

Significance
This paper reports the first structure of WT-human glucose transporter 1 (hGLUT1), to our knowledge, cocrystallized with inhibitors. The structures provide a template to develop therapeutic inhibitors applicable to cancers, because cancer cells become dependent on greatly increased glucose consumption. This dependence results in up-regulation of glucose transporter expression, especially hGLUT1. The bound inhibitors include the natural compound cytochalasin B and two of a series of previously undescribed organic compounds that bind in the submicromolar range. Our results emphasize that modulation of glucose import by hGLUTs should focus on making good interaction points for compounds and that the actual chemical backbone of the inhibitor is of less importance.
Keywords: X-ray structure, glucose facilitator, human MFS transporter, cytochalasin B, GLUT inhibitor
Abstract
Cancerous cells have an acutely increased demand for energy, leading to increased levels of human glucose transporter 1 (hGLUT1). This up-regulation suggests hGLUT1 as a target for therapeutic inhibitors addressing a multitude of cancer types. Here, we present three inhibitor-bound, inward-open structures of WT-hGLUT1 crystallized with three different inhibitors: cytochalasin B, a nine-membered bicyclic ring fused to a 14-membered macrocycle, which has been described extensively in the literature of hGLUTs, and two previously undescribed Phe amide-derived inhibitors. Despite very different chemical backbones, all three compounds bind in the central cavity of the inward-open state of hGLUT1, and all binding sites overlap the glucose-binding site. The inhibitory action of the compounds was determined for hGLUT family members, hGLUT1–4, using cell-based assays, and compared with homology models for these hGLUT members. This comparison uncovered a probable basis for the observed differences in inhibition between family members. We pinpoint regions of the hGLUT proteins that can be targeted to achieve isoform selectivity, and show that these same regions are used for inhibitors with very distinct structural backbones. The inhibitor cocomplex structures of hGLUT1 provide an important structural insight for the design of more selective inhibitors for hGLUTs and hGLUT1 in particular.
Human glucose transporter 1 (hGLUT1) belongs to a family of homologous sugar transporters or cotransporters found in both prokaryotes and eukaryotes (1). It is a uniporter that transports glucose from the ECM into cells (2, 3). hGLUT1 is the ubiquitous glucose transporter in the human body and is absolutely essential for cell viability (4).
The increased demand for glucose in cancer cell lines, as well as in benign and malignant tumor cells, results in up-regulation of glucose transporter expression, especially so for hGLUT1 (5). Cancerous cells grow and multiply faster than normal cells, resulting in an acutely increased demand for energy (6). The up-regulation of hGLUT1 upon increased demand for glucose highlights it as an important prognostic indicator in several cancer types (7–10), as well as a potential new target for therapeutic inhibitors.
Previous biochemical studies have shown that cytochalasin B and forskolin inhibit hGLUT1-mediated sugar transport in RBCs by binding at or close to the hGLUT1 sugar export site (11, 12). Recent structural studies on hGLUTs have been focused toward elucidating the glucose-binding site. The apo structure of hGLUT1 was recently reported at 3.2 Å (13). It features an inward-open conformation that shows the standard canonical fold of a major facilitator superfamily transporter. It was designed with two point mutations, N45T and E329Q, to facilitate crystallization. This structure was a milestone in the field, and it serves as a blueprint for development of therapeutic agents because it marks the putative binding site for the glucose moiety.
The structure of another glucose transporter, hGLUT3, bound to the α- and β-anomers of glucose was very recently reported in an outward-occluded state (14). The glucose-binding site was predicted to be similar to the site proposed for hGLUT1, and many of the residues lining the central cavity were conserved.
Here, we report the structure determination of WT-hGLUT1 cocrystallized with three inhibitors, including cytochalasin B. Cytochalasin B is a cell-permeable mycotoxin and features a macrocyclic ring structure (15) (Fig. 1A). It has been used extensively in the literature of hGLUTs, and hGLUT1 in particular, and this work presents, for the first time to our knowledge, the binding mode of this essential tool in the field. Two inhibitors never described before in the literature, glucose transporter-inhibitor 1 (GLUT-i1) and glucose transporter-inhibitor 2 (GLUT-i2), are based on a Phe-amide core scaffold and were identified in a high-throughput screening campaign for hGLUT1 inhibitors described here (Fig. 1 A–C). The structures are all in an “inward-facing” conformation at a resolution of 2.9–3.0 Å and provide a clear model for interactions with residues that mainly belong to the glucose substrate-binding site (Fig. 1D). The crystal structures define the inhibitor-binding modes in the central ligand-binding site and highlight that very distinct inhibitors interact with the same residues in the structure. Thus, our results emphasize that inhibitor design for hGLUTs should focus on providing a good interaction point for such residues and that the actual chemical backbone of the inhibitor is of less importance.
Fig. 1.

Chemical structures of inhibitors used in this study and their binding sites in hGLUT1. Cytochalasin B (A), GLUT-i1 (B), and GLUT-i2 (C) were used for cocrystallization with WT-hGLUT1. (D) Cartoon of hGLUT1 viewed cytoplasmic side down, with bound cytochalasin B shown as red sticks in the central cavity. Helices are shown as rods labeled using the conventional numbering scheme for major facilitator superfamily transporters, and are color-coded as follows: N-terminal domain, yellow; C-terminal domain, orange; intracellular (IC) domain, green.
Results
Characterization of WT-hGLUT1.
Full-length hGLUT1 with thrombin-cleavable C-terminal 10× His tag was expressed in Saccharomyces cerevisiae (16), as detailed in Materials and Methods. Despite a known glycosylation site at residue N45, the protein was not found to be glycosylated because no change in the bandwidth or migration position was observed on SDS/PAGE after treatment with peptide: N-glycosidase F (PNGase F) (Fig. S1). Hence, all our studies, functional or structural, were carried out with full-length hGLUT1.
Fig. S1.
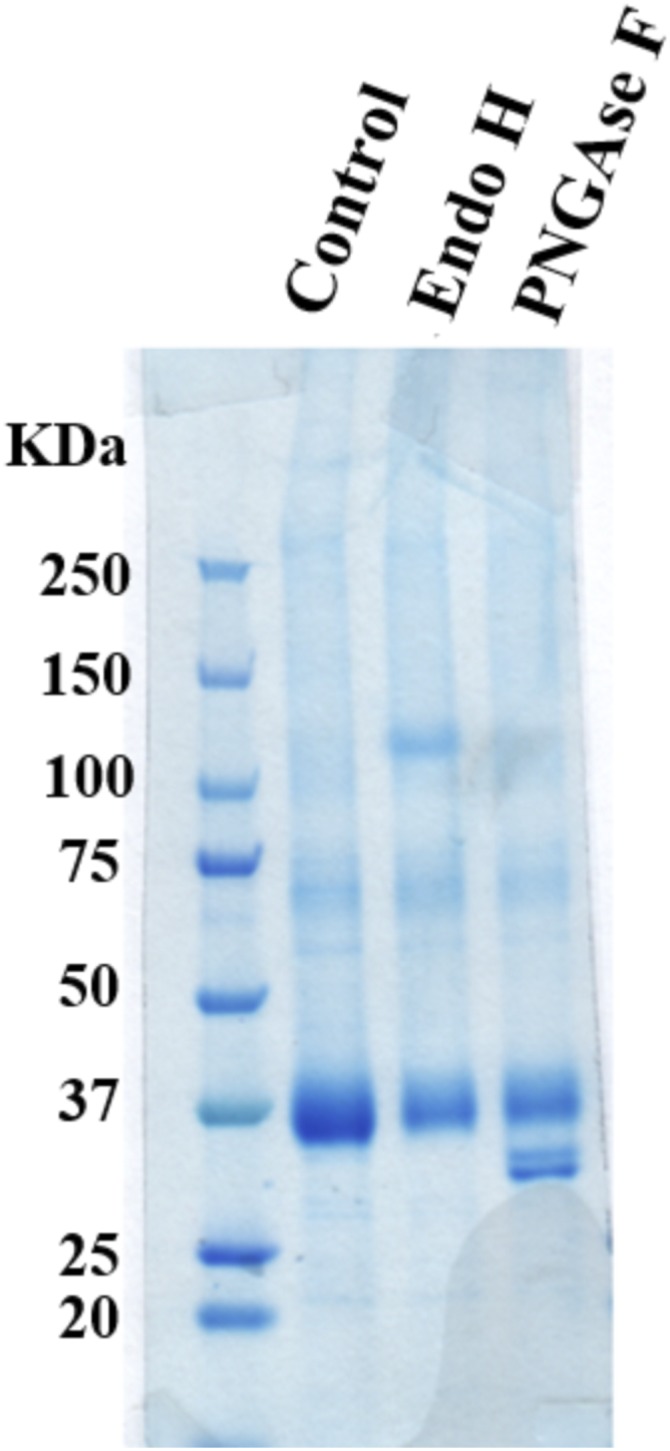
Glycosidase digestion pattern of hGLUT1 expressed in yeast. Lane 1, protein marker; lane 2, undigested; lane 3, endoglycosidase H (EndoH); lane 4, peptide: N-glycosidase F (PNGase). No difference in bandwidth or band density was observed between EndoH and PNGase F digestion, thus supporting lack of glycosylation of hGLUT1 expressed in yeast.
We identified the Phe amide compound class of GLUT-i1 and GLUT-i2 (Fig. 1 B and C) in a high-throughput screen (HTS) for inhibitors of hGLUT1 by indirect measurement of hGLUT1 inhibition (SI Materials and Methods). Crystallization screens were attempted for WT-hGLUT1 with various representatives of this HTS hit cluster and with several well-known compounds that were shown to be inhibitors of glucose uptake. The IC50s of cytochalasin B (Fig. 1A), as well as the GLUT-i1/i2 (Fig. 1 B and C), on HEK 293 cells were determined against hGLUT1-4 using an indirect cell-based assay in which ATP levels were measured using luminescence of luciferin. All compounds were shown to be very effective inhibitors of hGLUT1, with IC50 values in the nanomolar range (Table 1). All three compounds had submicromolar IC50s against hGLUT1 and hGLUT4, but 10- to 100-fold higher IC50s for hGLUT2 and intermediate IC50s against hGLUT3 (Table 1). We observed that cytochalasin B and GLUT-i1 both competed for the same binding pocket of hGLUT1 when tested against increasing concentrations of glucose (Table 2 and SI Materials and Methods).
Table 1.
IC50 values of inhibition of glucose transport in the presence of cytochalasin B, GLUT-i1, and GLUT-i2 for hGLUT family members hGLUT1–4 (n = 3)
| Inhibitor | Target | IC50, μM ± SD | Interacting residues* |
| Cytochalasin B | Glut1 | 0.110 ± 0.38 | Thr137, Gln282, Asn288, Gly384, Trp388, and Asn411 |
| Glut2 | 2.120 ± 0.640 | Ser169 and Asn443 | |
| Glut3 | 0.144 ± 0.110 | Thr135, Trp386, Asn409, and Thr28 | |
| Glut4 | 0.294 ± 0.113 | Ser153, Gln177, Trp404, and Asn427 | |
| GLUT-i1 | Glut1 | 0.267 ± 0.133 | Gln161 and Trp388 |
| Glut2 | 56 ± 13.6 | Trp420 | |
| Glut3 | 5.2 ± 1.1 | Trp386 | |
| Glut4 | 0.195 ± 0.066 | Gln299 and Trp404 | |
| GLUT-i2 | Glut1 | 0.140 ± 0.072 | Phe379 and Trp388 |
| Glut2 | >30 | Phe323 and Trp420 | |
| Glut3 | 7.7 ± 1.35 | Asn286, Phe377, Asn409, and Trp386 | |
| Glut4 | 0.090 ± 0.08 | Asn304, Phe307, Trp404, Asn427, and Asn431 |
Interactions with GLUT1 are derived from X-ray structures. Interactions with GLUT2–4 are derived from modeling.
Table 2.
IC50 values (μM) of inhibition of glucose transport in the presence of increasing concentrations of glucose
| hGLUT1 inhibitor | Glucose, 0.02 mM | Glucose, 0.1 mM | Glucose, 1 mM |
| Cytochalasin B | 0.11 | 0.5 | 2.2 |
| GLUT-i1 | 0.27 | 1.3 | 5.1 |
Crystal Structure of hGLUT1 with Cytochalasin B.
The crystal structure of hGLUT1 bound to cytochalasin was determined by molecular replacement using the published structure of apo-hGLUT1 as a search model. hGLUT1 lies in the same inward-open conformation as the previously reported apo-hGLUT1 double mutant (13) (rmsd = 0.6 Å for 447 aligned Cαs). Thus, in our hands, the mutations were not necessary to obtain well-diffracting crystals that capture the inward-facing conformation.
Cytochalasin B is a macrolide ring fused to a nine-membered bicyclic ring, and density for the bicyclic ring system and the macrolide ring was clear in the Fo-Fc map calculated after refinement of the protein (Fig. S2A). Our fit of cytochalasin B to density (Fig. 2A) was supported by the fact that Trp388 and Trp412 in transmembrane (TM) 10 and TM11, respectively, surround the binding site and contribute to hydrophobic interactions with the ligand (Fig. 3A). These residues have been shown to be labeled upon exposure to photoactivatable, radioactively labeled cytochalasin B in several biochemical studies (17, 18).
Fig. S2.

Unbiased electron density for the inhibitors in the three hGLUT1 complexes. The inhibitor-binding sites are shown as sticks: carbon, orange; oxygen, red; nitrogen, blue; fluorine, cyan. The bromine atom in GLUT-i2 is shown as a dark red sphere. Residues that contact the inhibitors are labeled. The views are the same as in Fig. 2. The likelihood-weighted difference density after refinement of the protein alone, before inhibitors were added to the structure, is shown with green contours, with contour levels of 2.1σ cytochalasin B (A) 2.4σ GLUT-i1 (B), and 2.5σ GLUT-i2 (C). This figure was prepared using the PyMOL Molecular Graphics System, version 1.4.1 (Schrödinger).
Fig. 2.

Electron density maps for hGLUT1–inhibitor complexes. Approximately equivalent views of the inhibitor binding sites (cytoplasmic side down) for three hGLUT1 complexes overlaid with 1σ likelihood-weighted 2Fo-Fc density, shown with blue contours. The complexes are plotted as sticks with the following color code: carbon, orange (protein) or green (inhibitor); oxygen, red; nitrogen, blue; bromine, dark red; and fluorine, cyan. Inhibitor-binding residues are labeled. Cytochalasin B (A), GLUT-i1 (B), and GLUT-i2 (C) are shown. This figure was prepared using PyMOL (The PyMOL Molecular Graphics System, Version 1.4.1; Schrödinger).
Fig. 3.

Interactions between inhibitors and hGLUT1. (Left) Schematics generated by LIGPLOT+ showing hGLUT1 residues that contact the inhibitors. The eyelash motif indicates a hydrophobic contact, and broken green lines indicate possible hydrogen bonds, with the hydrogen bond lengths shown in green. Residues that contact at least two of the three inhibitors are circled. (Right) Structures of the active sites of the complexes. The views are approximately the same as in Fig. 2. The inhibitors are shown as sticks. The protein is shown as semitransparent gold ribbons with side chains that contact the inhibitor shown as sticks using the following color code: carbon, gold (protein) or blue (inhibitor); oxygen, red; nitrogen, blue; bromine, dark red; and fluorine, green. Broken red lines denote hydrogen bonds. Cytochalasin B (A), GLUT-i1 (B), and GLUT-i2 (C) are shown. This figure was prepared using the UCSF Chimera package.
Crystal Structure of hGLUT1 with GLUT-i1.
Similar to the cytochalasin B complex structure, hGLUT1 lies in an inward-open conformation, with an rmsd of 0.6 Å for 447 Cαs aligned to the apo form (13). With GLUT-i1, unbiased difference electron density (phased on the protein alone) revealed a trilobed density in the central cavity (Fig. S2B). The strong ligand density from the Phe amide looked quite distinct in shape from cytochalasin B. GLUT-i1, being a nearly threefold symmetrical molecule, can be fitted with various permutations of its three rings into this density, but with one best orientation, which refined to a free R-factor of 29.5% (Fig. 2B). In the published apo-hGLUT1 structure, a detergent molecule that included a glucose moiety, n-nonyl-β-d-glucopyranoside (β-NG), occupied the binding site (13). Because we also used β-NG to produce crystals, our refinement included an assessment of the possibility that β-NG occupied the observed density, either partially or completely. There is density at the positions expected for the glucose-ring substituents (Fig. S3A), but placing glucose in the site produced no density for the nonyl chain in the residual Fo-Fc density (Fig. S3B), and joint refinement of partially occupied β-NG and GLUT-i1 yielded an occupancy of 0.8 for GLUT-i1 compared with an occupancy of 0.2 for β-NG, no improvement in the free R-factor, and extra positive difference density in several parts of the inhibitor-binding site. We conclude that β-NG is not bound. Rather, the positive difference density at the flurophenyl ring-binding site may result from unmodeled, partially occupied water molecules.
Fig. S3.

Density for hGLUT1–GLUT-i1 after refinement suggests partial occupancy of β-NG. (A) Likelihood-weighted 2Fo-Fc density (blue contours) and difference density (green contours, 3.5σ density; red contours, −3.5σ density) after refinement of hGLUT1–GLUT-i1. The complex is shown as sticks (carbon, magenta; oxygen, red; nitrogen, blue; fluorine, cyan). (B) mFo-DFc density is consistent with the oxygen substituents of the glucose moiety of a partially occupied β-NG molecule, shown modeled into the binding site identified in PDB ID code 4PYP as sticks with white carbons and red oxygens. However, there is no density for the nonyl chain of β-NG, and GLUT-i1 refines to almost full occupancy, so β-NG is most likely a minor component of the complex if it is present. This figure was prepared using the PyMOL Molecular Graphics System, version 1.4.1.
Crystal Structure of hGLUT1 with GLUT-i2.
To confirm the fit of the nearly symmetrical GLUT-i1 to density, we crystallized hGLUT1 in the presence of GLUT-i2, a bromine-containing variant of GLUT-i1. The model was built and refined to 3.0 Å. Due to the additional bromine atom, this compound is less symmetrical and generated strong additional electron density corresponding to the bromine (Fig. S2C). The refined bromine B-factor was high, and we did not observe a peak above noise level for the bromine in an anomalous difference map. When the B-factor of the bromine was set to the B-factor of the ring atom to which it was attached, the bromine occupancy that yielded a clean Fo-Fc map was 0.6. We speculate that the bromine might have partially detached from the inhibitor during data collection due to the high-energy synchrotron X-ray beam. The inhibitor-binding site overlaps the binding site of GLUT-i1. GLUT-i1/i2 binding primarily relies on hydrophobic contacts with hGLUT1 (Fig. 3 B and C).
Difference Maps Between Three Structures.
To investigate the orientation of these inhibitor compounds in the binding site further, we generated (Fo1-Fo2)αcalc difference maps between the three ligand-bound structures, where Fo1 and Fo2 are observed amplitudes for two compared structures. All three crystal structures were highly isomorphous; hence, these maps sensitively show the differences at the inhibitor-binding site. Because GLUT-i1 and GLUT-i2 both belong to the same class of compounds and are very similar in structure, the map clearly shows the presence of high integrated electron density for bromine at the standard contour levels of 3σ (Fig. S4A). Cytochalasin B minus GLUT-i1 shows the large negative difference electron density for each of the three phenyl rings and positive density for the cytochalasin B (Fig. S4B). Similar difference densities were found in the map between cytochalasin B and GLUT-i2 (Fig. S4C). The difference maps thus clearly support our placement of inhibitors in the maps.
Fig. S4.

Cross-eyed stereo plots of (Fo1-Fo2)αcalc maps between all pairs of the inhibitor complexes. The views are the same as in Fig. 2. (A) Difference maps between hGLUT1–GLUT-i2 (Fo1) and hGLUT1–GLUT-i1 (Fo2), with αcalc from hGLUT1–GLUT-i2. Green contours indicate positive (2.7σ) density (density for hGLUT1–GLUT-i2 not present in hGLUT1–GLUT-i1), and red contours indicate negative (−2.7σ) density (density for hGLUT1–GLUT-i1 not present in hGLUT1–GLUT-i2). The two structures are overlaid on the map and shown as sticks (carbon, blue for hGLUT1–GLUT-i2 and cyan for hGLUT1–GLUT-i1; oxygen, red; nitrogen, blue; fluorine, cyan; bromine, dark red). (B) Difference maps between hGLUT1-cytochalasin B (Fo1) and hGLUT1–GLUT-i1 (Fo2), with αcalc from hGLUT1-cytochalasin B. Green and red contours indicate 3σ positive and negative density, respectively. The two structures are overlaid on the map and shown as sticks. hGLUT1-cytochalasin B has orange carbons, and hGLUT1–GLUT-i1 has cyan carbons. (C) Difference maps between hGLUT1-cytochalasin B (Fo1) and hGLUT1–GLUT-i2 (Fo2), with αcalc from hGLUT1-cytochalasin B. Green and red contours indicate 3σ positive and negative density, respectively. The two structures are overlaid on the map and shown as sticks. hGLUT1-cytochalasin B has orange carbons, and hGLUT1–GLUT-i1 has blue carbons. This figure was prepared using the PyMOL Molecular Graphics System, version 1.4.1.
Homology Modeling of hGLUT2-4.
We sought to determine whether the inhibitor–hGLUT1 interactions revealed in the crystal structures accounted for the differences in IC50s of the inhibitors vs. other hGLUT family members. We generated homology models of hGLUT2, hGLUT3, and hGLUT4 in an inward-open conformation based on the crystal structures of ligand-bound hGLUT1. Compounds were docked into the models, and ΔG free energies were calculated using the molecular mechanics generalized Born surface area (MM-GBSA) algorithm for binding to each of the four hGLUTs (Table S1). Docking calculations were able to reproduce the hGLUT1 crystallographic conformation successfully with an rmsd of 0.46 Å for cytochalasin B, 0.28 Å for GLUT-i1, and 0.21 Å for GLUT-i2. This consistency supports the notion that the crystal structure of hGLUT1 can be relevant for the design of better inhibitors of glucose uptake. Other docked conformations of these three compounds were ranked poorly due to the disruption of key contacts, especially with four crucial amino acids in the binding site: Asn411, Thr137, Gln282, and Trp388. The crystallographic conformations are favorable due to hydrogen bonds and the π–π stacking network in the binding site (Figs. 4 and 5 and Fig. S5).
Table S1.
Predicted binding energies of hGLUT1–4 with the three inhibitors
| Predicted binding energies, kcal/mol | ||||
| GLUT inhibitor | GLUT1 | GLUT2 | GLUT3 | GLUT4 |
| Cytochalasin B | −79.63 | −71.97 | −75.48 | −84.53 |
| GLUT-i1 | −72.99 | −57.41 | −57.74 | −77.46 |
| GLUT-i2 | −64.45 | −57.65 | −61.40 | −71.79 |
These values were calculated using the Prime/MM-GBSA module implemented in Schrödinger. These predictions were based on receptor–ligand complexes obtained from molecular docking and the ΔG binding energy of each compound in the receptor’s binding site.
Fig. 4.

Cytochalasin B docked into hGLUT1–4. The protein structures are homology models based on the crystal structures of hGLUT1 and hGLUT3, and are shown as ribbons with side chains that contact cytochalasin B, shown as sticks. Hydrogen bonds are denoted with broken yellow lines. The views are equivalent for all four proteins (cytoplasmic side down). (A) hGLUT1, color code: carbons, orange (protein); oxygen, red; nitrogen, blue. (B) hGLUT2, color code: carbons, magenta (protein); oxygen, red; nitrogen, blue. (C) hGLUT3 color code: carbons, teal; oxygen, red; nitrogen, blue. (D) hGLUT4, color code: carbons, green; oxygen, red; nitrogen, blue. Cytochalasin B is shown with yellow carbons for the four proteins.
Fig. 5.

GLUT-i1 docked into hGLUT1–4. The protein structures are homology models based on the crystal structures of hGLUT1 and hGLUT3, and are shown as ribbons with side chains that contact cytochalasin B, shown as sticks. Hydrogen bonds are denoted with broken yellow lines. The views are equivalent for all four proteins (cytoplasmic side down). (A) hGLUT1, color code: carbons, orange (protein); oxygen, red; nitrogen, blue. (B) hGLUT2, color code: carbons, magenta; oxygen, red; nitrogen, blue. (C) hGLUT3, color code: carbons, teal (protein); oxygen, red; nitrogen, blue. (D) hGLUT4, color code: carbons, green (protein); oxygen, red; nitrogen, blue. GLUT-i1 is shown with purple carbons for the four proteins with fluorine in cyan.
Fig. S5.

GLUT-i2 docked into hGLUT1–4. The protein structures are homology models based on the crystal structures of GLUT1 and GLUT3, and are shown as ribbons with side chains that contact GLUT-i2 shown as sticks. Hydrogen bonds are denoted with broken yellow lines. The views are equivalent for all four proteins (cytoplasmic side down). (A) hGLUT1, color code: carbons, orange (protein); oxygen, red; nitrogen, blue. (B) hGLUT2, color code: carbons, magenta (protein); oxygen, red; nitrogen, blue. (C) hGLUT3, color code: carbons, teal (protein); oxygen, red; nitrogen, blue. (D) hGLUT4, color code: carbons, green (protein); oxygen, red; nitrogen, blue. GLUT-i2 is shown with the following color code: carbons, olive green; oxygen, red; nitrogen, blue; and bromine, dark red, for the four proteins.
Many of the amino acid side chains and their disposition in the four hGLUTS are conserved. However, hGLUT2 shows more hydrophobic interactions than hGLUT1, hGLUT3, and hGLUT4, signaled by substitution of a His (His311 in hGLUT2) for Gln279 (hGLUT1) in TM7. His311 changes the conformation of Trp388 due to the increased steric hindrance of His. Because the indole of Trp388 forms conserved hydrophobic contacts with all compounds, the observed altered conformation in hGLUT2 may result in generally weaker binding of these compounds to hGLUT2. Thr137 (hGLUT1) is substituted by Ser in hGLUT2 and hGLUT4, and both residues form polar interactions with the OH group of cytochalasin B (Fig. 4).
Calculated predicted binding energies showed a preference of GLUT-i1 and GLUT-i2 for hGLUT1 and hGLUT4, whereas hGLUT2 and hGLUT3 presented similar and lower binding energies (Table S1). The predicted ΔG energy values, although not directly comparable to observed binding energies, indicate that the ranking of these compounds is in line with their experimentally determined IC50s (Table 1). The interactions with cytochalasin B gave calculated energies that reiterate the generally weaker binding of cytochalasin B to hGLUT2. In hGLUT2, the cytochalasin B is positioned closer to Ser169 (TM4) due to Trp388 being pushed toward the inside of the pocket. In this position, the compound loses hydrogen bond contacts between the azole carbonyl and amino acids, Q314 and N320 (Fig. 4).
SI Materials and Methods
Protein Expression and Purification.
A S. cerevisiae expression construct based on p423_GAL1 was codon-optimized for the Homo sapiens glucose transporter GLUT1 (UniProt accession no. P11166). Transformed S. cerevisiae (strain DSY-5) was grown in a 15-L culture vessel (Biostat C15L; Sartorius AG) to high density by fed batch and harvested after a 16-h induction using Gal. Harvested yeast were washed in cold water, spun down, and resuspended in lysis buffer [200 mM Tris (pH 7.0), 600 mM NaCl, 100 mM phenylmethylsulfonyl fluoride] before lysis by bead beating using 0.5-mm glass beads. The homogenate was centrifuged for 25 min at 21,600 × g, followed by sedimentation of membranes by ultracentrifugation at 185,000 × g for 150 min. Membrane pellets were resuspended in buffer containing [25 mM Tris (pH 7.5), 150 mM NaCl, 5% glycerol] before being frozen in liquid nitrogen in 3-g aliquots. Although there is one glycosylation site known for hGLUT1 (N45T), we observed that this protein does not appear to be glycosylated in yeast. The absence of glycosylation was confirmed using endoglycosidase H (EndoH) and peptide: N-glycosidase F (PNGase F) digestion. Therefore, we proceeded with purification of WT-hGLUT1 from yeast membranes. Three-gram membranes were solubilized for 2 h in membrane buffer using 1 g of β-DDM [1:10 (wt/wt) ratio] and 0.5 mM TCEP in a total volume of 50 mL. The unsolubilized material was removed by ultracentrifugation at 185,000 × g for 20 min. The supernatant was filtered using a 1.2-μm filter. Twenty millimolar imidazole (pH 7.5) was added, and the solubilized membranes were loaded on a preequilibrated 1-mL Ni-NTA column (GE Healthcare) at 1 mL⋅min−1. After loading, the column was washed with 20 column volumes of W75 buffer [membrane buffer supplemented with 75 mM imidazole (pH 7.5), 0.5 mM TCEP, 25 mM Tris (pH 7.5), 0.1% DDM], and the protein was eluted in 10 mL of G buffer [25 mM MES (pH 6.0), 150 mM NaCl, 5% glycerol, 0.5 mM TCEP, 0.4% β-NG supplemented with 500 mM imidazole (pH 7.5)]. To remove purification tags, bovine thrombin was added and the sample was dialyzed for 16 h against 200 mL of G buffer. A normal yield was 2 mg of pure hGLUT1 from 3 g of membrane. After dialysis, the sample was concentrated using a spin column (50-kDa cutoff; Amicon) to a concentration of 10 mg/mL before setting up for crystallization trials.
Synthesis of GLUT-i1 and GLUT-i2.
Unless otherwise stated, all chemicals were purchased from Sigma–Aldrich or Merck and used without further purification. Peptide synthesis was carried out in an Apex 396 parallel synthesizer from AAPPTec using Rink-Amide resin (0.68 mmol/g) following the standard Fmoc strategy. All amino acid residues were, if not further specified, l-amino acid residues. Ultraperformance liquid chromatography (UPLC) analyses were performed using an Acquity UPLC BEH C18 instrument (50 × 2.1 mm, 1.7 μm) and following conditions [solvent A: water + 0.1% formic acid (vol/vol); solvent B: acetonitrile; gradient: 0–1.6 min 1–99% B, 1.6–2.0 min 99% B; flow: 0.8 mL/min; temperature: 60 °C]. The analytical HPLC system used was a Waters Acquity UPLCMS SingleQuad equipped with UV multiwavelength (scan: 210–400 nm). Preparative HPLC was performed using an XBrigde C18 (100 × 30 mm, 5 μm) and the following conditions [solvent A: water + 0.1% formic acid (vol/vol); solvent B: acetonitrile; gradient: 0–8 min 10–100% B, 8–10 min 100% B; flow: 50 mL/min; temperature: 25 °C). The preparative HPLC system used was a Waters autopurification system with pump 2545, sample manager 2767, motorized valve (CFO), diode array detector DAD 2996, evaporative light scattering detector ELSD 2424, a single quadrupole mass detector (SQD), with a diode array detector (DAD) detection scan range of 210–400 nm, and an electrospray ionization mass spectrometer (ESI-MS) with electrospray positive (ESI+) and negative (ESI−) scan ranger of 160–1,000 m/z. NMR spectra were recorded on 1H NMR spectra recorded with a Bruker AVANCE-300 and 400 MHz and a Bruker DRX-600 NMR spectrometer. Chemical shifts (δ) are expressed in parts per million relative to tetramethylsilane. Coupling constants (J) are expressed in hertz, and the following abbreviations are used to describe the appearance of peak patterns: s (singlet), d (doublet), t (triplet), and combinations thereof (e.g., dd, doublet of doublet), as well as m (multiplet) and br (broad signal). The purity of all compounds was judged to be in the 95% range according to the 1H NMR, with no chemical impurity detectable.
GLUT-i1.
The GLUT-i1 was partly synthesized via solid-phase peptide synthesis. One hundred fifty-six milligrams (0.25 mmol) of the Rink amide resin was placed in a peptide synthesizer. The amounts of reagents and building blocks in all reactions were calculated based on the amount of resin. After swelling the resin, a 0.5 M solution of N-[(9H-fluoren-9-ylmethoxy)carbonyl]-4-fluoro-l-Phe in N,N-dimethylformamide (4 eq), 0.5 M solution of O-(benzotriazol-1-yl)-N,N,N,N-tetramethyluronium hexafluorophosphate in DMF (4 eq), 0.5 M solution of 1-hydroxybenzotriazole in N,N-dimethylformamide (4 eq), and 1.0 M solution of N,N-diisopropylethylamine (4 eq) in N,N-dimethylformamide were added to the resin and shaken for 90 min at 25 °C. This step was repeated with a reaction time of 60 min. The resin was washed with N,N-dimethylformamide (twice), dichloromethane (twice), and N,N-dimethylformamide (twice). The resin was treated with 20% piperidine in N,N-dimethylformamide (vol/vol) for 5 min. This step was repeated with a reaction time of 20 min. The resin was washed with N,N-dimethylformamide (twice), dichloromethane (twice), and N,N-dimethylformamide (twice). Afterward, a 0.5 M solution of (3-hydroxyphenyl)acetic acid in N,N-dimethylformamide (8 eq), 0.5 M solution of O-(benzotriazol-1-yl)-N,N,N,N-tetramethyluronium hexafluorophosphate in N,N-dimethylformamide (8 eq), 0.5 M solution of 1-hydroxybenzotriazole in N,N-dimethylformamide (8 eq), and 1.0 M solution of N,N-diisopropylethylamine (8 eq) in N,N-dimethylformamide was added to the resin and shaken for 24 h at 25 °C. The resin was washed with N,N-dimethylformamide (twice), dichloromethane (twice), and N,N-dimethylformamide (twice) and dried with nitrogen flow. Subsequent treatment of the resin with a 2% solution of TFA in dichloromethane (4 mL), filtration, addition of two drops of toluene, and concentration in a vacuum afforded the 4-fluoro-N-[(1S)-1-phenylethyl]-l-phenylalaninamide as a crude intermediate. This intermediate was redissolved in 2 mL of N,N-dimethylformamide, and 250 μL of a 1 M solution of (benzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate in N,N-dimethylformamide, 70 μL (0.40 mmol) of N,N-diisopropylethylamine, and 277 μL (0.16 mmol) of (1S)-1-phenylethanamine were added. The reaction mixture was stirred for 24 h at 25 °C. The crude product in N,N-dimethylformamide was injected on preparative RP-HPLC, and lyophilized to get 15 mg (35 μmol, 14%) of 4-fluoro-Nalpha-[(3-hydroxyphenyl)acetyl]-N-[(1S)-1-phenylethyl]-l-phenylalaninamide. For UPLC-MS, the following were used: Retention time (Rt) = 1.11 min; MS (ESI+): m/z = 421 (M + H)+; 1H-NMR (400 MHz, DMSO-d6): δ 1.24 ppm (d, J = 6.82 Hz, 1 H), 1.33 ppm (d, J = 7.07 Hz, 2 H), 2.64–2.77 ppm (m, 1 H), 2.81–2.93 ppm (m, 1 H), 3.26–3.30 ppm (m, 1 H), 3.32–3.36 ppm (m, 1 H), 4.46–4.59 ppm (m, 1 H), 4.81–4.94 ppm (m, 1 H), 6.50–6.64 ppm (m, 3 H), 6.93–7.08 ppm (m, 3 H), 7.11–7.17 ppm (m, 3 H), 7.18–7.33 ppm (m, 4 H), 8.18 ppm (d, J = 8.34 Hz, 1 H), 8.43 ppm (d, J = 8.08 Hz, 1 H), and 9.22–9.25 ppm (m, 1 H).
GLUT-i2.
Five hundred milligrams (1.45 mmol) of 2-bromo-N-(tert-butoxycarbonyl)-l-Phe was dissolved in 10 mL of tetrahydrofuran, and 907 mg (1.74 mmol) of (benzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate, 304 μL (1.74 mmol) of N,N-diisopropylethylamine, and 18 mg (0.15 mmol) of 4-dimethylaminopyridine were added. The reaction mixture was stirred 15 min at 25 °C before 277 μL (2.18 mmol) of (1S)-1-phenylethanamine was added. The stirring was continued for 24 h at 25 °C. After evaporation, the crude product was purified via chromatography over 100 g of silica gel (1:1 hexane/ethyl acetate) to get 541 mg (1.21 mmol, 83%) of 2-bromo-Nalpha-(tert-butoxycarbonyl)-N-[(1S)-1-phenylethyl]-l-phenylalaninamide as a white solid (intermediate 1). For UPLC-MS, the following were used: Rt = 1.32 min; MS (ESI+): m/z = 449 (M + H)+; 1H-NMR (300 MHz, DMSO-d6): δ 1.32 ppm (s, 9 H), 1.36 ppm (d, J = 7.16 Hz, 3 H), 2.79–2.94 ppm (m, 1 H), 3.00–3.14 ppm (m, 1 H), 4.23–4.39 ppm (m, 1 H), 4.84–5.01 ppm (m, 1 H), 6.97 ppm (d, J = 8.85 Hz, 1 H), 7.09–7.35 ppm (m, 8 H), 7.57 ppm (d, J = 7.91 Hz, 1 H), and 8.23 ppm (d, J = 7.72 Hz, 1 H).
Five hundred forty-one mg (1.21 mmol) of intermediate 1 was dissolved in a mixture of 15 mL of dichloromethane and 1.40 mL of TFA. The solution was stirred for 4 h at 25 °C and concentrated under reduced pressure. The residue was redissolved in toluene and evaporated again. This step was repeated three times to get 549 mg (98%) of 2-bromo-N-[(1S)-1-phenylethyl]-l-phenylalaninamide (intermediate 2) as a white foam. For UPLC-MS, the following were used: Rt = 0.81 min; MS (ESI+): m/z = 349 (M + H)+; 1H-NMR (400 MHz, DMSO-d6): δ 1.37 ppm (d, J = 7.07 Hz, 3 H), 2.87–3.03 ppm (m, 2 H), 3.93–4.06 ppm (m, 1 H), 4.82–4.96 ppm (m, 1 H), 7.00–7.11 ppm (m, 4 H), 7.20–7.34 (m, 3 H) ppm, 7.39–7.47 ppm (m, 2 H), 8.24 ppm (br s, 2 H), and 8.78 (d, J = 7.83 Hz, 1 H).
Five hundred forty-nine mg (1.19 mmol) of intermediate 2 was dissolved in 15 mL of tetrahydrofuran, and 743 mg (1.43 mmol) of (benzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate, 249 μL (1.43 mmol) of N,N-diisopropylethylamine, and 15 mg (0.12 mmol) of 4-dimethylaminopyridine were added. The reaction mixture was stirred 15 min at 25 °C before 297 mg (1.79 mmol) of (4-methoxyphenyl) acetic acid was added. The stirring was continued for 24 h. After evaporation, the residue was redissolved in ethyl acetate and washed with saturated sodium bicarbonate solution. The organic layer was dried over sodium sulfate and concentrated under reduced pressure. The crude product was purified via chromatography over 100 g of silica gel (9:1 dichloromethane/methanol) to get 518 mg (88%) of 2-bromo-Nalpha-[(4-methoxyphenyl)acetyl]-N-[(1S)-1-phenylethyl]-l-phenylalaninamide. For UPLC-MS, the following were used: Rt = 1.29 min; MS (ESI+): m/z = 497 (M + H)+; 1H-NMR (300 MHz, DMSO-d6): δ 1.33 ppm (d, J = 6.97 Hz, 3 H), 2.89 ppm (dd, J = 14.13, 9.04 Hz, 1 H), 3.09 ppm (dd, J = 14.13, 5.65 Hz, 1 H), 3.35 ppm (s, 2 H), 3.72 ppm (s, 3 H), 4.62–4.73 ppm (m, 1 H), 4.85–4.97 ppm (m, 1 H), 6.81 ppm (d, J = 8.67 Hz, 2 H), 7.07 ppm (d, J = 8.67 Hz, 2 H), 7.13–7.30 ppm (m, 8 H), 7.50–7.58 ppm (m, 1 H), 8.27 ppm (d, J = 8.67 Hz, 1 H), and 8.35 ppm (d, J = 8.10 Hz, 1 H).
Ultrahigh-Throughput Screen with hGLUT1.
A combination of small-molecule inhibitors of mitochondrial electron transport chain and glucose catabolism synergistically suppresses ATP production (35, 36). For ultrahigh-throughput screening, CHO-K1 cells were stably transfected with hGLUT1 and a constitutively expressing luciferase as described previously (37). Cells were seeded in 1,536 microtiter plates with a density of 1,000 cells per well and starved for 24 h in glucose-free DMEM in the presence of 1% FCS. Before measurements, cells were incubated for 30 min at 37 °C in the presence of 10 μM rotenone to block oxidative phosphorylation fully. Test compounds and caged luciferin were loaded simultaneously. Before application of 0.5 mM glucose and corresponding activation of hGLUT1, basal ATP was indirectly measured by luciferase activity to identify effects on cellular ATP levels independent of glucose. Ten-minute kinetic luciferase recordings after application of 500 μM glucose allowed the investigation of compound-induced inhibition of hGLUT1.
Glucose Competition.
For the glucose competition, DLD1 cells were treated with trypsin and seeded into 384 plates at a density of 4,000 cells per well. The cells were then cultured overnight in glucose-free media containing 1% FCS to reduce intracellular ATP levels. After 16 h, the cells were incubated with a different glucose concentration (0.1; 1 and 10 mM, respectively) together with compound (30 μM to 1 nM) and 1 μM rotenone for 15 min. The CellTiter-Glo Luminescent Cell Viability Assay from Promega was then used to measure ATP levels.
Discussion
We succeeded in crystallizing full-length WT-hGLUT1 with cytochalasin B, a natural product inhibitor (Fig. 1A). We also crystallized hGLUT1 with GLUT-i1 and GLUT-i2, which belong to a Phe amide class of compounds and inhibit cellular glucose uptake with IC50 values of 267 nM and 140 nM, respectively (Fig. 1 B and C and Table 1). Even though GLUT-i1, with a three-ring structure of 420 Da, is much larger than glucose (180 Da), the 5-fluoro-phenyl ring nicely overlaps the glucose-binding site. The glucose site interacts predominantly with the C-terminal domain of hGLUT1. As in other secondary transporters, the binding site of this uniporter is specific for its natural substrates (19). By using intermediate water molecules, glucose achieves selectivity within a much larger binding site than the substrate. The resulting relatively weak binding site [Kd ∼ 35.7 mM (20)] may allow efficient glucose transport without blockade.
The phenyl ring of GLUT-i1 binds between helix TM11 in the C-terminal domain and helix TM4 in the N-terminal domain, and 2-hydroxy-phenyl is sandwiched between His160 in TM5 and Trp388 in TM10 (Fig. 3B). Trp388 is a conserved residue in the hGLUT family located in the mobile C-terminal segment of the discontinuous helix TM10. In the glucose-bound occluded states of hGLUT3 vs. unbound hGLUT3, the counterpart of Trp388 rotates into the channel and forms a hydrogen bond to the glucose substrate (14). GLUT-i1 could therefore block the movement of Trp388, and block rotation of the segment of TM10 into the central cavity.
In our WT structure, these interactions that displace TM10 may have compensated for the changes made by the mutation E325Q that was introduced to fix the conformation of apo-hGLUT1. Binding of the large inhibitor molecule can prop open the central cavity and inhibit the alternate access mechanism, thus inhibiting glucose transport. Trp388 in TM10 is a key binding determinant for all three inhibitors; thus, the inhibitors compete with glucose at the central substrate-binding site. The inhibitors likely cannot make similar hydrophobic or π–π interactions with Trp388 when the hGLUT transporters are in outward-open conformations because in the outward-open conformation of the closely related hGLUT3, the face of the Trp388 indole ring is inaccessible (14). It is widely accepted that cytochalasin B binds only to inward-open conformations of hGLUT1 (12).
Despite their different chemical structures, cytochalasin B, GLUT-i1, and GLUT-i2 overlap each other and the proposed glucose substrate site in the central cavity. The published outward-occluded structure of hGLUT3 shows that bound glucose overlaps the cyclohexane ring of cytochalasin B in our model of hGLUT3 based on our structure of cytochalasin B-hGLUT1. The macrolide ring overlaps the linkers between the three rings in GLUT-i1 and GLUT-i2 (Fig. S6). An overlay of all three inhibitors in the binding site shows that there are conserved π–π interactions with hGLUT1 (Fig. S6). The main interactions observed with GLUT-i1 are π–π stacking interactions of the phenyl and phenol groups with conserved Trp388 and of the fluorophenyl group with Phe379, whereas for cytochalasin B, there are hydrogen bond interactions with Thr137 and Trp388. Hydrophobic interactions with Asn411 and Trp412 are prominent binding determinants for all three inhibitors (Fig. S6). Additionally, there is a π–π interaction between GLUT-i1/i2 and His160 that is not used in the case of cytochalasin B. Most of these residues are conserved across hGLUT1-4 with few variations, (hGLUT2, hGLUT3, and hGLUT4 display 83%, 93%, and 85% sequence identity, respectively, to hGLUT1); however, the compounds show some selectivity between these proteins, inviting the quest for differences between the hGLUTs that can be exploited for generation of more selective compounds.
Fig. S6.

Overlapping inhibitor-binding sites in hGLUT1. The three hGLUT1-inhibitor structures were superposed, and the inhibitors were plotted as sticks with the following color code: Cytochalasin B, yellow; GLUT-i1, blue; GLUT-i2, red. The protein from hGLUT1–GLUT-i1 is plotted with ribbons, and the side chains that contact the inhibitor in all three complexes are shown as sticks and labeled. The C-terminal domain of hGLUT1 is pale blue, and the N-terminal domain is pale cyan. Helices are numbered with the conventional numbering for the major facilitator superfamily transporter fold. This figure was prepared using Chimera.
Trp388 and Trp412 of hGLUT1 are conserved across several homologous sugar transporters. The corresponding Trps of Escherichia coli GalP, Trp371, and Trp395, contribute to fluorescence quenching upon cytochalasin B binding. Their mutation to Phe results in decreased affinity for cytochalasin B and, in the case of W371F, decreased affinity of the inward-facing side of GalP for D-gal (21). These results are consistent with our hGLUT1-cytochalasin B structure and suggest similar cytochalasin B-binding modes in GalP and hGLUT1.
E. coli hGLUT1 homologs AraE and GalP are inhibited by cytochalasin B, whereas XylE is not. Highly conserved Asn394 (Asn411 in hGLUT1) was identified as a likely determinant of cytochalasin B binding because it is substituted by Gln in XylE, and mutation of Asn394 to Gln in GalP resulted in a 40- to 50-fold decrease in inhibition by cytochalasin B (22). Asn411 is 3.4 Å from cytochalasin B in our hGLUT1 complex structure. A Gln at this position would impinge on the cytochalasin B-binding site, altering the binding mode and probably perturbing the interactions with Trp412. Thus, XylE cannot make key interactions with cytochalasin B that are responsible for high-affinity binding of this inhibitor to GalP and AraE, explaining the lack of inhibition.
A comparison of the charged residues in XylE (a symporter) with their equivalents in hGLUT1 (a uniporter), and other hGLUT family members, reveals that Asp27 in XylE becomes Asn in the hGLUTs (except in hGLUT2), and the homologous residue to Glu206 in XylE is always uncharged in the hGLUTs. Thus, the residues that likely distinguish uniporters from secondary transporters lie outside the substrate-binding site. The conservation of the substrate-binding site reinforces the notion that the uniporters, like the secondary transporters, are capable of mediating alternate access to either side of the membrane. Substrates can bind with equal affinity from either side, commensurate with the uniporter/transporter–substrate complex, reaching the same lowest energy bound state that may lie anywhere between the outward-open and inward-open states.
In summary, we report inhibitor-bound crystal structures of WT-hGLUT1. We determined the structures with bound cytochalasin B and inhibitors GLUT-i1 and GLUT-i2. Several cross-checks established the binding modes. IC50s were determined for compounds to four well-characterized hGLUT family members. Docking of these compounds to hGLUT1 successfully reiterated the experimentally observed binding modes. We generated homology models for hGLUT2, hGLUT3, and hGLUT4, and calculated the computed binding energies of the compounds to them. These predicted values follow the same trend as the experimentally observed activities, and thus provide insight into the basis of selectivity within the hGLUT family. This study advances the understanding of the hGLUT-binding site, which helps in the design and optimization of more specific inhibitors for the hGLUT family.
Materials and Methods
Materials.
The cytochalasin B, tris(2-carboxyethyl)phosphine (TCEP), and buffers were obtained from Sigma–Aldrich. The nickel-nitrilotriacetic acid (Ni-NTA) resin columns (1 mL) were from GE Biosciences. The n-dodecyl-β-d-maltopyranoside (β-DDM) and β-NG were obtained from Affymetrix. GLUT-i1 and GLUT-i2 were synthesized as described in SI Materials and Methods. All other materials were of reagent grade and were obtained from commercial sources.
GLUT Isoform Specificity Testing.
For specificity testing between hGLUT1, hGLUT2, hGLUT3, and hGLUT4, we used DLD1 cells (for hGLUT1), DLD1Glut1−/− (Horizon Discovery; for hGLUT3), CHO-hGLUT2, and CHO-hGLUT4 (hGLUT2 and hGLUT4) cells in combination with an oxidative phosphorylation inhibitor (1 μM rotenone). Cell lines were maintained in DMEM/Ham’s F12 with Glutamax (Thermofisher), supplemented with 10% (vol/vol) FCS (Sigma) and 1% (vol/vol) penicillin-streptomycin solution (Gibco) under standard conditions. The cells were treated with trypsin and seeded into 384 plates at a density of 4,000 cells per well. The cells were then cultured overnight in glucose-free media containing 1% FCS to reduce intracellular ATP levels. For hGLUT1/hGLUT2/hGLUT3, after 16 h, the cells were incubated with an appropriate glucose concentration, or fructose concentration in the case of hGLUT2 (0.1 M glucose for hGLUT1, 0.3 M glucose for hGLUT3, and 30 mM fructose for hGLUT2, respectively), with or without compounds and 1 μM rotenone for 15 min. The CellTiter-Glo Luminescent Cell Viability Assay from Promega was then used to measure ATP levels.
For hGLUT4, after 16 h, the medium was removed and cells were adapted to KCl-free tyrode buffer for 3 h. Compounds and rotenone were added, and after 20 min, cells were incubated with glucose (0.1 M final concentration) for 15 min. The CellTiter-Glo Luminescent Cell Viability Assay was then used to measure ATP levels.
Protein Expression and Purification.
For large-scale production of hGLUT1 for purification and crystallization trials, the full-length WT-human hGLUT1 was expressed in S. cerevisiae. A detailed description is provided in SI Materials and Methods. In brief, the protein was solubilized from yeast cell membranes in the presence of 2% (wt/vol) β-DDM at 4 °C for 2 h and purified using a deca-His affinity tag on an Ni-NTA column in the presence of 0.1% (wt/vol) β-DDM. The protein was eluted from the column in the buffer containing 25 mM MES (pH 6.0), 150 mM NaCl, 5% glycerol, 0.4% (wt/vol) β-NG, 0.5 mM TCEP, and 500 mM imidazole. The sample was dialyzed overnight in the same buffer along with thrombin but no imidazole. The sample was collected and concentrated to about 10 mg/mL.
Crystallization.
Cytochalasin B, GLUT-i1, and GLUT-i2 were added to the purified protein (10 mg/mL) at a concentration of 1 mM. The ligands were mixed well and left on ice for at least 30 min before setting up the crystallization trays. The samples contained 1–2% DMSO at the time of crystallization setup. Crystals were grown at 4 °C by vapor diffusion in 200 nL + 200 nL sitting drops in 96-well plates with a reservoir containing 30–40% (vol/vol) PEG 400, 100 mM MES (pH 5.5–6.5), and 50–200 mM NaCl or MgCl2. Crystals were harvested from the trays and frozen directly in liquid nitrogen for data collection.
Data Processing.
All datasets were collected at Lawrence Berkeley National Laboratory Advanced Light Source Beamline 8.3.1 at +103 K and at a wavelength of either 1.115 Å (cytochalasin B and GLUT-i1 complexes) or the bromine absorption edge, λ = 0.909Å (GLUT-i2 complex). The data were processed in space group C2 using MOSFLM and AIMLESS from the CCP4 suite (GLUT-i1) (23–25) or the HKL2000 packages in space group C2 (GLUT-i2 and cytochalasin B) (26). The selected resolution cutoffs were based on correlation coefficient CC1/2 values (27). Complete datasets to a resolution of 3.0 Å were obtained from single crystals for complexes with the GLUT-i2 and cytochalasin B. A complete 2.9-Å dataset for GLUT-i1 was obtained by averaging data from three crystals. The structures were solved by molecular replacement in PHENIX (28) using hGLUT1 Protein Data Bank (PDB) ID code 4PYP as the search model (13). As in 4PYP, residues 1–8 and 456–504 were not visible in density maps and are not part of our structures. Coot was used for density fitting (29), and refinement was done with phenix.refine using a refinement strategy of individual sites and individual ADP against a maximum likelihood target (28). Geometric constraints for the inhibitors were prepared using phenix.eLBOW (28). Hydrogen atoms were included in riding positions during refinement to minimize clashes. The MolProbity server was used for structure validation (30). The final structure of the cytochalasin B complex was refined at a resolution of 3.0 Å to R/Rfree = 25.0%/28.8%. The final structure of the GLUT-i1 complex was refined at a resolution of 2.9 Å to R/Rfree = 23.2%/28.2%. The final structure of the GLUT-i2 complex was refined at a resolution of 3.0 Å to R/Rfree = 22.5% /27.2%. Data collection and refinement statistics are compiled in Table S2.
Table S2.
Data collection and refinement statistics
| Refinement statistics | hGLUT1-cytochalasin B | hGLUT1–GLUT-i1 | hGLUT1–GLUT-i2 |
| Data collection* | |||
| PDB ID code | 5EQI | 5EQG | 5EQH |
| Space group | C2 | C2 | C2 |
| Cell dimensions | |||
| a, b, c; Å | 122.4, 102.3, 68.1 | 121.3, 102.5, 68.1 | 121.5, 102.4, 68.7 |
| β, ° | 100.1 | 99.5 | 99.0 |
| Resolution, Å | 50.0–3.0 (3.05–3.0)† | 78.0–2.9 (3.0–2.9) | 50.0–3.0 (3.05–3.0) |
| Rpim‡ | 6.7 (68.3) | 8.9 (168) | 4.9 (69.9) |
| CC1/2, % | 99 (47) | 99 (27) | 99 (35) |
| I/σI | 13.8 (0.8) | 8.4 (0.6) | 13.7 (0.5) |
| Completeness, % | 98.8 (89.1) | 99.5 (99.5) | 89.1 (39.4) |
| Redundancy | 2.9 (2.0) | 6.8 (6.1) | 7.0 (2.9) |
| Refinement | |||
| Resolution, Å | 49.3–3.0 (3.09–3.00) | 67.2–2.9 (3.05–2.9) | 41.9–3.0 (3.08–2.99) |
| No. of reflections | 14,631 (1,105) | 17,392 (2,481) | 13,452 (538) |
| Rwork/Rfree | 25.0/28.8 (38.3/43.7) | 23.2/28.2 (38.1/40.8) | 22.5/27.2 (29.4/35.3) |
| No. of atoms | |||
| Protein | 3,463 | 3,463 | 3,463 |
| Ligand/ion | 31 | 32 | 36 |
| Water | 0 | 0 | 0 |
| B-factors | |||
| Protein | 100 | 91 | 88 |
| Ligand/ion | 109 | 98 | 99 |
| rmsd | |||
| Bond lengths, Å | 0.003 | 0.003 | 0.003 |
| Bond angles, ° | 0.72 | 0.63 | 0.69 |
| Ramachandran angles | |||
| Most favored, % | 94 | 94 | 93 |
| Outliers | 2 | 3 | 3 |
CC1/2, correlation coefficient1/2; I/σI, average ratio of a measured intensity to its standard deviation; Rpim, precision-indicating global R-factor; Rwork/Rfree, least-squares crystallographic agreement factor for reflections used in refinement/agreement factor for subset of reflections not included in refinement.
Number of crystals used for the datasets of cytochalasin B, GLUT-i1, and GLUT-i2 were 1, 3, and 1, respectively.
Values for highest-resolution shell are given in parentheses.
Rpim is taken from Weiss (38).
Molecular Modeling, Homology Models.
Homology models for hGLUT2, hGLUT3, and hGLUT4 were built based on multiple templates, including hGLUT1 structure with cytochalasin B, GLUT-i1, and β-NG [PDB ID code 4PYP] using the Prime software implemented in Schrödinger (release 2015-1 and Maestro, version 10.1; Schrödinger) and validated using QMean (31) and Procheck (32).
Three-dimensional complexes were prepared using the protein preparation wizard function implemented in Schrödinger, which consists of adding hydrogen atoms, assigning partial charges using the OPLS-2005 force field, and assigning protonation states. A final restrained minimization was carried out using the OPLS-2005 force field.
Molecular Modeling, Rigid Receptor Docking.
Conformations and tautomeric states were assigned to the ligands by following the ligand preparation protocol implemented in Schrödinger with default settings, generating a maximum of 32 conformations for each compound.
The ligand in the hGLUT1-4 crystal structures and models was set as a centroid to build a grid box following Schrödinger default parameters. The Glide function (33) was used to dock an ensemble of ligand conformations in the grid-defined binding site. A maximum of 10 poses per ligand were retained and scored using the standard precision function. No constraints were applied in the docking studies.
Molecular Modeling, Prime/MM-GBSA Calculations.
Prime/MM-GBSA calculations were carried out on receptor–ligand complexes obtained from the molecular docking. The free binding energy (ΔGbind) is calculated for each molecular species (protein, complex, and ligand) according to the following equation:
The ΔGbind is a sum of nonbonded electrostatic interactions (coulombs), van der Waals, internal strain, and solvation energy terms. These parameters are calculated using the VSGB2.0 implicit solvent model with the OPLS-2005 force field implemented in Prime (34). The entropy term related to the ligand or protein was not included in the calculations. However, the solvent entropy term is included in the VSGB2.0 implicit solvent model.
Acknowledgments
We thank John Pak for expert help with initial data analysis and J. Holton and G. Meigs for assistance with synchrotron data collection at the Advanced Light Source. We thank the University of California Office of the President, Multicampus Research Programs and Initiatives Grant MR‐15-338599 and the Program for Breakthrough Biomedical Research, which is partially funded by the Sandler Foundation, for support of Beamline 8.3.1. Chimera has been developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by National Institute of General Medical Sciences Grant P41-GM103311). This work was supported by NIH Grant R37 GM024485, the Danish Council for Independent Research (Grant DFF-4002-00052), and the European Research Council (Grant 637372).
Footnotes
Conflict of interest statement: R.C.H, P.B., I.H., T.M., and H.S. are or have been employees and stockholders of Bayer AG.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org [PDB ID codes 5EQI (hGLUT1 with inhibitor cytochalasin B), 5EQG (hGLUT1 with inhibitor GLUT-i1), and 5EQH (hGLUT1 with inhibitor GLUT-i2)].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1603735113/-/DCSupplemental.
References
- 1.Henderson PJ, Maiden MC. Homologous sugar transport proteins in Escherichia coli and their relatives in both prokaryotes and eukaryotes. Philos Trans R Soc Lond B Biol Sci. 1990;326(1236):391–410. doi: 10.1098/rstb.1990.0020. [DOI] [PubMed] [Google Scholar]
- 2.Mueckler M, et al. Sequence and structure of a human glucose transporter. Science. 1985;229(4717):941–945. doi: 10.1126/science.3839598. [DOI] [PubMed] [Google Scholar]
- 3.Olson AL, Pessin JE. Structure, function, and regulation of the mammalian facilitative glucose transporter gene family. Annu Rev Nutr. 1996;16:235–256. doi: 10.1146/annurev.nu.16.070196.001315. [DOI] [PubMed] [Google Scholar]
- 4.Santer R, Klepper J. Disorders of glucose transport. In: Saudubray JM, van den Berghe G, Walter JH, editors. Inborn Metabolic Diseases: Diagnosis and Treatment. Springer; New York: 2011. pp. 175–181. [Google Scholar]
- 5.Macheda ML, Rogers S, Best JD. Molecular and cellular regulation of glucose transporter (GLUT) proteins in cancer. J Cell Physiol. 2005;202(3):654–662. doi: 10.1002/jcp.20166. [DOI] [PubMed] [Google Scholar]
- 6.Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 7.Amann T, Hellerbrand C. GLUT1 as a therapeutic target in hepatocellular carcinoma. Expert Opin Ther Targets. 2009;13(12):1411–1427. doi: 10.1517/14728220903307509. [DOI] [PubMed] [Google Scholar]
- 8.Shim BY, et al. Glucose transporter 1 (GLUT1) of anaerobic glycolysis as predictive and prognostic values in neoadjuvant chemoradiotherapy and laparoscopic surgery for locally advanced rectal cancer. Int J Colorectal Dis. 2013;28(3):375–383. doi: 10.1007/s00384-012-1542-3. [DOI] [PubMed] [Google Scholar]
- 9.Ramani P, Headford A, May MT. GLUT1 protein expression correlates with unfavourable histologic category and high risk in patients with neuroblastic tumours. Virchows Arch. 2013;462(2):203–209. doi: 10.1007/s00428-012-1370-4. [DOI] [PubMed] [Google Scholar]
- 10.Airley RE, Mobasheri A. Hypoxic regulation of glucose transport, anaerobic metabolism and angiogenesis in cancer: Novel pathways and targets for anticancer therapeutics. Chemotherapy. 2007;53(4):233–256. doi: 10.1159/000104457. [DOI] [PubMed] [Google Scholar]
- 11.Sergeant S, Kim HD. Inhibition of 3-O-methylglucose transport in human erythrocytes by forskolin. J Biol Chem. 1985;260(27):14677–14682. [PubMed] [Google Scholar]
- 12.Carruthers A, Helgerson AL. Inhibitions of sugar transport produced by ligands binding at opposite sides of the membrane. Evidence for simultaneous occupation of the carrier by maltose and cytochalasin B. Biochemistry. 1991;30(16):3907–3915. doi: 10.1021/bi00230a015. [DOI] [PubMed] [Google Scholar]
- 13.Deng D, et al. Crystal structure of the human glucose transporter GLUT1. Nature. 2014;510(7503):121–125. doi: 10.1038/nature13306. [DOI] [PubMed] [Google Scholar]
- 14.Deng D, et al. Molecular basis of ligand recognition and transport by glucose transporters. Nature. 2015;526(7573):391–396. doi: 10.1038/nature14655. [DOI] [PubMed] [Google Scholar]
- 15.Jung CY, Rampal AL. Cytochalasin B binding sites and glucose transport carrier in human erythrocyte ghosts. J Biol Chem. 1977;252(15):5456–5463. [PubMed] [Google Scholar]
- 16.Hays FA, Roe-Zurz Z, Stroud RM. Overexpression and purification of integral membrane proteins in yeast. Methods Enzymol. 2010;470:695–707. doi: 10.1016/S0076-6879(10)70029-X. [DOI] [PubMed] [Google Scholar]
- 17.Inukai K, et al. Replacement of both tryptophan residues at 388 and 412 completely abolished cytochalasin B photolabelling of the GLUT1 glucose transporter. Biochem J. 1994;302(Pt 2):355–361. doi: 10.1042/bj3020355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Katagiri H, et al. Substitution of leucine for tryptophan 412 does not abolish cytochalasin B labeling but markedly decreases the intrinsic activity of GLUT1 glucose transporter. J Biol Chem. 1991;266(12):7769–7773. [PubMed] [Google Scholar]
- 19.Augustin R. The protein family of glucose transport facilitators: It’s not only about glucose after all. IUBMB Life. 2010;62(5):315–333. doi: 10.1002/iub.315. [DOI] [PubMed] [Google Scholar]
- 20.Lachaal M, Spangler RA, Jung CY. Adenosine and adenosine triphosphate modulate the substrate binding affinity of glucose transporter GLUT1 in vitro. Biochim Biophys Acta. 2001;1511(1):123–133. doi: 10.1016/s0005-2736(01)00272-3. [DOI] [PubMed] [Google Scholar]
- 21.McDonald TP, Walmsley AR, Martin GE, Henderson PJ. The role of tryptophans 371 and 395 in the binding of antibiotics and the transport of sugars by the D-galactose-H+ symport protein (GalP) from Escherichia coli. J Biol Chem. 1995;270(51):30359–30370. doi: 10.1074/jbc.270.51.30359. [DOI] [PubMed] [Google Scholar]
- 22.McDonald TP, Walmsley AR, Henderson PJ. Asparagine 394 in putative helix 11 of the galactose-H+ symport protein (GalP) from Escherichia coli is associated with the internal binding site for cytochalasin B and sugar. J Biol Chem. 1997;272(24):15189–15199. doi: 10.1074/jbc.272.24.15189. [DOI] [PubMed] [Google Scholar]
- 23.Leslie AG. The integration of macromolecular diffraction data. Acta Crystallogr D Biol Crystallogr. 2006;62(Pt 1):48–57. doi: 10.1107/S0907444905039107. [DOI] [PubMed] [Google Scholar]
- 24.Winn MD, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67(Pt 4):235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Evans P. Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr. 2006;62(Pt 1):72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 26.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. In: Abelson JN, Simon MI, Carter CW Jr, Sweet RM, editors. Methods in Enzymology. Vol 276. Academic; New York: 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 27.Karplus PA, Diederichs K. Linking crystallographic model and data quality. Science. 2012;336(6084):1030–1033. doi: 10.1126/science.1218231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Adams PD, et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 30.Davis IW, et al. MolProbity: All-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35(Web Server issue):W375–W383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Benkert P, Kunzli M, Schwede T. QMEAN server for protein model quality estimation. Nucleic Acids Res. 2009;37(Web Server issue):W510–W514. doi: 10.1093/nar/gkp322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morris AL, MacArthur MW, Hutchinson EG, Thornton JM. Stereochemical quality of protein structure coordinates. Proteins. 1992;12(4):345–364. doi: 10.1002/prot.340120407. [DOI] [PubMed] [Google Scholar]
- 33.Friesner RA, et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47(7):1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 34.Li J, et al. The VSGB 2.0 model: A next generation energy model for high resolution protein structure modeling. Proteins. 2011;79(10):2794–2812. doi: 10.1002/prot.23106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ulanovskaya OA, Cui J, Kron SJ, Kozmin SA. A pairwise chemical genetic screen identifies new inhibitors of glucose transport. Chem Biol. 2011;18(2):222–230. doi: 10.1016/j.chembiol.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ulanovskaya OA, et al. Synthesis enables identification of the cellular target of leucascandrolide A and neopeltolide. Nat Chem Biol. 2008;4(7):418–424. doi: 10.1038/nchembio.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Craig FF, Simmonds AC, Watmore D, McCapra F, White MR. Membrane-permeable luciferin esters for assay of firefly luciferase in live intact cells. Biochem J. 1991;276(Pt 3):637–641. doi: 10.1042/bj2760637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weiss MS. Global indicators of X-ray data quality. J Appl Crystallog. 2001;34:130–135. [Google Scholar]