

Abstract
Progressive HIV-1 infection leads to both profound immune suppression and pathologic inflammation in the majority of infected individuals. While adaptive immune dysfunction, as evidenced by CD4+ T cell depletion and exhaustion, has been extensively studied, less is known about the functional capacity of innate immune cell populations in the context of HIV-1 infection. Given the broad susceptibility to opportunistic infections and the dysregulated inflammation observed in progressive disease, we hypothesized that there would be significant changes in the innate cellular responses. Using a cohort of patients with multiple samplings before and after antiretroviral therapy (ART) initiation, we demonstrated increased responses to innate immune stimuli following viral suppression, as measured by the production of inflammatory cytokines. Plasma viral load itself had the strongest association with this change in innate functional capacity. We further identified epigenetic modifications in the TNFA promoter locus in monocytes that are associated with viremia, suggesting a molecular mechanism for the observed changes in innate immune function following initiation of ART. These data indicate that suppression of HIV-1 viremia is associated with changes in innate cellular function that may in part determine the restoration of protective immune responses.
A comprehensive characterization of innate immune function in HIV patients initiating ART shows that viral load strongly associates with changes in innate functional capacity.
Introduction
The pathogenesis of HIV-1 infection is marked by both dysregulated and pathologic immune activation and profound immune deficiency. The underlying determinants of this paradoxical state of poor control of pathogens and ongoing inflammation are incompletely understood.
There are obvious and measurable effects of HIV-1 infection on the adaptive immune system, with significant and largely nonspecific activation of T cells (1, 2), increased markers of exhaustion (3), and, ultimately, profound depletion of CD4+ T cells. Clinically, disease progression is defined by susceptibility to a diverse array of pathogens. The pathophysiology points to a broad immune deficiency, with CD4+ T cells contributing directly, but also serving as markers of further disruption to the immune system. This is consistent with the observation that the CD4 count is a less robust indicator of susceptibility to opportunistic infections in the HIV-1–uninfected population (4) and that suppression of viremia alone can ameliorate susceptibility to some infections (5, 6) and improve vaccine responses (7). Drawing from the clinical phenotype, we hypothesized that, in addition to its pleiotropic effects on adaptive immunity, HIV-1 infection leads to an upstream disruption in innate immune function that contributes to susceptibility to opportunistic infections.
The innate immune system mediates initial responses to pathogen insults and further directs expansion and recruitment of antigen-specific adaptive responses. This control is mediated by signaling through stereotyped, germline-encoded receptors (pattern recognition receptors [PRRs]), which identify pathogen-associated molecular patterns (PAMPs) that differentiate between self and foreign antigens. The receptors are both specific for molecular signals and compartmentalized to ensure a proper context of response and include TLRs (8), C-type lectin receptors (CLRs) (9), nucleotide-binding oligomerization domain–like (NOD-like) receptors (10), and RIG-I–like receptors (RLRs) (11). These receptor families are classically described to detect generic signals, without the features of pathogen-specific memory that are characteristic of adaptive immunity. However, despite the lack of clonal antigen specificity, there is clear evidence that an initial inflammatory stimulus can alter subsequent responses, leading to decreased (tolerance) or enhanced (training) cytokine production (reviewed in refs. 12, 13).
Recent work has focused attention on the role of epigenetic modifications in the delineation of altered innate phenotypes such as endotoxin (LPS) tolerance and trained immunity. Multiple mechanisms govern a concentration and context-dependent effect of LPS on the capacity of monocytes to respond to secondary stimuli (14, 15). Genomic regions in monocytes that become accessible to transcription factors after stimulation comprise a group of latent enhancers that prepare a cell for distinct secondary responses (16). Innate training, whereby monocytes are primed for more robust secondary responses (17), likewise shows a strong signature of epigenetic marks (18). Thus, while the lifespan of innate cellular populations such as monocytes is limited, it is clear that the response to a pathogen can be influenced by a distinct, preceding exposure. We hypothesized that the environment conditioned by untreated HIV-1 infection, with high levels of virus particles, an inflammatory milieu of cytokines, and products of microbial translocation, would induce analogous alterations in innate cellular function.
HIV-1 infection has several effects on innate immune pathogen recognition and activation. Primary infection leads to a robust cytokine response with induction of IFN-stimulated genetic programs and a state of immune activation (19). Both direct sensing of virus (20–23) and indirect consequences of infection (i.e., increased microbial translocation; ref. 24) contribute to this innate immune activation. Taken together, previous work has offered important insights and has clearly pointed to disruptions in the innate immune system as a foundation of HIV-1 pathogenesis. However, the precise mechanisms by which HIV-1 infection alters immune responses to secondary pathogen challenges are incompletely defined. In order to directly assess the effects of HIV-1 on innate functional capacity, we performed a longitudinal analysis in patients initiating antiretroviral therapy (ART). By sampling each individual at a range of viral loads, we sought to identify changes in innate functional capacity linked to the suppression of viremia and determine the molecular basis of these changes.
Results
ART initiation dramatically alters adaptive immune activation.
To address the question of whether there are relevant changes in immune functional status early after initiation of ART, plasma and peripheral blood mononuclear cell (PBMC) samples from HIV-1–infected individuals (n = 8), both before and after the initiation of ART, were analyzed (Table 1 and Supplemental Table 1; supplemental material available online with this article; doi:10.1172/jci.insight.85433DS1). For each patient, 3–5 time points were analyzed from the time period during which full suppression was achieved (range, 8–24 months). We first examined T cell activation, which has previously been shown to be associated with viral load and disease progression, to establish the adaptive immune activation characteristics of this cohort. CD4+ and CD8+ T cell activation was assessed by coexpression of HLA-DR and CD38, and both were highly correlated with declining viral load (Figure 1, A–C). Consistent with the short time frame for the sample collection, reconstitution of the CD4+ T cell compartment, as measured by the increase in absolute CD4+ T cell counts, was modest but was associated with viral load decline (Figure 1D). Despite minimal changes in CD4+ T cell counts, these individuals had profound changes in measures of immune activation with suppression of HIV-1 viremia.
Table 1. Demographic and clinical characteristics of the study subjects.
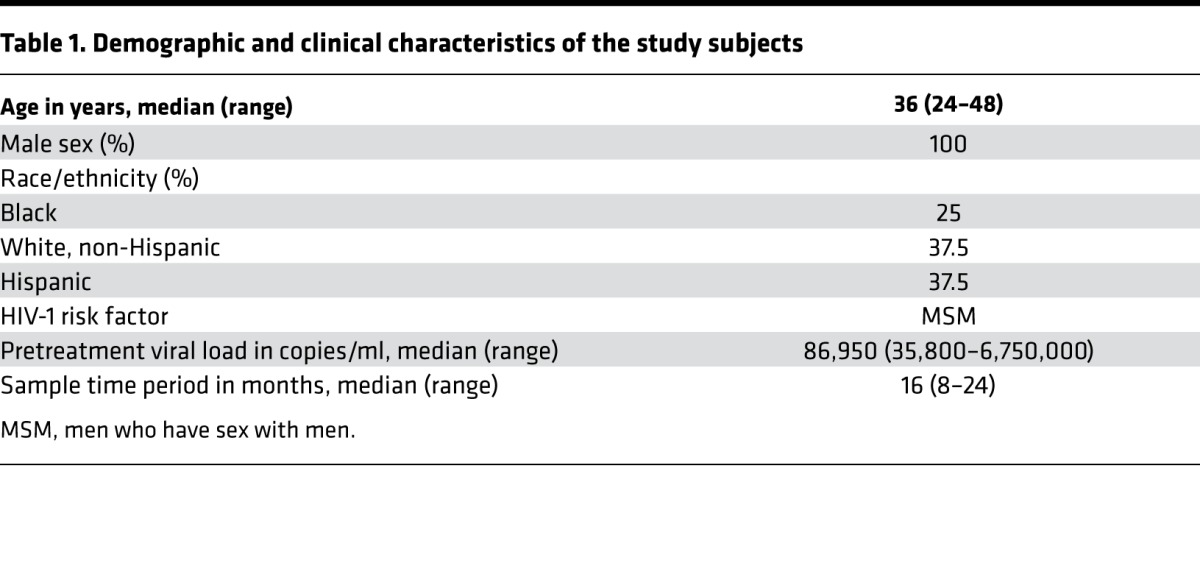
Figure 1. T cell activation correlates with declining viral load.

CD8+ (A) and CD4+ (B) T cell activation as defined by CD38 and HLA-DR coexpression correlated with declining viral load after initiation of ART. (C) Representative flow cytometric results for T cell activation with corresponding plasma viral load measures. (D) Recovery of CD4+ T cell count as related to viral load. Each color represents the measures for an individual patient over time (n = 8). r and P values for rank-based relationship, accounting for repeated measures.
Plasma viral load is associated with bulk PBMC responses to innate stimuli.
With the clear changes in adaptive immune status during the initiation of therapy as a background, we next characterized the innate immune responsiveness of PBMCs from these patients. Cryopreserved PBMCs from time points before and after viral suppression were thawed and cultured for 24 hours with agonists of TLR4 (LPS), TLR2 (heat-killed Listeria monocytogenes [HKLM]), dectin 1 (curdlan), or of the NOD pathway. Supernatants were collected and analyzed for cytokine levels via multiplex analysis. All results represent the background subtracted from the unstimulated control wells, with values below the background set to zero. By univariate analysis, there was an inverse relationship between viral load and cytokine production from PBMCs for several analytes (Table 2). All cytokines measured are shown for LPS, while for curdlan and NOD, only significant values are indicated; no significant correlations were seen for HKLM. For LPS and curdlan stimulation, TNF-α, IL-1β, and CCL20 all had negative associations with viral load at a P value threshold of 0.01, and for NOD stimulation, CCL20 and IL-6 were both inversely related to viral load (Table 2). As cytokines are biologically interrelated, we further explored these data set using a multivariate modeling approach. Partial least-squares discriminant analysis (PLSDA) was used to build a model that could correctly categorize the samples as derived from either a viremic or suppressed time point. A single canonical value consisting of weighted cytokine values could appropriately classify the samples as derived from either viremic or suppressed time points for LPS, curdlan, and NOD stimulation conditions, with a prediction accuracy of more than 80% (Supplemental Figure 1). These exploratory analyses suggested distinct patterns in cytokine production in samples from either viremic or suppressed time points, prompting further investigation
Table 2. Univariate analysis of cytokine levels in culture supernatants.
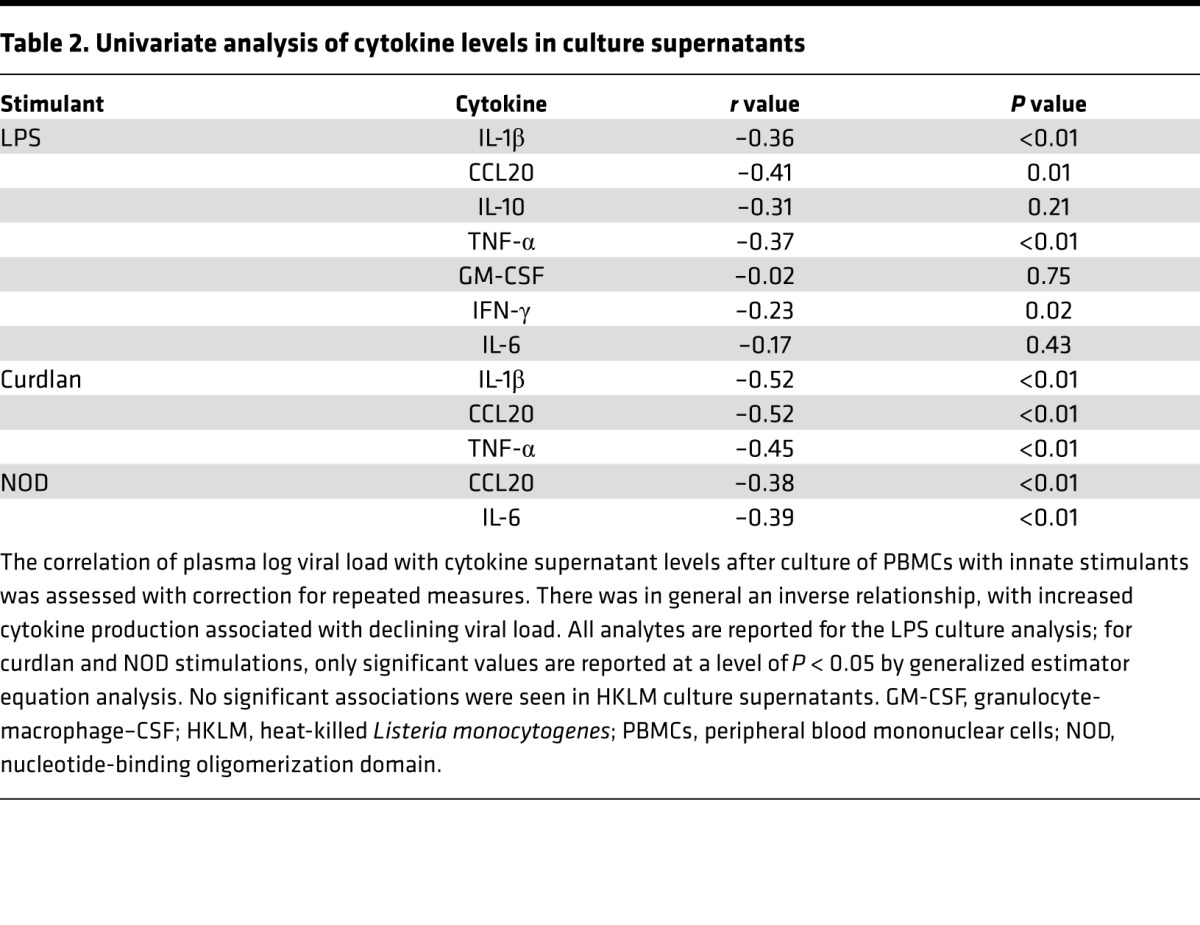
Intracellular cytokine staining identifies an inverse relationship between cytokine production and plasma viral load.
The data from the culture supernatants reflect both individual cellular responses to stimuli as well as the distribution of cellular subsets represented in a particular PBMC sample. As the percentage of specific cell populations (e.g., monocytes, T cells) may shift over time and with initiation of ART, we next sought to directly characterize the individual responses of monocyte and myeloid DC (mDC) populations.
PBMCs were cultured from the same time points and with stimulants as detailed above, but in the presence of a protein export inhibitor for 20 hours, and were then harvested for intracellular cytokine staining (Figure 2A). With this analysis, we identified significant associations between log viral load and stimulus-specific production of cytokines from both monocytes and mDCs, indicating that the differences in culture supernatants were not solely the result of different subset distributions. This relationship was strongest for LPS-induced cytokine production, highlighted by the increased TNF-α production by monocytes with a declining viral load (Figure 2, B and C). Samples from HIV-1–infected patients who did not start ART were also cultured and stained for cytokine production (IL-1β and TNF-α) as a control to assess for variation without a therapeutic intervention and did not show a pattern of change over time (Figure 2D). We next sought to determine which pattern of cytokine production was more consistent with a healthy immune status. A single measurement from the viremic subjects (first time point for ART-treated subjects, highest viral load time point for untreated subjects) was compared with a sample from a single time point following viral suppression (last time point examined) and with samples from a group of uninfected control subjects. The viremic samples had significantly less production of TNF-α from monocytes following LPS stimulation than was seen in samples at the suppressed time point or those from uninfected controls, and there was no significant difference between the suppressed and uninfected measurements (Figure 2E).
Figure 2. Cytokine production increases as viral load declines with ART.

(A) Representative intracellular cytokine staining of monocytes and mDCs following LPS stimulation for a single individual over a range of viral load measurements (n = 8 individuals total, with 35 individual time points analyzed). (B) Monocyte TNF-α production after LPS stimulation increased (left y axis, blue line) as viral load declined (right y axis, red line). (C) Monocyte TNF-α production after LPS stimulation correlated with plasma log viral load (rank-based statistics). (D) PBMCs from untreated, HIV-1–infected subjects did not show a pattern of change over time in cytokine secretion following LPS stimulation in vitro. Monocyte TNF-α production after LPS stimulation (blue line, left y axis), log viral load (right y axis, red line). (E) Comparison of monocyte TNF-α production in samples from a single viremic and a single suppressed time point per subject with monocyte TNF-α production in uninfected control subjects. *P < 0.01 and **P = 0.02, by Mann-Whitney U test. Data represent the mean and SD. ART, antiretroviral therapy; mDCs, myeloid DCs; PBMCs, peripheral blood mononuclear cells.
There were additional relationships between declining viral load and responses to other stimuli in the treated cohort (Table 3). Taken together, these data demonstrate that the magnitude of inflammatory cytokine response to ex vivo stimulation through several innate pathways increases with viral suppression. The increased response after viral suppression at the individual cell level is consistent with the increased cytokine production observed with bulk PBMC stimulation (Table 2) and is in the context of declining markers of T cell activation (Figure 1).
Table 3. Correlation of plasma log viral load with cytokine production.
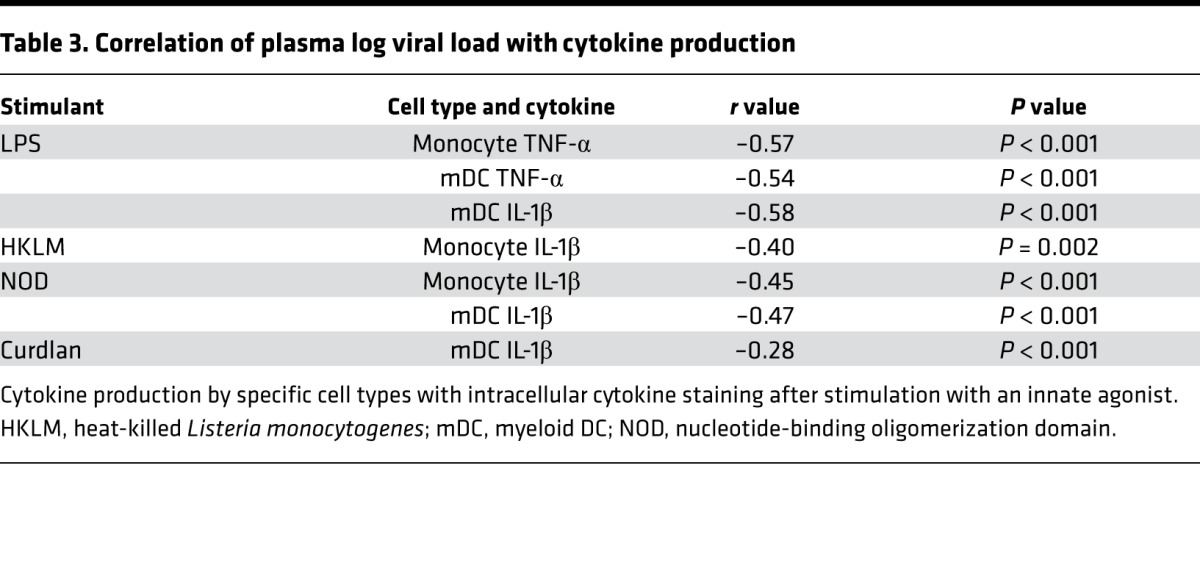
Plasma inflammatory profile and markers of microbial translocation.
We analyzed the in vivo environment from which the PBMCs were sampled to identify conditioning factors that might drive the differences in immune cell function. Specifically, plasma samples matched to the PBMCs were tested for inflammatory mediators.
We measured LPS and soluble CD14 (sCD14) levels, markers of microbial translocation; given the differences in ex vivo responses to stimulation with LPS and the known phenomenon of endotoxin tolerance, plasma levels of LPS were of particular interest. However, the plasma LPS levels showed only a weak relationship to cytokine production: plasma LPS related to LPS-stimulated production of TNF-α by monocytes, with an r value of –0.19 (P = 0.10). Of note, in this sample set, plasma LPS levels were not correlated with plasma viral load. However, sCD14 did show an association with LPS-induced TNF-α from monocytes (r = –0.28, P < 0.01), suggesting that this axis may indeed be influencing monocyte function.
In the context of the altered functional phenotype of monocytes and given the previous reports of aberrant monocyte activation in HIV-1 infection, we measured plasma sCD163 levels. CD163 is a scavenger receptor that is expressed on monocytes and macrophages and cleaved by metalloproteinases that are upregulated by inflammation, leading to increased circulating levels of the soluble ectodomain (25). sCD163 was associated with LPS-induced TNF-α (monocyte TNF-α, r = –0.23, P = 0.03) and IL-1β production (mDC IL-1β, r = –0.56, P < 0.01; monocyte IL-1β, r = –0.38, P = 0.02). sCD163 was also correlated with plasma viral load (r = 0.42, P < 0.01), consistent with high viral load conditioning an environment that drives monocyte activation.
We also measured a panel of inflammatory cytokines (IL-1β, IFN-γ, IL-10, IL-6, IL-7, MCP-1, IP-10, IFN-α2, MIP-1β, TNF-α). However, despite the relationship of IP-10 to contemporaneous viral load, no consistent pattern of correlation between innate functional capacity and plasma inflammatory cytokines was found by univariate analysis.
With the limited univariate relationships, we applied a multivariate modeling approach using the measured plasma factors to predict the innate functional responses. Specifically, partial least-squares regression (PLSR) analysis was performed to try to identify a multivariate predictor of innate function, using LPS-stimulated monocyte production of TNF-α as our outcome variable. This type of model generates a variable importance in projection (VIP) score, which indicates the variable’s contribution to the predictive composite variable. In an overall model, plasma viral load was the strongest contributor to the predictive composite variable (Figure 3). The 2 analytes with the next highest VIP scores were sCD163 and LPS. Of note, the effect of viral load was critical for modeling performance; if the viral load was excluded from analysis, the remaining analytes could not be used to build a model to predict innate function.
Figure 3. Multivariate modeling of plasma analytes identified log viral load as the most significant contributor to the prediction of innate immune function.

PLSR was performed to generate a composite variable of plasma analytes to predict innate immune responses as indicated by LPS-induced TNF-α from monocytes. Log viral load dominated the model with the highest variable importance in projection (VIP) score, followed by sCD163, LPS, and IP-10. A model without viral load included could not predict the outcome variable.
Taken together, these analyses indicate that plasma viral load is the strongest plasma predictor of the ex vivo functional responses, although there is a weaker relationship to monocyte activation (sCD163). We were not able to detect relationships with other plasma inflammatory mediators, possibly because the power to detect these associations was limited by the sample numbers.
Mechanism of impaired ex vivo cytokine production.
We further studied the monocyte response to LPS stimulation, which was strongly associated with plasma viral load (Figure 3), to determine the mechanisms of altered responses to innate stimuli following ART initiation. Monocyte subsets have been shown to have distinct cytokine secretion capacity and homing potential (26, 27) and an altered distribution in HIV-1 infection that is associated with disease progression (28, 29). Monocytes were stained for surface markers and divided into classical (CD14+16–), intermediate (CD14+16+), and nonclassical (CD14dim16+) subsets (representative flow data, Figure 4A). Further, expression of the homing markers was consistent with the subset divisions, with highest expression of CX3CR1 and lowest expression of CCR2 on the nonclassical monocytes (Figure 4A). There was no significant difference in the distribution of these subsets between viremic and suppressed PBMC samples, and the distribution of mDC subsets was comparable as well (Figure 4B and Supplemental Figure 2). Taken together, these data suggest that the altered functional profiles were not the result of a change in the pattern of subset distributions between the 2 time points analyzed.
Figure 4. Monocyte populations from viremic and suppressed samples are identical in subset distribution and NF-κB translocation but show distinct epigenetic programs.

(A) Monocyte subsets were identified by CD14 and CD16, and homing potential was assessed by CCR2 and CX3CR1. (B) No significant differences in monocyte subset distribution between viremic and suppressed time points were noted (n = 7 individuals for all). S, suppressed; V, viremic. (C) Nuclear localization of NF-κB in CD14+ monocytes was assessed by ImageStream at baseline and after LPS stimulation. Both viremic and suppressed samples showed substantial nuclear translocation after LPS stimulation, as reflected by an increase of similar magnitude in the similarity score for both samples (right panel, P > 0.05, by Mann Whitney U test). Representative images from a low (black triangle) and high (black diamond) similarity score are shown. From left to right (original magnification, ×40): bright-field image; blue = DAPI nuclear stain; green = NF-κB; yellow = CD14; merge. (D) ChIP for specific histones, followed by qPCR for regions of the TNF-α promoter identified significant enrichment of H4Ac and H3K4me3 across the promoter. Data represent the fold change in suppressed samples compared with viremic samples. Data shown are representative of 1 of 3 experiments; summary data are shown in Supplemental Figure 3. qPCR, quantitative PCR; stim, stimulation.
As defects in TLR4 responses have been linked to changes in receptor expression, adaptor protein recruitment, and transcription factor activation (12), we subsequently assessed NF-κB activation as a downstream indicator of LPS recognition and initial signaling pathways. PBMCs from viremic and suppressed time points were analyzed by ImageStream for nuclear localization of NF-κB at baseline and after stimulation with LPS in CD14+ monocytes. For each sample, a similarity score was calculated, reflecting the colocalization of staining for the nucleus and NF-κB and indicating the degree of translocation. Monocytes derived from both viremic and suppressed samples showed substantial nuclear translocation after LPS stimulation (Figure 4C). There were no significant differences between the baseline similarity scores and the change following LPS stimulation, indicating that this pathway does not account for the differences in cytokine production.
Given the differences in cytokine production, despite evidence of intact signaling as indicated by translocation of NF-κB, we sought to identify epigenetic modifications affecting promoter accessibility. Monocytes were enriched from cryopreserved PBMCs, and equal numbers of cells from each study subject were analyzed at both viremic and suppressed time points. We performed ChIP using Abs against acetylated histone 4 (H4Ac), H3K27me3, H3K4me3, and H3K9me1. Four regions in the TNFA promoter were amplified by quantitative PCR (qPCR) to assess for fold enrichment of the promoter sequences in the IP compared with input DNA. A fold change between the samples derived from viremic and suppressed time points was calculated for each promoter site. We observed marked enrichment in H4 acetylation and H3K4 trimethylation across the TNFA promoter in monocytes from the viral suppression time point (Figure 4D). In contrast, there was no significant difference in H3K9me1 or H3K27me3 at these sites and minimal changes in GAPDH between the 2 groups of samples (Figure 4D). These data were consistent across 3 independent IPs from 2 distinct sample preparations (summary data in Supplemental Figure 3). This pattern of increased H4Ac and H3K4me3 connotes an active or poised transcriptional state in monocytes derived from time points after viral suppression.
Discussion
HIV-1 infection progresses to broad susceptibility to opportunistic pathogens and dysregulated inflammation, strongly implicating an upstream disruption in innate immune function in addition to a well-characterized loss of CD4+ T cells. Consistent with this model, our data identify a substantial increase in functional responses to innate immune stimuli following viral suppression. These changes in functional capacity were most closely tied to a decline in plasma viral load, mirrored the decline in adaptive immune activation, and were related to soluble markers of monocyte activation. Despite these differences in ex vivo responses, there were no differences in monocyte subset distribution, and signaling capacity was intact on the basis of measures of NF-κB translocation. Instead, significant changes were identified in the epigenetic modifications at the TNFA promoter of monocytes derived from samples collected at viremic and suppressed time points. These finding suggest that HIV-1 infection and high viral load condition an in vivo environment that leads to suppressed innate immune responses to pathogen challenge by altering the epigenetic profile of these cells. These histone modifications, in the context of an intact LPS-signaling axis through NF-κB, provide a molecular mechanism for the differences seen in ex vivo cytokine production.
The consequences of the inflammatory milieu of HIV-1 infection for innate immune function are likely to contribute to the immune deficiency in advanced disease as well as to comorbid complications in treated disease (30, 31). Previous studies delineated changes in frequency and maturation state of plasmacytoid and mDCs and monocytes (32–35). Deficits of IFN-α production have also been described and shown to correlate with viral load and, in some cases, with clinical status (36–38). However, ex vivo functional studies have primarily relied on cross-sectional comparisons of healthy control subjects with HIV-1–infected subjects on or off ART. These studies have generated conflicting data, with reports of stimulus-specific innate responses that are blunted (35, 39–41) or preserved (42, 43) and of increased spontaneous production of cytokines (43, 44). These variations are likely due to the different signaling pathways and cell types interrogated, the different disease stages compared, and the cross-sectional study designs (45). The limited studies of the innate cellular compartment after initiation of ART showed increased capacity to prime CD4 proliferation with therapy (46) and recovery of some cytokine responses dependent on the initial virologic status (33, 47). The data presented here advance the understanding of innate immune function in HIV-1 infection and identify deficits in function that are ameliorated by ART; the longitudinal design of this study allows isolation of differences that may otherwise be difficult to identify due to interindividual variability.
The strong relationship of innate function with viral load reported here is consistent with the clinical data that demonstrate that individuals with viral suppression are less susceptible to specific opportunistic infections (5, 6) and have a lower incidence of AIDS-related morbidities even before full recovery of their CD4+ T cell counts (48). The data are also intriguing from the perspective of the immune reconstitution inflammatory syndrome (IRIS), a clinical constellation of symptoms of immune activation and inflammation without evidence of a new infectious insult (reviewed in ref. 49). The pathophysiology of this process is incompletely understood, and, of note, while a low CD4+ T cell count prior to therapy has been associated with an increased incidence of IRIS (50), the symptoms typically occur before full reconstitution of the T cell compartment,within 2 months after initiation of therapy (51). The kinetics would be consistent with a conditioning effect on the innate cellular pool that is gradually washed out with cell turnover; monocyte lifespan is typically on the order of days, but can be prolonged through induction of antiapoptotic pathways in the setting of inflammation (52). An individual on effective ART could repopulate the circulating pool of cells with monocytes that have not been conditioned by a high viral load over the course of weeks to months following initiation of ART. The data presented here suggest that such turnover leads to a population of cells with higher functional capacity and may contribute to a robust inflammatory response to pathogens.
Adding to the clinical picture described above is the recent body of work demonstrating the capacity of innate cellular reprogramming to shape subsequent immune responses. In vitro and in vivo studies in mice and humans have defined a phenomenon distinct from antigen-specific memory, whereby innate responses can be tuned to a secondary challenge. This can manifest as either a tolerized, refractory phenotype, as seen in endotoxin-induced tolerance (12, 15, 53–55), or as a priming or innate training response, as seen after exposure to Candida albicans and bacille Calmette-Guérin (17, 56). In addition to changes in receptor expression, compartmentalization, and signaling protein levels, transcriptional control of gene expression is an important mechanism of functional reprogramming (57, 58). Recent work has elegantly demonstrated that epigenetic modifications can unmask distinct enhancers that drive the production of inflammatory mediators (16) and that epigenomic differences in trained and tolerized monocytes likely underlie the phenotypic differences of these monocytes (18). Importantly, there is evidence that these phenotypes can persist over months (59) and that these core signatures can direct pharmacologic intervention points (18).
This phenomenon of an epigenetic program altering innate responses to bacterial stimuli has not been previously described in the setting of HIV-1 infection. The high incidence of bacterial sepsis as a cause of death in the HIV-1–infected population and the significant morbidity related to IRIS underscore the significance of identifying disruptions in innate immunity in order to direct interventions (58). The data presented here show that the suppression of HIV-1 viremia conditions an epigenetic pattern of poised functionality at the TNFA promoter; H4 acetylation confers enhanced chromatin accessibility (60), and H3K4me3 occupancy and transcription of TNF-α have been strongly correlated in models of innate training (17). In vitro stimulation of human alveolar macrophages with HIV-1 single-stranded RNA has previously been shown to induce similar histone modifications and promote production of TNF-α (61), but the data presented here are the first report to our knowledge of ex vivo evidence of epigenetic innate programming in the setting of HIV-1 infection. The changes reported here do not map directly to a described phenomenon of either tolerance or training, and further work will be necessary to define the extent of epigenetic changes outside the TNFA promoter region. While our study is limited by the relatively small sample size, it has the significant strengths of sampling from the complex in vivo environment of virus, microbial products, and inflammatory cytokines, along with a comprehensive characterization of each individual.
The in vivo environment of viremic HIV-1 infection is characterized by high levels of viral particles, inflammatory cytokines, and products of microbial translocation, all of which may contribute to innate cellular reprogramming. The direct causes of the epigenetic changes observed in our study are not clear, although the strong correlation between the functional profile and the viral load is striking. Direct stimulation of monocytes by viral components is a possibility, although the in vivo frequency with which the viral products access the intracellular signaling pathways (e.g., TLR7/8 and RIG-I) is unknown. Recent reports have identified the metabolic shift to increased glucose consumption and lactate production through HIF-1α as a critical step in the induction of trained immunity through β glucan (62). Of note, HIV-1 infection increases flux through the glycolytic pathways in activated CD4+ T cells, thereby enhancing virion production (63), and has been shown to activate the mTOR pathway in CD8+ T cells (61). Although speculative, this suggests that the environment induced by high-level viremia, inflammatory cytokines, and microbial products may converge on metabolic changes to alter innate function in the setting of untreated HIV-1 infection. Other downstream inflammatory mediators, including type I IFNs, may contribute to the conditioning environment, although these associations were not detected in our study, possibly due to the interindividual variability of these measures and the small number of study subjects.
Apart from their role in pathogen control, monocytes have been linked to several other pathways of immunopathogenesis in HIV-1 infection. Innate immune activation, as measured by CRP, IL-6, and sCD14 levels, and activation of the coagulation cascade, indicated by elevated D-dimer levels, have been associated with all-cause mortality in chronic, treated HIV-1 infection (64, 65). In addition, these soluble biomarkers are predictive of AIDS or death when measured prior to initiation of therapy (66). More recently, monocyte phenotypes have been shown to be associated with these biomarkers (67, 68), and, further, these monocyte phenotypes have been linked to cardiovascular disease, an important determinant of non-AIDS mortality (69). Given the clear alterations in the TNFA promoter identified here, it is reasonable to hypothesize that altered functional programs in monocytes during treated disease may contribute to pathologic expression of other inflammatory mediators. A detailed analysis of the epigenomic landscape at multiple gene loci in viremic and suppressed HIV-1 infection may offer insight into both the mechanisms of pathology in treated disease and the etiologic drivers of inflammatory markers including CRP, D-dimer, and IL-6.
In conclusion, these studies identify epigenetic modifications at the TNFA promoter in monocytes that are linked to altered functional capacity in the setting of HIV-1 viremia. This mechanism for impaired innate immunity in untreated HIV-1 infection may also contribute to dysregulated responses after initiation of ART and points to potential new targets for intervention.
Methods
Study subjects.
HIV-1–infected individuals were enrolled in the study and were followed with serial visits for phlebotomy and collection of clinical parameters. The first group of 8 subjects included patients with clinical information, cryopreserved PBMCs, and plasma from visits both before and after the initiation of ART, within a time frame of 8 to 24 months of clinical follow-up. Variable durations of time elapsed between visits, ranging from 1.5 weeks to more than 6 months after suppression, with closer sampling done during the time of viral load decline upon ART initiation. The second group included patients diagnosed with HIV infection who were not started on therapy, but had 3–4 serial samples with clinical data, along with cryopreserved PBMCs and plasma. Clinical data including plasma HIV viral load and CD4+ T cell counts were assessed using standard clinical assays. Subjects without HIV-1 infection were enrolled from 2 cohorts. The first was a cohort of healthy volunteers who underwent intermittent blood sampling, and the second was a group of men self-identified as being at high risk for HIV-1 infection, who were not taking preexposure prophylaxis. Frozen PBMCs from these 2 cohorts were used to establish a baseline monocyte response to LPS stimulation in the absence of HIV-1 infection.
T cell and monocyte characterization.
CD4 and CD8 T cell activation was measured by flow cytometry on a 4-laser LSR II (BD Biosciences). T cell populations were defined as CD14– CD3+ and either CD4+ or CD8+. Activation was quantified on the basis of the percentage of CD4+ or CD8+ cells that were double positive for CD38 and HLA-DR. The T cell activation panel included the following Abs: CD14 APC-Cy7, HLA-DR–PerCP, CD4 Pacific Blue, CD3 Alexa Fluor 700, CD8 PE-Cy7, and CD38 PE (all from BD Biosciences). The monocyte panel consisted of CD3, CD19, and CD56 — all on FITC (BD) and CD14 BUV 395, CD16 BV510, CCR2 PE, CX3CR1 APC, CD11c PEcf594, CD80 BV421, CD86 BV605, HLA-DR BV785, CD123 PECy7, and eFluor 780 fixable viability dye (BioLegend). Data were acquired on a 4 laser LSR II and analyzed with FlowJo software. Additional details are provided in Supplemental Table 2.
Bulk PBMC stimulation.
Bulk PBMC responses to innate immune stimuli were assessed by culturing thawed PBMCs with innate stimuli including LPS, a TLR4 agonist; HKLM, a TLR2 agonist; and N-muramyl glycopeptide, a NOD pathway agonist (all from Invivogen); or curdlan, a dectin 1 agonist (Sigma-Aldrich). After a 24-hour culture, supernatants were harvested and stored at –80 degrees for batched analysis. Culture supernatants were assessed for cytokine production by multiplex assay using Luminex technology (granulocyte-macrophage–CSF, MIP-3α, IFN-γ, IL-10, IL-6, IL-1β, and TNF-α) and kits from EMD Millipore.
Intracellular cytokine staining.
To assess the functional capacity of individual cell types for the production of cytokines after a stimulus, cultures of the patient-derived PBMCs with each stimulant were treated with addition of a protein export inhibitor at time zero (brefeldin A from Sigma-Aldrich, 5 μg/ml). Samples were harvested after a 20-hour culture and assessed for cytokine production by intracellular cytokine staining. Cell populations were defined as follows: all populations were gated on singlet events in the live cell gate, were negative for lineage markers (CD3, CD19, CD56), and were positive for HLA-DR. From this cell population, monocytes were identified as the CD14+ cell population, and mDCs were CD11c+. Intracellular staining for IL-1β and TNF-α was performed and the results quantified as a percentage of cells positive for cytokine production (gating strategy is shown in Supplemental Figure 4). The following Abs were used: CD3, CD19, and CD56, all on Alexa Fluor 700; CD123 PE-Cy5, CD14 APC-Cy7, and TNF-α PE-Cy7(all from BD); HLA-DR Pacific Blue and CD11c APC (Biolegend); and IL-1β PE (eBioscience). Additional details in are provided in Supplemental Table 2.
Plasma markers of innate immune activation.
Plasma samples matched to the selected PBMCs were assessed for cytokine patterns by multiplex assay using the Luminex technology. IL-1β, IFN-γ, IL-10, IL-6, and IL-7 were measured by a high-sensitivity assay, and MCP-1, IP-10, IFN-α2, MIP-1β, and TNF-α were measured with regular-sensitivity assays. All kits were from EMD Millipore, and the data were acquired on a Bio-Rad instrument. ELISA was used to determine sCD163 (Trillium Diagnostica) and sCD14 (R&D Systems) levels, and the limulus amebocyte lysate (LAL) assay (Pierce Biotechnology/Thermo Scientific) was used to assess plasma levels of LPS.
ImageStream.
NF-κB localization was assessed using ImageStream X technology (Amnis, EMD Millipore). Cryopreserved PBMCs were thawed, rested, and analyzed as either untreated or following a 1-hour stimulation with 100 ng/ml LPS. Samples were surface stained with CD14 PE (BioLegend) and fixed and permeabilized with the Foxp3/Transcription Factor Staining Buffer Set (eBioscience) for intracellular staining of NF-κB p65. Abs used included rabbit polyclonal NF-κB/p65 with secondary detection by goat anti-rabbit IgG FITC (Santa Cruz Biotechnology Inc.). DAPI (1 μg/ml) was added to each sample immediately prior to use for nuclear staining. Images were acquired from single, focused, DAPI+CD14+ cells hierarchically gated on an ImageStream X Mark II. NF-κB nuclear translocation was measured by similarity score, a log-transformed Pearson’s correlation coefficient comparing pixel values of the DAPI and NF-κB stains, using IDEAS software (Amnis, EMD Millipore).
ChIP.
Epigenetic modifications at the TNFA promoter were assessed by ChIP. Cryopreserved PBMCs were thawed from 1 viremic and 1 suppressed time point for each patient and enriched for monocytes using an AutoMACS Monocyte Isolation Kit (Miltenyi Biotec). Equal numbers of monocytes from each patient were pooled into viremic and suppressed samples. ChIP was performed using a Magna ChIP HiSens Chromatin Immunoprecipitation Kit (EMD Millipore) with the following Abs: H3K4me3, H3K9me1, H3K27me3, H4Ac, and IgG (EMD Millipore). The Ab clones are listed in Supplemental Table 3. qPCR was performed using primers for 4 regions of the TNFA promoter that have been previously described (60). Enrichment was calculated by comparison of IP samples with input DNA, and background IP was subtracted out on the basis of enrichment of nonspecific IgG. Fold change in histone occupancy was calculated between viremic and suppressed samples.
Statistics.
Univariate analysis of relationships between individual cytokines and viral load was performed using Spearman’s nonparametric correlations, estimated by a resampling procedure appropriate for repeated measurements of individuals (70); with this method, a randomly chosen value from each subject was analyzed as an independent data set. This approach was repeated several times, generating an average of the associated values as a point estimate. Statistical significance was assessed by a generalized estimating equations model applied to the corresponding ranks of the data, with a P value of less than 0.05 considered significant. Multivariate modeling was performed using PLSDA to identify patterns of cytokines produced after stimulation with innate agonists. Samples were designated as being from a viremic or suppressed (viral load <400) time point, and a canonical variable composed of weighted measures of cytokines was used to discriminate between the different viral load conditions. All input variables were mean centered and variance scaled to reduce bias introduced by measures with higher absolute values or greater variance. The misclassification rate was used as the measure of model accuracy. Robustness was assessed by testing performance on sets of data after iterative exclusion of random subsets of measures. PLSR was used to identify multivariate patterns of plasma analytes in relation to the continuous variable of monocyte TNF-α production. Again, values were mean centered and variance scaled. Model performance is reflected in the percentage of variance accounted for by the model, and the contribution of individual components to the model prediction is reflected in the VIP score. Model validation was performed with K-fold assessment; data were partitioned into K folds (subsets of data, here K = 7), and each subset was used to validate the model fit to the overall data set, fitting a total of 7 models. All analysis was done with JMP Pro software (SAS Institute).
Study approval.
Study subjects were enrolled in protocols approved by the IRB of Massachusetts General Hospital, and all subjects provided written informed consent prior to their participation in the study.
Author contributions
Experiments were conceived and designed by EPS and MA, with input from JJC, WGB, and AL. Experiments were performed by EPS, AL, and CM, and data were analyzed by EPS. The patient cohort was designed and maintained by ER, MA, and TMA. Statistical analysis was done by EPS, CDP, and RJB. The manuscript was written by EPS and MA, with review by and contributions of all the authors.
Supplementary Material
Acknowledgments
We are very grateful to the study participants and to Kim Lane, Molly Amero, Sue Bazner, and Graham McMahon for maintaining the cohort and assisting in sample selection. We thank Martin Hirsch and Christian Korner for their critical review of the manuscript. This work was supported by grants from the NIH (R01 AI 078784-01 A2, to M. Altfeld; 5 PO1 AI074415-04, to M. Altfeld and T.M. Allen; and T32 AI007387, to E.P. Scully).
Footnotes
J. Judy Chang’s present address is: Doherty Institute for Infection and Immunity, The University of Melbourne, Melbourne, Australia.
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:JCI Insight. 2016;1(3):e85433. doi:10.1172/jci.insight.85433.
Contributor Information
Ainsley Lockhart, Email: ainsley.lockhart@gmail.com.
Christine D. Palmer, Email: christine.palmer03@alumni.imperial.ac.uk.
Chelsey Musante, Email: chelseymusante@gmail.com.
Eric Rosenberg, Email: erosenberg1@partners.org.
J. Judy Chang, Email: judy.chang@unimelb.edu.au.
Ronald J. Bosch, Email: ronbosch@sdac.harvard.edu.
Marcus Altfeld, Email: marcus.altfeld@hpi.uni-hamburg.de.
References
- 1.Giorgi JV, et al. CD8+ lymphocyte activation at human immunodeficiency virus type 1 seroconversion: development of HLA-DR+ CD38– CD8+ cells is associated with subsequent stable CD4+ cell levels. J Infect Dis. 1994;170(4):775–781. doi: 10.1093/infdis/170.4.775. [DOI] [PubMed] [Google Scholar]
- 2.Doisne JM, et al. CD8+ T cells specific for EBV, cytomegalovirus, and influenza virus are activated during primary HIV infection. J Immunol. 2004;173(4):2410–2418. doi: 10.4049/jimmunol.173.4.2410. [DOI] [PubMed] [Google Scholar]
- 3.El-Far M, et al. T-cell exhaustion in HIV infection. Curr HIV/AIDS Rep. 2008;5(1):13–19. doi: 10.1007/s11904-008-0003-7. [DOI] [PubMed] [Google Scholar]
- 4.Overgaard UM, Helweg-Larsen J. Pneumocystis jiroveci pneumonia (PCP) in HIV-1-negative patients: a retrospective study 2002–2004. Scand J Infect Dis. 2007;39(6–7):589–595. doi: 10.1080/00365540601150497. [DOI] [PubMed] [Google Scholar]
- 5.Costiniuk CT, Fergusson DA, Doucette S, Angel JB. Discontinuation of Pneumocystis jirovecii pneumonia prophylaxis with CD4 count < 200 cells/microL and virologic suppression: a systematic review. PLoS One. 2011;6(12):e85433. doi: 10.1371/journal.pone.0028570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mocroft A, et al. Is it safe to discontinue primary Pneumocystis jiroveci pneumonia prophylaxis in patients with virologically suppressed HIV infection and a CD4 cell count < 200 cells/microL? Clin Infect Dis. 2010;51(5):611–619. doi: 10.1086/655761. [DOI] [PubMed] [Google Scholar]
- 7.Evison J, Farese S, Seitz M, Uehlinger DE, Furrer H, Muhlemann K. Randomized, double-blind comparative trial of subunit and virosomal influenza vaccines for immunocompromised patients. Clin Infect Dis. 2009;48(10):1402–1412. doi: 10.1086/598193. [DOI] [PubMed] [Google Scholar]
- 8.Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34(5):637–650. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 9.Osorio F, Reis e Sousa C. Myeloid C-type lectin receptors in pathogen recognition and host defense. Immunity. 2011;34(5):651–664. doi: 10.1016/j.immuni.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 10.Elinav E, Strowig T, Henao-Mejia J, Flavell RA. Regulation of the antimicrobial response by NLR proteins. Immunity. 2011;34(5):665–679. doi: 10.1016/j.immuni.2011.05.007. [DOI] [PubMed] [Google Scholar]
- 11.Loo YM, Gale M., Jr Immune signaling by RIG-I-like receptors. Immunity. 2011;34(5):680–692. doi: 10.1016/j.immuni.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Biswas SK, Lopez-Collazo E. Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends Immunol. 2009;30(10):475–487. doi: 10.1016/j.it.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 13.Netea MG, Quintin J, van der Meer JW. Trained immunity: a memory for innate host defense. Cell Host Microbe. 2011;9(5):355–361. doi: 10.1016/j.chom.2011.04.006. [DOI] [PubMed] [Google Scholar]
- 14.Kondo T, Kawai T, Akira S. Dissecting negative regulation of Toll-like receptor signaling. Trends Immunol. 2012;33(9):449–458. doi: 10.1016/j.it.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 15.Morris MC, Gilliam EA, Li L. Innate immune programing by endotoxin and its pathological consequences. Front Immunol. 2014;5:e85433. doi: 10.3389/fimmu.2014.00680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ostuni R, et al. Latent enhancers activated by stimulation in differentiated cells. Cell. 2013;152(1-2):157–171. doi: 10.1016/j.cell.2012.12.018. [DOI] [PubMed] [Google Scholar]
- 17.Quintin J, et al. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe. 2012;12(2):223–232. doi: 10.1016/j.chom.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saeed S, et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science. 2014;345(6204):e85433. doi: 10.1126/science.1251086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stacey AR, et al. Induction of a striking systemic cytokine cascade prior to peak viremia in acute human immunodeficiency virus type 1 infection, in contrast to more modest and delayed responses in acute hepatitis B and C virus infections. J Virol. 2009;83(8):3719–3733. doi: 10.1128/JVI.01844-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beignon AS, et al. Endocytosis of HIV-1 activates plasmacytoid dendritic cells via Toll-like receptor-viral RNA interactions. J Clin Invest. 2005;115(11):3265–3275. doi: 10.1172/JCI26032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heil F, et al. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. 2004;303(5663):1526–1529. doi: 10.1126/science.1093620. [DOI] [PubMed] [Google Scholar]
- 22.Meier A, et al. MyD88-dependent immune activation mediated by human immunodeficiency virus type 1-encoded Toll-like receptor ligands. J Virol. 2007;81(15):8180–8191. doi: 10.1128/JVI.00421-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tomkowicz B, Lee C, Ravyn V, Cheung R, Ptasznik A, Collman RG. The Src kinase Lyn is required for CCR5 signaling in response to MIP-1beta and R5 HIV-1 gp120 in human macrophages. Blood. 2006;108(4):1145–1150. doi: 10.1182/blood-2005-12-012815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brenchley JM, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med. 2006;12(12):1365–1371. doi: 10.1038/nm1511. [DOI] [PubMed] [Google Scholar]
- 25.Moller HJ. Soluble CD163. Scand J Clin Lab Invest. 2012;72(1):1–13. doi: 10.3109/00365513.2011.626868. [DOI] [PubMed] [Google Scholar]
- 26.Ziegler-Heitbrock L, et al. Nomenclature of monocytes and dendritic cells in blood. Blood. 2010;116(16):e74–e80. doi: 10.1182/blood-2010-02-258558. [DOI] [PubMed] [Google Scholar]
- 27.Ziegler-Heitbrock L. The CD14+ CD16+ blood monocytes: their role in infection and inflammation. J Leukoc Biol. 2007;81(3):584–592. doi: 10.1189/jlb.0806510. [DOI] [PubMed] [Google Scholar]
- 28.Thieblemont N, Weiss L, Sadeghi HM, Estcourt C, Haeffner-Cavaillon N. CD14lowCD16high: a cytokine-producing monocyte subset which expands during human immunodeficiency virus infection. Eur J Immunol. 1995;25(12):3418–3424. doi: 10.1002/eji.1830251232. [DOI] [PubMed] [Google Scholar]
- 29.Han J, et al. CD14(high)CD16(+) rather than CD14(low)CD16(+) monocytes correlate with disease progression in chronic HIV-infected patients. J Acquir Immune Defic Syndr. 2009;52(5):553–559. doi: 10.1097/QAI.0b013e3181c1d4fe. [DOI] [PubMed] [Google Scholar]
- 30.Lederman MM, Funderburg NT, Sekaly RP, Klatt NR, Hunt PW. Residual immune dysregulation syndrome in treated HIV infection. Adv Immunol. 2013;119:51–83. doi: 10.1016/B978-0-12-407707-2.00002-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hunt PW. HIV and inflammation: mechanisms and consequences. Curr HIV/AIDS Rep. 2012;9(2):139–147. doi: 10.1007/s11904-012-0118-8. [DOI] [PubMed] [Google Scholar]
- 32.Anthony DD, et al. Selective impairments in dendritic cell-associated function distinguish hepatitis C virus and HIV infection. J Immunol. 2004;172(8):4907–4916. doi: 10.4049/jimmunol.172.8.4907. [DOI] [PubMed] [Google Scholar]
- 33.Chehimi J, et al. Persistent decreases in blood plasmacytoid dendritic cell number and function despite effective highly active antiretroviral therapy and increased blood myeloid dendritic cells in HIV-infected individuals. J Immunol. 2002;168(9):4796–4801. doi: 10.4049/jimmunol.168.9.4796. [DOI] [PubMed] [Google Scholar]
- 34.Schmidt B, Fujimura SH, Martin JN, Levy JA. Variations in plasmacytoid dendritic cell (PDC) and myeloid dendritic cell (MDC) levels in HIV-infected subjects on and off antiretroviral therapy. J Clin Immunol. 2006;26(1):55–64. doi: 10.1007/s10875-006-8401-3. [DOI] [PubMed] [Google Scholar]
- 35.Tilton JC, et al. Human immunodeficiency virus viremia induces plasmacytoid dendritic cell activation in vivo and diminished alpha interferon production in vitro. J Virol. 2008;82(8):3997–4006. doi: 10.1128/JVI.01545-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Feldman S, et al. Decreased interferon-alpha production in HIV-infected patients correlates with numerical and functional deficiencies in circulating type 2 dendritic cell precursors. Clin Immunol. 2001;101(2):201–210. doi: 10.1006/clim.2001.5111. [DOI] [PubMed] [Google Scholar]
- 37.Siegal FP, Fitzgerald-Bocarsly P, Holland BK, Shodell M. Interferon-alpha generation and immune reconstitution during antiretroviral therapy for human immunodeficiency virus infection. AIDS. 2001;15(13):1603–1612. doi: 10.1097/00002030-200109070-00002. [DOI] [PubMed] [Google Scholar]
- 38.Soumelis V, et al. Depletion of circulating natural type 1 interferon-producing cells in HIV-infected AIDS patients. Blood. 2001;98(4):906–912. doi: 10.1182/blood.V98.4.906. [DOI] [PubMed] [Google Scholar]
- 39.Conry SJ, et al. Impaired plasmacytoid dendritic cell (PDC)-NK cell activity in viremic human immunodeficiency virus infection attributable to impairments in both PDC and NK cell function. J Virol. 2009;83(21):11175–11187. doi: 10.1128/JVI.00753-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jiang W, Lederman MM, Salkowitz JR, Rodriguez B, Harding CV, Sieg SF. Impaired monocyte maturation in response to CpG oligodeoxynucleotide is related to viral RNA levels in human immunodeficiency virus disease and is at least partially mediated by deficiencies in alpha/beta interferon responsiveness and production. J Virol. 2005;79(7):4109–4119. doi: 10.1128/JVI.79.7.4109-4119.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nowroozalizadeh S, et al. Studies on toll-like receptor stimuli responsiveness in HIV-1 and HIV-2 infections. Cytokine. 2009;46(3):325–331. doi: 10.1016/j.cyto.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 42.Martinson JA, et al. Dendritic cells from HIV-1 infected individuals are less responsive to toll-like receptor (TLR) ligands. Cell Immunol. 2007;250(1–2):75–84. doi: 10.1016/j.cellimm.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tilton JC, et al. Diminished production of monocyte proinflammatory cytokines during human immunodeficiency virus viremia is mediated by type I interferons. J Virol. 2006;80(23):11486–11497. doi: 10.1128/JVI.00324-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yonkers NL, Rodriguez B, Asaad R, Lederman MM, Anthony DD. Systemic immune activation in HIV infection is associated with decreased MDC responsiveness to TLR ligand and inability to activate naive CD4 T-cells. PLoS One. 2011;6(9):e85433. doi: 10.1371/journal.pone.0023884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chang JJ, et al. Differential regulation of toll-like receptor pathways in acute and chronic HIV-1 infection. AIDS. 2012;26(5):533–541. doi: 10.1097/QAD.0b013e32834f3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gompels M, Patterson S, Roberts MS, Macatonia SE, Pinching AJ, Knight SC. Increase in dendritic cell numbers, their function and the proportion uninfected during AZT therapy. Clin Exp Immunol. 1998;112(2):347–353. doi: 10.1046/j.1365-2249.1998.00590.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chehimi J, et al. Baseline viral load and immune activation determine the extent of reconstitution of innate immune effectors in HIV-1-infected subjects undergoing antiretroviral treatment. J Immunol. 2007;179(4):2642–2650. doi: 10.4049/jimmunol.179.4.2642. [DOI] [PubMed] [Google Scholar]
- 48.Lundgren JD, et al. Inferior clinical outcome of the CD4+ cell count-guided antiretroviral treatment interruption strategy in the SMART study: role of CD4+ Cell counts and HIV RNA levels during follow-up. J Infect Dis. 2008;197(8):1145–1155. doi: 10.1086/529523. [DOI] [PubMed] [Google Scholar]
- 49.French MA. HIV/AIDS: immune reconstitution inflammatory syndrome: a reappraisal. Clin Infect Dis. 2009;48(1):101–107. doi: 10.1086/595006. [DOI] [PubMed] [Google Scholar]
- 50.Muller M, Wandel S, Colebunders R, Attia S, Furrer H, Egger M. Immune reconstitution inflammatory syndrome in patients starting antiretroviral therapy for HIV infection: a systematic review and meta-analysis. Lancet Infect Dis. 2010;10(4):251–261. doi: 10.1016/S1473-3099(10)70026-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Murdoch DM, Venter WD, Van Rie A, Feldman C. Immune reconstitution inflammatory syndrome (IRIS): review of common infectious manifestations and treatment options. AIDS Res Ther. 2007;4:e85433. doi: 10.1186/1742-6405-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Parihar A, Eubank TD, Doseff AI. Monocytes and macrophages regulate immunity through dynamic networks of survival and cell death. J Innate Immun. 2010;2(3):204–215. doi: 10.1159/000296507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.de Vos AF, Pater JM, van den Pangaart PS, de Kruif MD, van ‘t Veer C, van der Poll T. In vivo lipopolysaccharide exposure of human blood leukocytes induces cross-tolerance to multiple TLR ligands. J Immunol. 2009;183(1):533–542. doi: 10.4049/jimmunol.0802189. [DOI] [PubMed] [Google Scholar]
- 54.Draisma A, Pickkers P, Bouw MP, van der Hoeven JG. Development of endotoxin tolerance in humans in vivo. Crit Care Med. 2009;37(4):1261–1267. doi: 10.1097/CCM.0b013e31819c3c67. [DOI] [PubMed] [Google Scholar]
- 55.Munoz C, Carlet J, Fitting C, Misset B, Bleriot JP, Cavaillon JM. Dysregulation of in vitro cytokine production by monocytes during sepsis. J Clin Invest. 1991;88(5):1747–1754. doi: 10.1172/JCI115493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kleinnijenhuis J, et al. Bacille Calmette-Guerin induces NOD2-dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc Natl Acad Sci U S A. 2012;109(43):17537–17542. doi: 10.1073/pnas.1202870109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hargreaves DC, Horng T, Medzhitov R. Control of inducible gene expression by signal-dependent transcriptional elongation. Cell. 2009;138(1):129–145. doi: 10.1016/j.cell.2009.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Foster SL, Hargreaves DC, Medzhitov R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature. 2007;447(7147):972–978. doi: 10.1038/nature05836. [DOI] [PubMed] [Google Scholar]
- 59.Ishii M, et al. Epigenetic regulation of the alternatively activated macrophage phenotype. Blood. 2009;114(15):3244–3254. doi: 10.1182/blood-2009-04-217620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mehta S, Jeffrey KL. Beyond receptors and signaling: epigenetic factors in the regulation of innate immunity. Immunol Cell Biol. 2015;93(3):233–244. doi: 10.1038/icb.2014.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Han X, et al. Epigenetic regulation of tumor necrosis factor alpha (TNFα) release in human macrophages by HIV-1 single-stranded RNA (ssRNA) is dependent on TLR8 signaling. J Biol Chem. 2012;287(17):13778–13786. doi: 10.1074/jbc.M112.342683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cheng SC, et al. mTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science. 2014;345(6204):e85433. doi: 10.1126/science.1250684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chignalia AZ, et al. p120-catenin expressed in alveolar type II cells is essential for the regulation of lung innate immune response. Am J Pathol. 2015;185(5):1251–1263. doi: 10.1016/j.ajpath.2015.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kuller LH, et al. Inflammatory and coagulation biomarkers and mortality in patients with HIV infection. PLoS Med. 2008;5(10):e85433. doi: 10.1371/journal.pmed.0050203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sandler NG, et al. Plasma levels of soluble CD14 independently predict mortality in HIV infection. J Infect Dis. 2011;203(6):780–790. doi: 10.1093/infdis/jiq118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Boulware DR, et al. Higher levels of CRP, D-dimer, IL-6, and hyaluronic acid before initiation of antiretroviral therapy (ART) are associated with increased risk of AIDS or death. J Infect Dis. 2011;203(11):1637–1646. doi: 10.1093/infdis/jir134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Funderburg NT, et al. Increased tissue factor expression on circulating monocytes in chronic HIV infection: relationship to in vivo coagulation and immune activation. Blood. 2010;115(2):161–167. doi: 10.1182/blood-2009-03-210179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wilson EM, et al. Monocyte-activation phenotypes are associated with biomarkers of inflammation and coagulation in chronic HIV infection. J Infect Dis. 2014;210(9):1396–1406. doi: 10.1093/infdis/jiu275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Funderburg NT, et al. Shared monocyte subset phenotypes in HIV-1 infection and in uninfected subjects with acute coronary syndrome. Blood. 2012;120(23):4599–4608. doi: 10.1182/blood-2012-05-433946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Follmann D, Proschan M, Leifer E. Multiple outputation: inference for complex clustered data by averaging analyses from independent data. Biometrics. 2003;59(2):420–429. doi: 10.1111/1541-0420.00049. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.