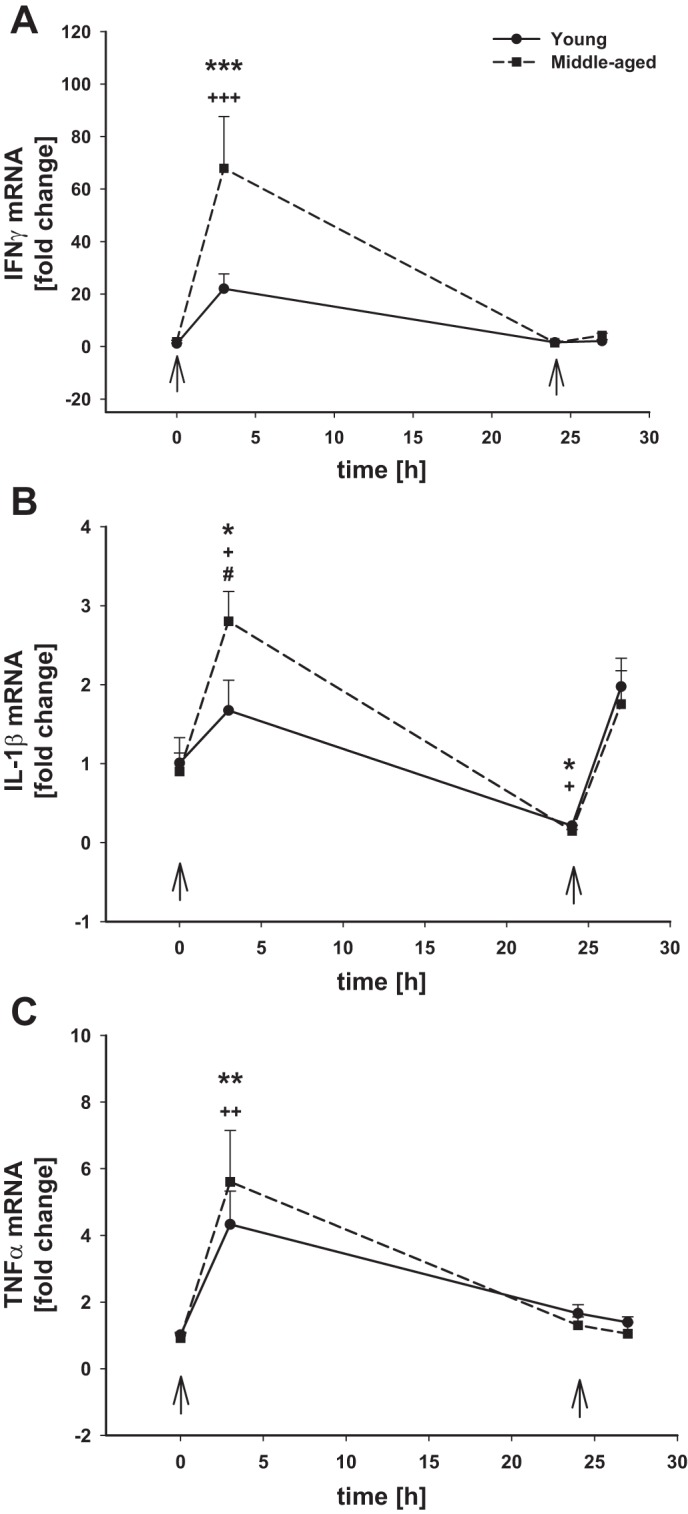
Inflammatory gene mRNA levels in the spleen after intraperitoneal LPS injection. Young adult and middle-aged mice were injected with either PBS or LPS (5 mg/kg ip) at times 0 and 24 h (arrows). Spleens were harvested at 0, 3, and 24 h after the 1st LPS injection and 3 h after a 2nd PBS or LPS injection (24+3 h). qRT-PCR was used to evaluated the expression of Ifnγ (A), Il-1β (B), and Tnfα (C). 1 symbol, P < 0.05; 3 symbols, P < 0.01; 3 symbols, P < 0.001; *time point vs. baseline (0 h) in young adult mice; +time point vs. baseline (0 h) in middle-aged mice; #P = 0.059, young adult vs. middle-aged mice at the indicated time point.
DISCUSSION
In the present study we investigated how microglial responses to acute and repeated systemic inflammation are affected by aging. Our data suggest that microglial responses to systemic inflammation experienced at critical periods beginning in middle age may have profound consequences on CNS vulnerability to neurodegenerative and other neural disorders associated with neuroinflammation and microglial activation. Microglial senescence, dystrophy, increased expression of proinflammatory cytokines, and dysregulated immune responses are evident in very old (>20 mo) mice and rats, but some transitioning to the inflammatory phenotype is already present in 12 mo old animals (6, 14, 27, 30, 38, 48, 54, 59). Here we show that some exacerbated proinflammatory responses occur as early as 9–10 mo of age and that these exaggerated responses are evident in both splenocytes and microglia, suggesting that age-dependent central and peripheral immune responses are affected earlier than previously thought.
Although there were no significant age-related differences detected in basal gene expression or in the number of microglia in the young and middle-aged adult brain, we found two major differences in microglial responses to acute systemic inflammation induced by LPS between young and middle-aged animals. First, after LPS injection, there was about a 30% increase in the number of CD11b+ cells in the CNS in young but not older mice. That CD45 levels after LPS treatment were unchanged in CD11b+ cells suggests that proliferation of resident microglia, rather than infiltration of peripheral macrophages, is likely responsible for this increase in microglial cell number. Since an increase in microglial number was not observed to the same degree in middle-aged mice following LPS treatment, microglia from the older mice may have a decreased proliferative capacity, but additional studies are needed to further test this idea. Second, microglia from the older mice mounted a significantly stronger inflammatory response to the first peripheral LPS challenge than microglia from the younger mice. But by 24 h after a single LPS injection, the expression of all proinflammatory genes examined was significantly downregulated in both young and middle-aged mice, suggesting that the inflammatory response was transient and well regulated in animals of both ages. Tnfα and Il-1β mRNA levels appeared to remain higher than baseline in the middle-aged mice, perhaps suggesting that the age related changes in the regulation of neuroinflammation are already beginning.
Because microglial inflammatory responses in the aging brain are prolonged and often dysregulated (27, 38), we were interested in whether priming microglia with acute systemic inflammation might reveal possible age-dependent differences in their responses to subsequent activation with a second dose of LPS. Repeated stimulation of microglia with LPS has been done in immortalized BV2 cells, and a senescent phenotype involving heterochromatic foci and replication arrest was observed (58); proinflammatory genes were not assessed. However, in primary microglia (neonatal-derived), LPS induction of TNF-α and iNOS was successively decreased after each LPS challenge (1). Our observations here in adult microglia in vivo indicate that microglial proinflammatory responses to the second LPS challenge are just as strong as the first. Moreover, we found that microglia from both young and middle-aged mice mounted equally strong proinflammatory responses to a second LPS challenge. Some differences were noted in the expression of the M2a markers arginase-1 and Ym1 in response to the first and the second LPS injections in mice of both ages; they were higher after the second dose than the first. In macrophages, this phenotype is associated with tissue repair and growth stimulation (18). The upregulation of these M2a molecules in microglia after the second LPS dose suggests that microglia may be polarizing toward the anti-inflammatory phenotype, perhaps to promote tissue healing after repeated inflammatory episodes. Interestingly, the Il-10 response in young and middle-aged adult microglia were not different after either LPS dose, suggesting that the exacerbated IL-10 signaling that has been reported in geriatric mice (18–20 mo) (23) does not yet occur in the middle-aged (9–10 mo old) animals used here despite the disparities in some proinflammatory cytokines that are already apparent at this age. These data suggest dichotomous regulation of pro- and anti-inflammatory cytokines during aging.
The LPS model of systemic inflammation used here is a common inflammatory stimulus used in rodent neuroimmune aging research, and our results are readily comparable with existing literature from older aged animals. Also important, LPS treatment induces a similar profile of inflammatory markers and mirrors the exaggerated microglial responses observed during aging in other models of neuroinflammation that are induced, for example, by beta amyloid overexpression (25, 29, 32) or ischemia (26, 31, 40). Lastly, LPS is clinically relevant in its own right since gram-negative bacterial infections are the most common in geriatric populations (16, 34, 35). The present results using this model of inflammation provide important information for future mechanistic studies aimed at elucidating the relationship between chronic inflammation and altered neural immune system function with age.
Sex is a risk factor for many neurodegenerative disorders (41, 42, 51). We have previously shown that basal microglial expression of the cytokines evaluated here did not differ between males and females at similar ages (4 and 12 mo) (14), and our unpublished observations in independent male and female primary microglial cultures indicate that LPS responsiveness of the genes assessed here does not differ by sex. However, in light of the present results demonstrating that exaggerated microglial responses to inflammation are already evident at middle age, it will be necessary to assess females in future studies, to determine if sexually dimorphic interactions are revealed when age and systemic inflammation are combined in vivo.
Another interesting observation in our study is the downregulation of microglial expression of Tlr4 and the upregulation of Tlr2 after systemic administration of LPS. We found that Tlr2 mRNA was upregulated in microglia from both young and middle-aged mice. This is consistent with previous observations in microglia from geriatric (20–24 mo) BALB/c mice after systemic LPS administration (23); effects on Tlr4 were not presented. Exposure of macrophage or microglial cultures to LPS (which strongly binds TLR4 and weakly binds TLR2) downregulates Tlr4 and upregulates Tlr2 expression (12, 57). However, the changes we observed in microglial Tlr expression due to systemic LPS delivery in vivo are likely mediated by mechanisms independent of TLR4-LPS binding because peripherally administered LPS does not cross the blood brain barrier (BBB) (4, 47).
Whereas LPS does not directly cross the BBB, it can increase BBB permeability (4, 47). In addition, BBB epithelial cells can directly respond to LPS by producing large amounts of cytokines such as IL-1β that can be released into the CNS to propagate neuroinflammation (47, 52). We found that LPS injection resulted in a transient upregulation of the proinflammatory genes Ifnγ, Tnfα, and Il-1β in the spleen, surrogate measures of peripheral immune responses, which were higher in older animals, suggesting that LPS may also cause greater systemic inflammation in middle-aged animals like it does in the CNS. IFNγ is produced primarily by NK cells and Th1-polarizing CD4 and CD8 T cells that are important cellular activators of macrophages (44). IL-1β and TNF-α can be produced by many innate immune cell types, but macrophages are a major source of these inflammatory cytokines (49). Interestingly, the expression levels of Ifnγ and Tnfα were not upregulated in the spleen following the second LPS challenge like they were after the first; Il-1β was increased equally in mice of both ages after the second LPS dose.
It is interesting to consider that IL-1β may play an important role in microglial responsiveness to systemic inflammation since microglial responses in the CNS followed Il-1β expression in the spleen after both doses of LPS. IL-1β, associated with the pathology of many neurodegenerative diseases, can traverse the BBB (3) and propagate inflammatory responses within the CNS (13, 46). Of note, it has been suggested that there is an amplification of the immune response within the aged CNS that is independent of the peripheral immune system (39). Young adult inbred BALB/c mice (2–6 mo) had greater circulating levels of IL-1β than old mice (20–24 mo) following stimulation with systemic LPS, whereas brain IL-1β levels were higher in the geriatric mice (21). While our data also suggest an amplification of microglial activities (including Il-1β) in middle-aged mice, Il-1β mRNA levels in the spleen and microglia were similarly elevated. Reasons for this difference may involve age, as the mice in our study are considerably younger (9–10 mo old) than the geriatric mice used in the above study. Furthermore, the mice in our study were an outbred strain (ICR/CD1), so there may be also be strain-dependent differences in the interactions between the peripheral and central immune systems; we have previously reported important differences in microglial responses between inbred and outbred mouse strains (37).
Systemic inflammation is associated with many conditions such as atherosclerosis, obesity, metabolic syndrome, Type II diabetes, and others. Several epidemiologic studies suggest that populations affected by these diseases have significantly higher risks for neurodegenerative disorders (8, 10, 33), implying an important link between systemic inflammation and neurodegeneration. Indeed systemic inflammation induces cognitive dysfunction, memory disruption, neurodegeneration, and neuroinflammation and exacerbates neurodegenerative disorders such as Parkinson's and Alzheimer's diseases, multiple sclerosis, and amyotrophic lateral sclerosis (7, 9, 11, 17, 53). Understanding how interactions between the peripheral immune system and microglia are influenced by age is critical if we are to begin to identify novel targets to manipulate microglial activities during neurodegenerative disease.
Conclusion
In conclusion, our results demonstrate that exaggerated microglial inflammatory responses to acute systemic inflammation are already evident at middle-age (9–10 mo) in mice. These age-related microglial responses to an initial dose of LPS mirrored age-associated responses observed in the spleen, an important component of the peripheral immune system. We did not find augmented proinflammatory responses to a subsequent LPS challenge in microglia (or splenocytes) from either age group, suggesting that microglial senescence is not yet apparent at this age. We did note, however, that while M1 proinflammatory genes displayed age-dependent differences to the first LPS challenge, anti-inflammatory M2 genes did not, suggesting a previously unreported disparity in the effects of aging on M1 and M2 microglial gene expression. More studies are needed to fully understand how acute and chronic systemic inflammation, conditions that are highly prevalent in middle-aged and older populations, affect microglia in the aging CNS. We suggest that the exposure of microglia to systemic inflammation at critical periods, such as middle age, may set the stage for CNS vulnerability to neurodegeneration and other neural disorders associated with neuroinflammation.
GRANTS
This work was supported by National Institutes of Health Grants R01 HL-111598 and NS-085226 (J. J. Watters).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: M.N. and J.J.W. conception and design of research; M.N., A.L.S., and R.S.K. performed experiments; M.N., A.L.S., R.S.K., and J.J.W. analyzed data; M.N. and J.J.W. interpreted results of experiments; M.N. and J.J.W. prepared figures; M.N. and J.J.W. drafted manuscript; M.N. and J.J.W. edited and revised manuscript; M.N., A.L.S., R.S.K., and J.J.W. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Alyssa Gardner for technical assistance with real-time PCR.
REFERENCES
- 1.Ajmone-Cat MA, Nicolini A, Minghetti L. Prolonged exposure of microglia to lipopolysaccharide modifies the intracellular signaling pathways and selectively promotes prostaglandin E2 synthesis. J Neurochem 87: 1193–1203, 2003. [DOI] [PubMed] [Google Scholar]
- 2.Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O'Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T. Inflammation and Alzheimer's disease. Neurobiol Aging 21: 383–421, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Banks WA, Farr SA, La Scola ME, Morley JE. Intravenous human interleukin-1alpha impairs memory processing in mice: dependence on blood-brain barrier transport into posterior division of the septum. J Pharmacol Exp Ther 299: 536–541, 2001. [PubMed] [Google Scholar]
- 4.Banks WA, Robinson SM. Minimal penetration of lipopolysaccharide across the murine blood-brain barrier. Brain Behav Immun 24: 102–109, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barcia C, Ros CM, Ros-Bernal F, Gomez A, Annese V, Carrillo-de Sauvage MA, Yuste JE, Campuzano CM, de Pablos V, Fernandez-Villalba E, Herrero MT. Persistent phagocytic characteristics of microglia in the substantia nigra of long-term Parkinsonian macaques. J Neuroimmunol 261: 60–66, 2013. [DOI] [PubMed] [Google Scholar]
- 6.Bardou I, Brothers HM, Kaercher RM, Hopp SC, Wenk GL. Differential effects of duration and age on the consequences of neuroinflammation in the hippocampus. Neurobiol Aging 34: 2293–2301, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barrientos RM, Higgins EA, Biedenkapp JC, Sprunger DB, Wright-Hardesty KJ, Watkins LR, Rudy JW, Maier SF. Peripheral infection and aging interact to impair hippocampal memory consolidation. Neurobiol Aging 27: 723–732, 2006. [DOI] [PubMed] [Google Scholar]
- 8.Biessels GJ, Kappelle LJ. Increased risk of Alzheimer's disease in Type II diabetes: insulin resistance of the brain or insulin-induced amyloid pathology? Biochem Soc Trans 33: 1041–1044, 2005. [DOI] [PubMed] [Google Scholar]
- 9.Birnbaum G, Kotilinek L, Miller SD, Raine CS, Gao YL, Lehmann PV, Gupta RS. Heat shock proteins and experimental autoimmune encephalomyelitis. II: environmental infection and extra-neuraxial inflammation alter the course of chronic relapsing encephalomyelitis. J Neuroimmunol 90: 149–161, 1998. [DOI] [PubMed] [Google Scholar]
- 10.Casserly I, Topol E. Convergence of atherosclerosis and Alzheimer's disease: inflammation, cholesterol, and misfolded proteins. Lancet 363: 1139–1146, 2004. [DOI] [PubMed] [Google Scholar]
- 11.Chen J, Buchanan JB, Sparkman NL, Godbout JP, Freund GG, Johnson RW. Neuroinflammation and disruption in working memory in aged mice after acute stimulation of the peripheral innate immune system. Brain Behav Immun 22: 301–311, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chow JC, Young DW, Golenbock DT, Christ WJ, Gusovsky F. Toll-like receptor-4 mediates lipopolysaccharide-induced signal transduction. J Biol Chem 274: 10689–10692, 1999. [DOI] [PubMed] [Google Scholar]
- 13.Craft JM, Watterson DM, Hirsch E, Van Eldik LJ. Interleukin 1 receptor antagonist knockout mice show enhanced microglial activation and neuronal damage induced by intracerebroventricular infusion of human beta-amyloid. J Neuroinflamm 2: 15, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crain JM, Nikodemova M, Watters JJ. Microglia express distinct M1 and M2 phenotypic markers in the postnatal and adult central nervous system in male and female mice. J Neurosci Res 91: 1143–1151, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cribbs DH, Berchtold NC, Perreau V, Coleman PD, Rogers J, Tenner AJ, Cotman CW. Extensive innate immune gene activation accompanies brain aging, increasing vulnerability to cognitive decline and neurodegeneration: a microarray study. J Neuroinflamm 9: 179, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crossley KB, Peterson PK. Infections in the elderly. Clin Infect Dis 22: 209–215, 1996. [DOI] [PubMed] [Google Scholar]
- 17.Cunningham C, Campion S, Lunnon K, Murray CL, Woods JF, Deacon RM, Rawlins JN, Perry VH. Systemic inflammation induces acute behavioral and cognitive changes and accelerates neurodegenerative disease. Biol Psychiatry 65: 304–312, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.David S, Kroner A. Repertoire of microglial and macrophage responses after spinal cord injury. Nat Rev Neurosci 12: 388–399, 2011. [DOI] [PubMed] [Google Scholar]
- 19.Flurkey K, Currer J, Harrison D. The Mouse in Biomedical Research. Burlington, MA: American College of Laboratory Animal Medicine, Elsevier, 2007. [Google Scholar]
- 20.Frank MG, Barrientos RM, Biedenkapp JC, Rudy JW, Watkins LR, Maier SF. mRNA up-regulation of MHC II and pivotal pro-inflammatory genes in normal brain aging. Neurobiol Aging 27: 717–722, 2006. [DOI] [PubMed] [Google Scholar]
- 21.Godbout JP, Chen J, Abraham J, Richwine AF, Berg BM, Kelley KW, Johnson RW. Exaggerated neuroinflammation and sickness behavior in aged mice after activation of the peripheral innate immune system. FASEB J 19: 1329–1331, 2005. [DOI] [PubMed] [Google Scholar]
- 22.Harry GJ. Microglia during development and aging. Pharmacol Ther 139: 313–326, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Henry CJ, Huang Y, Wynne AM, Godbout JP. Peripheral lipopolysaccharide (LPS) challenge promotes microglial hyperactivity in aged mice that is associated with exaggerated induction of both pro-inflammatory IL-1β and anti-inflammatory IL-10 cytokines. Brain Behav Immun 23: 309–317, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jack CR Jr, Holtzman DM. Biomarker modeling of Alzheimer's disease. Neuron 80: 1347–1358, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jimenez S, Baglietto-Vargas D, Caballero C, Moreno-Gonzalez I, Torres M, Sanchez-Varo R, Ruano D, Vizuete M, Gutierrez A, Vitorica J. Inflammatory response in the hippocampus of PS1M146L/APP751SL mouse model of Alzheimer's disease: age-dependent switch in the microglial phenotype from alternative to classic. J Neurosci 28: 11650–11661, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee CH, Yoo KY, Choi JH, Park OK, Hwang IK, Kim SK, Kang IJ, Kim YM, Won MH. Neuronal damage is much delayed and microgliosis is more severe in the aged hippocampus induced by transient cerebral ischemia compared to the adult hippocampus. J Neurol Sci 294: 1–6, 2010. [DOI] [PubMed] [Google Scholar]
- 27.Lee DC, Ruiz CR, Lebson L, Selenica ML, Rizer J, Hunt JB Jr, Rojiani R, Reid P, Kammath S, Nash K, Dickey CA, Gordon M, Morgan D. Aging enhances classical activation but mitigates alternative activation in the central nervous system. Neurobiol Aging 34: 1610–1620, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2[-Delta Delta C(T)] method. Methods 25: 402–408, 2001. [DOI] [PubMed] [Google Scholar]
- 29.Lopez-Gonzalez I, Schlüter A, Aso E, Garcia-Esparcia P, Ansoleaga B, Llorens F, Carmona M, Moreno J, Fuso A, Portero-Otin M, Pamplona R, Pujol A, Ferrer I. Neuroinflammatory signals in Alzheimer disease and APP/PS1 transgenic mice: correlations with plaques, tangles, and oligomeric species. J Neuropathol Exp Neurol 74: 319–344, 2015. [DOI] [PubMed] [Google Scholar]
- 30.Ma W, Cojocaru R, Gotoh N, Gieser L, Villasmil R, Cogliati T, Swaroop A, Wong WT. Gene expression changes in aging retinal microglia: relationship to microglial support functions and regulation of activation. Neurobiol Aging 34: 2310–2321, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Manwani B, Liu F, Scranton V, Hammond MD, Sansing LH, McCullough LD. Differential effects of aging and sex on stroke induced inflammation across the lifespan. Exp Neurol 249: 120–131, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Minogue AM, Jones RS, Kelly RJ, McDonald CL, Connor TJ, Lynch MA. Age-associated dysregulation of microglial activation is coupled with enhanced blood-brain barrier permeability and pathology in APP/PS1 mice. Neurobiol Aging 35: 1442–1452, 2014. [DOI] [PubMed] [Google Scholar]
- 33.Misiak B, Leszek J, Kiejna A. Metabolic syndrome, mild cognitive impairment and Alzheimer's disease–the emerging role of systemic low-grade inflammation and adiposity. Brain Res Bull 89: 144–149, 2012. [DOI] [PubMed] [Google Scholar]
- 34.Mody L, Krein SL, Saint S, Min LC, Montoya A, Lansing B, McNamara SE, Symons K, Fisch J, Koo E, Rye RA, Galecki A, Kabeto MU, Fitzgerald JT, Olmsted RN, Kauffman CA, Bradley SF. A targeted infection prevention intervention in nursing home residents with indwelling devices: a randomized clinical trial. JAMA Int Med 175: 714–723, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Niederman MS, Bass JB Jr, Campbell GD, Fein AM, Grossman RF, Mandell LA, Marrie TJ, Sarosi GA, Torres A, Yu VL. Guidelines for the initial management of adults with community-acquired pneumonia: diagnosis, assessment of severity, and initial antimicrobial therapy. American Thoracic Society Medical Section of the American Lung Association. Am Rev Respir Dis 148: 1418–1426, 1993. [DOI] [PubMed] [Google Scholar]
- 36.Nikodemova M, Watters JJ. Efficient isolation of live microglia with preserved phenotypes from adult mouse brain. J Neuroinflammation 9: 147, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nikodemova M, Watters JJ. Outbred ICR/CD1 mice display more severe neuroinflammation mediated by microglial TLR4/CD14 activation than inbred C57Bl/6 mice. Neuroscience 190: 67–74, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Norden DM, Godbout JP. Review: microglia of the aged brain: primed to be activated and resistant to regulation. Neuropathol Appl Neurobiol 39: 19–34, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Norden DM, Muccigrosso MM, Godbout JP. Microglial priming and enhanced reactivity to secondary insult in aging, and traumatic CNS injury, and neurodegenerative disease. Neuropharmacology 96 A: 29–41, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Popa-Wagner A, Badan I, Walker L, Groppa S, Patrana N, Kessler C. Accelerated infarct development, cytogenesis and apoptosis following transient cerebral ischemia in aged rats. Acta Neuropathol (Berl) 113: 277–293, 2007. [DOI] [PubMed] [Google Scholar]
- 41.Pringsheim T, Jette N, Frolkis A, Steeves TD. The prevalence of Parkinson's disease: a systematic review and meta-analysis. Mov Disord 29: 1583–1590, 2014. [DOI] [PubMed] [Google Scholar]
- 42.Ramien C, Taenzer A, Lupu A, Heckmann N, Engler JB, Patas K, Friese MA, Gold SM. Sex effects on inflammatory and neurodegenerative processes in multiple sclerosis. Neurosci Biobehav Rev [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 43.Rao JS, Kellom M, Kim HW, Rapoport SI, Reese EA. Neuroinflammation and synaptic loss. Neurochem Res 37: 903–910, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schoenborn JR, Wilson CB. Regulation of interferon-gamma during innate and adaptive immune responses. Adv Immunol 96: 41–101, 2007. [DOI] [PubMed] [Google Scholar]
- 45.Schuitemaker A, van der Doef TF, Boellaard R, van der Flier WM, Yaqub M, Windhorst AD, Barkhof F, Jonker C, Kloet RW, Lammertsma AA, Scheltens P, van Berckel BN. Microglial activation in healthy aging. Neurobiol Aging 33: 1067–1072, 2012. [DOI] [PubMed] [Google Scholar]
- 46.Sheng JG, Mrak RE, Griffin WS. Glial-neuronal interactions in Alzheimer disease: progressive association of IL-1alpha+ microglia and S100beta+ astrocytes with neurofibrillary tangle stages. J Neuropathol Exp Neurol 56: 285–290, 1997. [PubMed] [Google Scholar]
- 47.Singh AK, Jiang Y. How does peripheral lipopolysaccharide induce gene expression in the brain of rats? Toxicology 201: 197–207, 2004. [DOI] [PubMed] [Google Scholar]
- 48.Streit WJ, Xue QS. Human CNS immune senescence and neurodegeneration. Curr Opin Immunol 29: 93–96, 2014. [DOI] [PubMed] [Google Scholar]
- 49.Striz I, Brabcova E, Kolesar L, Sekerkova A. Cytokine networking of innate immunity cells: a potential target of therapy. Clin Sci (Lond) 126: 593–612, 2014. [DOI] [PubMed] [Google Scholar]
- 50.Taylor RA, Sansing LH. Microglial responses after ischemic stroke and intracerebral hemorrhage. Clin Dev Immunol 2013: 746068, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ungar L, Altmann A, Greicius MD. Apolipoprotein E, gender, and Alzheimer's disease: an overlooked, but potent and promising interaction. Brain Imag Behav 8: 262–273, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Verma S, Nakaoke R, Dohgu S, Banks WA. Release of cytokines by brain endothelial cells: a polarized response to lipopolysaccharide. Brain Behav Immun 20: 449–455, 2006. [DOI] [PubMed] [Google Scholar]
- 53.Villaran RF, Espinosa-Oliva AM, Sarmiento M, De Pablos RM, Arguelles S, Delgado-Cortes MJ, Sobrino V, Van Rooijen N, Venero JL, Herrera AJ, Cano J, Machado A. Ulcerative colitis exacerbates lipopolysaccharide-induced damage to the nigral dopaminergic system: potential risk factor in Parkinson's disease. J Neurochem 114: 1687–1700, 2010. [DOI] [PubMed] [Google Scholar]
- 54.Wong WT. Microglial aging in the healthy CNS: phenotypes, drivers, and rejuvenation. Front Cell Neurosci 7: 22, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu J, Zhao Z, Sabirzhanov B, Stoica BA, Kumar A, Luo T, Skovira J, Faden AI. Spinal cord injury causes brain inflammation associated with cognitive and affective changes: role of cell cycle pathways. J Neurosci 34: 10989–11006, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xu H, Chen M, Forrester JV. Para-inflammation in the aging retina. Prog Retin Eye Res 28: 348–368, 2009. [DOI] [PubMed] [Google Scholar]
- 57.Yang RB, Mark MR, Gray A, Huang A, Xie MH, Zhang M, Goddard A, Wood WI, Gurney AL, Godowski PJ. Toll-like receptor-2 mediates lipopolysaccharide-induced cellular signalling. Nature 395: 284–288, 1998. [DOI] [PubMed] [Google Scholar]
- 58.Yu HM, Zhao YM, Luo XG, Feng Y, Ren Y, Shang H, He ZY, Luo XM, Chen SD, Wang XY. Repeated lipopolysaccharide stimulation induces cellular senescence in BV2 cells. Neuroimmunomodulation 19: 131–136, 2012. [DOI] [PubMed] [Google Scholar]
- 59.Zhang R, Kadar T, Sirimanne E, MacGibbon A, Guan J. Age-related memory decline is associated with vascular and microglial degeneration in aged rats. Behav Brain Res 235: 210–217, 2012. [DOI] [PubMed] [Google Scholar]