

ABSTRACT
The role of inflammation in tendon injury is uncertain and a topic of current interest. In vitro studies of tendon accelerated overload damage can serve as a valuable source of information on the early stages of tendinopathy. Viable fascicle bundles from bovine flexor tendons were subjected to cyclic uniaxial loading from 1–10% strain. Immuno‐staining for inflammatory markers and matrix degradation markers was performed on the samples after mechanical testing. Loaded samples exhibited visible extracellular matrix damage, with disrupted collagen fibers and fiber kinks, and notable damage to the interfascicular matrix. Inflammatory markers COX‐2 and IL‐6 were only expressed in the cyclically loaded samples. Collagen degradation markers MMP‐1 and C1,2C were colocalized in many areas, with staining occurring in the interfascicular matrix or the fascicular tenocytes. These markers were present in control samples, but staining became increasingly intense with loading. Little MMP‐3 or MMP‐13 was evident in control sections. In loaded samples, some sections showed intense staining of these markers, again localized to interfascicular regions. This study suggests that inflammatory markers may be expressed rapidly after tendon overload exercise. Interestingly, both inflammation and damage‐induced matrix remodeling seem to be concentrated in, or in the vicinity of, the highly cellular interfascicular matrix. © 2015 The Authors. Journal of Orthopaedic Research Published by Wiley Periodicals, Inc. J Orthop Res 33:889–897, 2015.
Keywords: tendon, fatigue, inflammation, matrix degradation, ECM damage, interfascicular matrix, collagen
Tendinopathy is a general term for chronic tendon disease1 characterized by a combination of pain, swelling, and impaired tendon performance.2 The aetiology of this disease remains to be elucidated and is likely to be multi‐factorial. Tendinopathy is often the result of damage accumulation during overuse, and may also involve either mechanical over‐stimulation or under‐stimulation of tenocytes and the related (imbalanced) remodeling in tendon tissue.1 Tendinopathy has often been considered a degenerative disease1 not associated with inflammation. However the role of inflammation within the initiation, progression, and resolution of tendinopathy remains unclear.3
Tendons have an inhomogeneous multilevel hierarchical composite structure. Considering the mesoscale, looking at the largest hierarchical subunits in tendon, it is possible to describe tendon as a composite material with two main phases (or matrix components). The phases are the fascicles (fascicular matrix, FM), which are longitudinal structures composed predominantly of uniaxially arranged type I collagen fibers, and the interfascicular matrix (IFM) or endotenon that fills the spaces between fascicles (see Fig. 1). IFM is a highly cellular matrix phase consisting of a range of non‐collagenous proteins including elastin, proteoglycans, and also minor collagens.4
Figure 1.
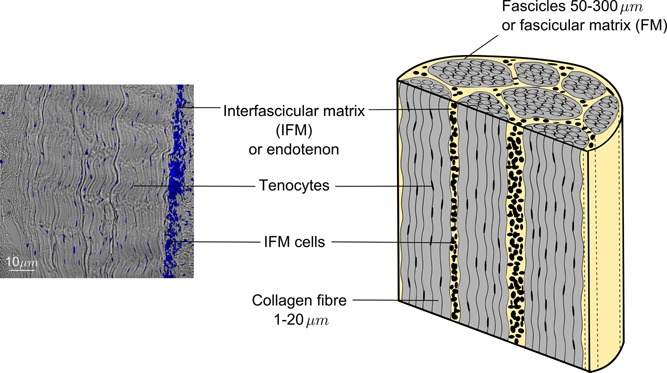
Schematic and histological section of a bovine flexor tendon, depicting the two phases (components) of the tendons structure at the meso‐scale: collagen‐rich fascicles, termed fascicular matrix (FM) and the surrounding endotendon, termed interfascicular matrix (IFM). Notice the different cell shapes and numbers within fascicles (tenocytes) and the IFM (IFM cells). Cell nuclei are stained with DAPI (blue).
The IFM has a more sparse fiber arrangement, with lower volume fractions of fibrillar material than fascicular matrix. Arrangement of fibers in the IFM is more network‐like, which suggests softer, and likely more complex mechanical behavior5, 6 as compared to the uniaxially arranged, mainly collagen type I, fibers in fascicles. The collagenous phase volume fraction and the orientation of fibers both have previously been shown to have a direct impact on tissue mechanics,7, 8, 9, 10 suggesting that the interfascicular matrix is likely more prone to damage and injury. This softer phase is thought to create sliding planes between fascicles that facilitate increased tendon extension and recoil.11 However, the exact role of the interfascicular matrix in tendon injury or remodeling remains uncertain.
Tendons are thought to undergo slow remodeling mediated by matrix metalloproteinases (MMPs) and disintegrins and metalloproteinases with thrombospondin motifs (ADAMTS), whose activity is inhibited though tissue inhibitors of metalloproteinases (TIMPs).1
These enzymes, and their inhibitors, have roles in extracellular matrix turnover in health but also in pathological conditions. However, it remains unclear how tendon turnover is mediated by the tendon cells population. The cell population in tendon is heterogeneous, consisting of elongated tenocytes between collagen fibers within the FM and IFM cells which are more rounded in shape (Fig. 1). However, there are as yet no specific markers to distinguish these types of cells from each other or from other connective tissue cells.1 It has been shown that cell shape is influenced by local mechanical properties,12, 13 and the two cell populations of tendon likely live in mechanically distinct environments (stiffer FM or softer IFM). In addition, the IFM regions are more highly cellular than FM regions. The turnover of non‐collagenous matrix has been shown to be faster than that of collagenous matrix in tendon. The greater quantity of non‐collagenous matrix in the IFM thus suggests that IFM cells may be more active in healthy turnover, but they may also play different roles in tendon injury and repair.14
The role of an inflammatory response in tendinopathy has been discussed in the literature, but no consensus has been achieved. How the inflammation results in damage accumulation and the initiation of tendinopathy is not easily investigated in human subjects, because the affected tendons can normally only be assessed scientifically once tendinopathic changes are advanced. Therefore, in vitro studies of the inflammatory response to induced tendon overload damage can serve as a valuable source of information on the early stages of tendinopathy. The goal of this project was to investigate the influence of cyclic overload exercise performed in vitro on the expression of inflammatory mediators and markers of matrix degradation in bovine tendons comparing the response of FM and IFM regions. We hypothesized that cyclic loading would result in an increase in the levels of the inflammatory markers COX‐2 and IL‐6, and would also increase levels of the matrix degrading enzymes MMP‐1, ‐3, and ‐13, resulting in increased collagen degradation. Further we hypothesized that the response to overload would be localized predominantly in the IFM.
METHODS
Samples and Loading Protocol
Deep digital flexor tendons from the forelimbs of skeletally mature adult cows aged 18 months (n = 3) were obtained from an abattoir immediately post‐mortem. As we aimed to investigate the matrix response from both collagen‐rich fascicles and the interfascicular matrix, fascicle bundles (n = 12) containing both types of matrix were dissected (Fig. 2). The fascicle bundles we tested in this study were on average 1.5 mm in diameter, incorporating a minimum of three fascicles. From each tendon, one fascicle bundle was assigned to each test group:
CON ‐ fresh control: snap frozen immediately after dissection;
STA ‐ static control: held at 1% strain for 24h;
300C ‐ cyclically loaded for 300 cycles (5min) from 1–10% uniaxial strain (1Hz) followed by 1% static strain for 24h;
1,800C ‐ cyclically loaded for 1,800 cycles (30min) from 1–10% uniaxial strain (1Hz) followed by 1% static strain for 24h.
Figure 2.
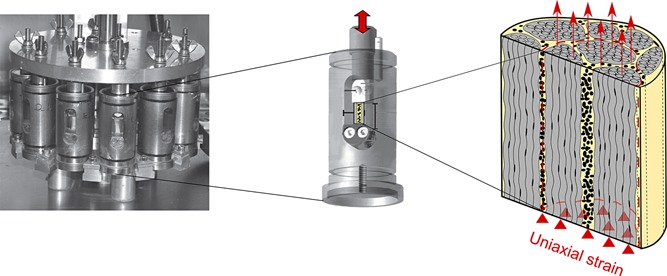
Loading system used to apply overload exercise to fascicle bundles.16 Test samples contain collagen‐rich fascicles (gray) and interfascicular matrix (yellow) which allows for later observation of the both regions' response to overload exercise.
Samples from groups 2–4 were placed into individual chambers of the multiple chamber system (Fig. 2), gripped with a gauge length of 10 mm. The cyclically loaded samples (groups 3–4) were subjected to large cyclic strains from 1–10%, using a Bose mechanical test system (Wintest v 4.0, Bose Coorporation, USA) housed in an incubator. The straining regimes have previously been shown to simulate overload.15 During testing the samples were maintained in sterile cell culture medium (DMEM) supplemented with penicillin (50 U/ml), streptomycin (0.05 mg/ml) and l‐glutamine (2 mM) and maintained in an incubator at 37 °C, 20% O2, 5% CO2.
After the end of the 24 h test period, samples were removed from the chambers and snap frozen in optimal cutting temperature (OCT compound, VWR) embedding medium (cooled in hexane on dry ice) and stored at −80 °C. Three 20 µm thick longitudinal cryo‐sections were cut from each sample and mounted on poly‐l‐lysine coated slides for further analysis.
Immuno‐staining
Dual immuno‐staining for the following three marker pairs was performed:
inflammatory markers ‐ cyclooxygenase (COX‐2) and interleukin (IL‐6).
matrix degradation markers: matrix metalloproteinase MMP‐1 and collagen degradation marker C1,2C.
matrix degradation markers: matrix metalloproteinases MMP‐3 and MMP‐13.
Details of the antibodies used are shown in Table 1.
Table 1.
Details of the Antibodies Used
| Marker | Target | Primary antibody | Concentration | Secondary antibody | Concentration | Validation |
|---|---|---|---|---|---|---|
| IL‐6 Interleukin‐6 | Inflammatory marker | Goat polyclonal anti‐IL6 (R&D systems AF1886) | 1:25 | Donkey anti‐goat 543 (red) | 1:500 | 16 |
| COX‐2 Cyclo‐oxygenase‐2 | Inflammatory marker | Mouse monoclonal anti‐Cox‐2 (Cayman Chemical 160112) | 1:50 | Donkey anti‐mouse 488 (green) | 1:500 | 17, 18 |
| MMP‐1 Matrix metallo‐proteinase‐1 | Fibrillar collagens degradation | Goat polyclonal anti‐MMP1 (Santa Cruz sc‐6837) | 1:50 | Donkey anti‐goat 543 (red) | 1:500 | 19 |
| C1,2C | Collagen type I and II degradation | Rabbit polyclonal anti‐C1,2C (Abcam 18898) | 1:20 | Donkey anti‐rabbit 488 (green) | 1:500 | 20, 21 |
| MMP‐3 Matrix metallo‐proteinase‐3 | Minor matrix proteins degradation (collagens III, IV, IX and X, proteoglycans, elastin) | Goat polyclonal anti‐MMP3 (Abcam 18898) | 1:25 | Donkey anti‐goat 543 (red) | 1:500 | 22 |
| MMP‐13 Matrix metallo‐proteinase‐13 | Fibrillar collagens degradation | Rabbit polyclonal anti‐MMP13 (Santa Cruz sc‐30073) | 1:50 | Donkey anti‐rabbit 488 (green) | 1:500 | 23 |
Both the inflammatory markers selected, IL‐624, 25 and COX‐226, 27 have previously been shown to be upregulated in tendinopathy. MMPs 1 and 13 were selected as they are known to be active against fibrillar collagens, including the predominant collagen type I.1, 28, 29, 30 MMP‐3 was selected as it degrades other minor proteins in tendon matrix, including collagens III, IV, IX, and X, and also proteoglycans and elastin.28, 29, 30, 31, 32, 33 The C1,2C (or COL 2 3/4C short) antibody detects the carboxy terminus of fragmented type I and II collagen, cleaved by collagenases MMP‐1, MMP‐8 or MMP‐13.21, 34, 35, 36, 37
Cryo‐sections were thawed and fixed with acetone and then rehydrated in phosphate buffered saline (PBS, Sigma–Aldrich) followed by tris buffered saline (TBS, Sigma–Aldrich, Dorset, United Kingdom) plus 0.1% Triton X‐100 (Sigma–Aldrich, Dorset, United Kingdom) to permeabilize cell membranes. Blocking buffer‐TBS with 1% bovine serum albumin, (BSA, Sigma–Aldrich, Dorset, United Kingdom) 10% donkey serum (Sigma–Aldrich, Dorset, United Kingdom) was applied to sections for 2 h at room temperature. After draining, the primary antibodies diluted in blocking buffer with 1% BSA were then applied. Concentrations of antibodies used are listed in Table 1. For negative controls, blocking buffer without the primary antibody was applied. Sections were incubated overnight at 4 °C. Next, sections were rinsed in TBS with 0.1% Triton, followed by the application of the fluorophore‐conjugated secondary antibodies, diluted to the concentration listed in Table 1 in blocking buffer, and incubated for 1 h at room temperature. This step was done in the dark to avoid photo‐bleaching. Finally, sections were rinsed in PBS, mounted with DAPI containing mounting medium (ProLong Gold, Life Technologies, Paisley, UK) and left overnight at 4 °C to allow distinction of the highly cellular IFM regions from less cellular fascicular regions. Negative control sections were also imaged, to confirm lack of binding of secondary antibodies.
Imaging, Image Processing, and Analysis
Sections were imaged with a confocal microscope (Leica TCS SP2) using a 20 × objective and 2 × digital zoom. Within each slide, at least one region was imaged, in an area including fascicular and interfascicular matrix. The region was selected using bright field imaging, ensuring an area incorporating both interfascicular matrix and fascicular matrix were selected, but blinding the selector to the extent of staining for any marker during region selection. Four matching images were taken of each region: bright field; blue channel (wavelength 351 nm) for visualization of DAPI showing cell nuclei; green channel (wavelength 488 nm); and red channel (wavelength 543 nm) used for each pair of dual antibodies investigated.
Semi‐quantitative analyses of the images were performed in Gimp 2.8 software (GNU Image Manipulation Program). Images were thresholded at a single level threshold (for a given stain) and overlaid for visualization and to facilitate a qualitative assessment of the extent and intensity of immuno‐staining within IFM and fascicles (FM). All images were assessed for the number of pixels falling above the threshold. The brightness of each pixel falling above the threshold was first measured after which an average image brightness was calculated. Stain intensity was calculated, defined as the stain brightness multiplied by the stained area.38
One hundred and eight stained sections have been imaged and evaluated from which mean stain intensity and standard deviation was determined for each antibody used across all load conditions. Some images were discarded based on features like prominent blood vessels in the field of view. Sections with a bright background or large artefactual dots visible across both green and red channels (possible dirt particles) were also discarded.
RESULTS
Tendon Matrix Damage
Signs of fibrils damage were visible in sections of tendons that were loaded for either 300 or 1,800 cycles (marked with white ellipses in Figures Fig. 3, Fig. 4,3, 4 and 5). fiber kinks and damage were visible and increased in severity with increasing load regime. Damage was also evident in the IFM as disruption of the structure.
Figure 3.

Typical static (a) and loaded (b, c) samples immunostained for inflammatory markers. Damage is evident in both the fascicles and the interfascicular matrix of the loaded samples (examples of damage— collagen fibers kinks and IFM disruption shown with white ellipses). Both inflammatory markers were only expressed in loaded samples, with COX‐2 expression increasing with the cycle number. Scale bar is 10 µm.
Figure 4.
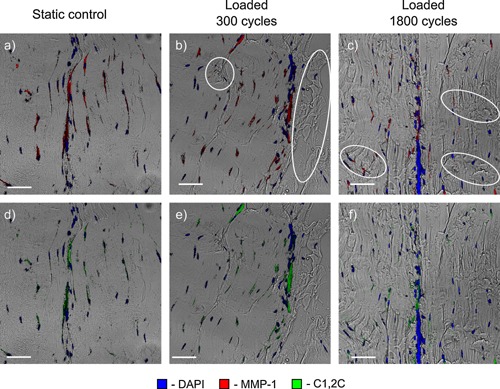
Typical static (a, d) and loaded (b, c, e, f) samples immunostained for collagen matrix degradation markers. MMP‐1 and C1,2C are colocalized and associated predominantly with the interfascicular matrix, in both control and loaded samples, indicating continual turnover of the interfascicular matrix. Damaged areas of fibers, fiber kinks, and the IFM damage are marked with white ellipses. Scale bar is 10 µm.
Figure 5.
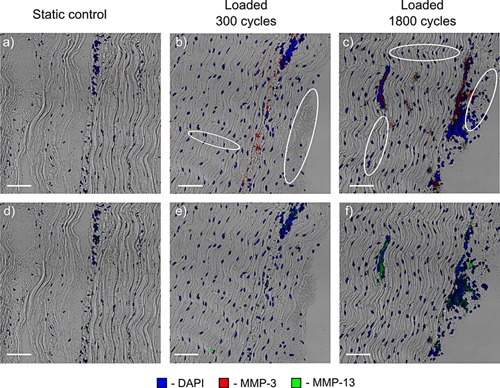
Typical static (a, d) and loaded (b, c, e, f) samples immunostained for matrix degradation markers. MMP‐13 is associated with areas in which there is a large aggregation of tenocytes and in the interfascicular matrix. MMP‐3 expression levels are higher and also remain mainly related to interfascicular matrix. Damaged areas of fibers and the IFM are marked with white ellipses. Scale bar is 10 µm.
A semi‐quantitative evaluation of the inflammatory and matrix degradation markers was carried out, as summarized in Figure 6 and discussed below.
Figure 6.
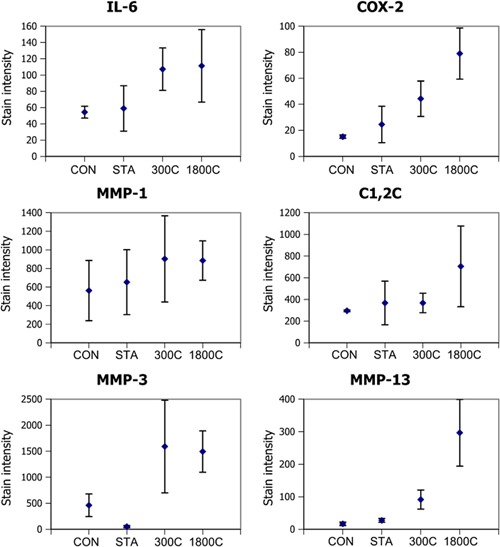
Changes in stain intensity for control samples (CON), statically loaded samples (STA), samples loaded for 300 cycles (300C) and samples loaded for 1,800 cycles (1,800C) calculated as an average across the whole sample. Test groups show the mean values for the stain intensity (defined as stain brightness multiplied by the stained area). Error bars show standard deviations and are shown for all data sets. Note the differences in the levels of stain for different antibodies (y‐axes), with the highest levels of expression for MMP‐3 and the lowest for the inflammatory markers (IL‐6 and COX‐2).
Inflammatory Markers
Inflammatory markers were only expressed in the cyclically loaded samples (see Fig. 3) with staining seen within both the FM, and IFM.
The intensity of IL‐6 staining was similar between the samples loaded for 300 and 1,800 cycles, while the areas of COX‐2 staining (see Fig. 3) increased substantially with increasing time of loading.
Matrix Degradation Markers
The matrix degrading enzyme MMP‐1 and degradation marker C1,2C were present in control samples (Fig. 4 a and d) as well as the loaded samples (Fig. 4 b, c, e, f), with the high levels of these markers in controls suggesting continuous tendon turnover.
MMP‐1 and C1,2C were colocalized in many areas therefore they are shown in separate images in Figure 4. Whilst staining for these antibodies was observed within the FM and often associated with tenocytes, it was predominantly localized to the IFM, suggesting that continual turnover is predominantly localized to the IFM.
MMP‐1 stain intensity (Fig. 6) showed a trend towards increasing with loading, as did the area stained with C1,2C. Standard deviations in MMP‐1 and C1,2C staining data were both high with different tendons showing large variability. However data indicated possible dose dependent staining, suggesting increased collagen breakdown with more overload.
By contrast, little MMP‐3 or MMP‐13 was evident in either control group. However, in loaded samples, some sections showed increased staining of these markers, again localized mainly to the IFM (see Fig. 5). While MMP‐3 staining showed a similar intensity in both loaded groups, the response appeared to be more strain dose dependent for MMP‐13 (Fig. 6).
Fold Change of Markers With Increasing Loading Time
Figure 7 presents the mean fold change in stain intensity (stain brightness multiplied with stained area) with respect to the fresh frozen controls (shown by the horizontal dotted line) for each stain and all load conditions. No visible changes were observed between fresh frozen and static control samples for any stain. Fold changes between fresh frozen and static controls were between 1.1 and 1.6 for all stains except MMP‐3, where static controls showed, on average, less staining than the freshly frozen controls.
Figure 7.
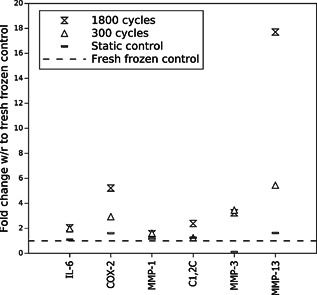
Mean fold change in stain intensity in the loaded samples (static controls, samples loaded for 300 and 1,800 cycles) with respect to fresh frozen controls (dotted line).
There are clear fold increases in inflammatory markers with strain in loaded samples: 2.0 for IL‐6 and 5.2 for COX‐2, for the samples loaded for 1,800 cycles (Fig. 7). The highest fold increase of 17.7 was observed for MMP‐13.
Inflammatory markers and matrix degrading enzymes MMP‐3 and MMP‐13 all showed a clear increase with load, suggesting a response to cyclical mechanical loading. By contrast, MMP‐1 and C1,2C were both evident in control samples and showed little change with loading, suggesting the constant turnover is less affected by the range of the overload strains applied in our in vitro experiments.
DISCUSSION
This study subjects viable bundles of fascicles from bovine flexor tendons to overload damage, adopting immunohistochemistry to investigate the cellular response to injury. The in vitro overload protocol adopted here has previously been validated, to ensure the generation of accelerated matrix damage,15 and data indicated a rapid inflammatory response from tendon cells directly after overload. An in vitro model of overuse such as this is unlikely to fully recapitulate the in vivo environment, but allows for precise control of the loading environment, which it is otherwise very difficult to achieve. Further, our in vitro model is likely to result in larger and more uniform straining across our fascicle bundles than would be seen in vivo, where strains are distributed across the whole tendon's hierarchical structure and along the whole length of the tendon. However, this accelerated fatigue model facilitates assessment of the cell response to local tendon damage.16
Immuno‐staining and semi‐quantification relies on sufficient antibodies having access to epitope binding sites within the tissue section. The structure of the two phases of tendon: FM and IFM is different, with densely packed collagenous fibers (mainly collagen type I) within the FM and sparser, network‐like fibrillar structure in the IFM. It is feasible that antibody binding epitopes were more accessible in the IFM than in the dense FM structure, which could lead to diminished staining in the FM phase of tendon. Also different antibodies are likely to have different binding affinities, which makes the comparison between different stains difficult. A detailed solution for this problem could be devised with molecular dynamics simulations of the particular biological surface and the immuno‐staining solution, but this was not in the scope of the work presented here.
This study suggests that inflammatory markers may be expressed rapidly and early after tendon overload exercise (within 24 h following 5 or 30 min of overload exercise in our in vitro model), indicating that inflammation may be an early occurrence in tendinopathy. We choose to study IL‐6 as this inflammatory mediator has previously been linked to tendinopathy24 and COX‐2 as increased levels of COX‐2 have been shown in patellar tendinopathy,26, 27 loaded human tendon cells39 and injured tendons.40 Other markers of inflammation such as IL‐1 were not included in this study but have previously been implicated in tendinopathic tendons41, 42 or bursa of the shoulder.25 In contrast, other studies have shown no inflammatory response related to pathological tendons.43
The levels of MMP‐1 increased with loading, correlating with previous studies, which have shown increased MMP‐1 expression in tendinopathic44, 45 or ruptured46 tendons. Further, MMP‐1 expression has recently been shown to increase with overload fatigue inducing exercise in equine tendons.47 Interestingly, the increased levels of MMP‐1 in the IFM colocalized with staining for the C1,2C marker that detects the fragmented type I and II collagen, indicating that the MMP‐1 present is in an active form.
The up‐regulation of MMP‐3 and MMP‐13 in the current study also agrees with previous work, showing up‐regulation of both these MMPs in pathological tendons.45, 47, 48, 49, 50 However, MMP‐3 has also been shown to be down‐regulated in both ruptured and tendinopathic tendons, as shown by the mRNA expression level.46 The difference in response may be attributed to both timing (post‐injury) and the method used for identification of tissue response.
Interestingly, in this study, both inflammation and damage‐induced matrix remodeling seem to be concentrated in or in the vicinity of the IFM. This could be partially explained by the high number of cells present in the IFM as compared to fascicles, with more cells available to respond to overload of the IFM. However, it is also possible that IFM cells and tenocytes differ in their phenotype and activity levels. Their respective roles in initiation, progression, and resolution of inflammation, and the consequences for tendinopathy are yet to be elucidated.
Further, the mechanical role of IFM in tendon extension remains unclear. However, it has been hypothesized to be instrumental in allowing fascicles to slide with respect to each other,11 allowing higher tendon deformations before the onset of fascicle damage. In physiological use, the IFM may thus be subjected to higher shear stresses then the fascicular matrix, and as a sparse, softer component of tendon, may be more prone to damage and injury. This could partially explain the higher turnover rates in this phase as compared to fascicular matrix (observed high levels of MMP‐1 and C1,2C in control samples). IFM may need to turnover more rapidly and respond more robustly to damage to ensure proper IFM and tendon function. Further characterization of the mechanobiology of the IFM is needed to fully understand its role.
Authors' Contribution
All authors have substantially contributed to research design, or the acquisition, analysis or interpretation of data, drafting the paper or revising it critically and have approved of the submitted version.
ACKNOWLEDGEMENTS
The work was funded by the Wellcome Trust (WT087112) and Arthritis Research UK (20262).
REFERENCES
- 1. Riley G. 2008. Tendinopathy‐from basic science to treatment. Nat Clin Pract Rheumatol 4:82–89. [DOI] [PubMed] [Google Scholar]
- 2. Maffulli N, Khan KM, Puddu G. 1998. Overuse tendon conditions: time to change a confusing terminology. Arthroscopy 14:840–843. [DOI] [PubMed] [Google Scholar]
- 3. Dakin SG, Dudhia J, Smith RKW. 2014. Resolving an inflammatory concept: the importance of inflammation and resolution in tendinopathy. Vet Immunol Immunopathol 158:121–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Thorpe CT, Birch HL, Clegg PD, et al. 2013. The role of the non‐collagenous matrix in tendon function. Int J Exp Pathol 94:248–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Storm C, Pastore JJ, MacKintosh FC, et al. 2005. Nonlinear elasticity in biological gels. Nature 435:191–194. [DOI] [PubMed] [Google Scholar]
- 6. Cioroianu AR, Spiesz EM, Storm C. 2013. An improved non‐affine arruda‐boyce type constitutive model for collagen networks. Biophys J 104:511. [Google Scholar]
- 7. Spiesz EM, Reisinger AG, Kaminsky W, et al. 2013. Computational and experimental methodology for site‐matched investigations of the influence of mineral mass fraction and collagen orientation on the axial indentation modulus of lamellar bone. J Mech Behav Biomed Mater 28:195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Spiesz EM, Reisinger AG, Roschger P, et al. 2014. Experimental validation of a mean filed model of mineralized collagen fiber arrays at two levels of hierarchy. J Mech Med Biol 14:1450013. [Google Scholar]
- 9. Spiesz EM, Roschger P, Zysset PK. 2012. Influence of mineralization and microporosity on tissue elasticity: experimental and numerical investigation on mineralized turkey leg tendons. Calcif Tissue Int 90:319–329. [DOI] [PubMed] [Google Scholar]
- 10. Spiesz EM, Kaminsky W, Zysset PK. 2011. A quantitative collagen fibers orientation assessment using birefringence measurements: calibration and application to human osteons. J Struct Biol 176:302–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Thorpe CT, Udeze CP, Birch HL, et al. 2012. Specialization of tendon mechanical properties results from interfascicular differences. J R Soc Interface 9:3108–3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Discher DE, Janmey Pl, Wang Y‐L. 2005. Tissue cells feel and respond to the stiffness of their substrate. Science 310:1139–1143. [DOI] [PubMed] [Google Scholar]
- 13. van Hoorn H, Harkes R, Spiesz EM, et al. 2014. The nanoscale architecture of force‐bearing focal adhesions. Nano Lett 14:4257–4262. [DOI] [PubMed] [Google Scholar]
- 14. Thorpe CT, Streeter I, Pinchbeck GL, et al. 2010. Aspartic acid racemization and collagen degradation markers reveal an accumulation of damage in tendon collagen that is enhanced with aging. J Biol Chem 285:15674–15681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shepherd JH, Riley GP, Screen HRC. 2014. Early stage fatigue damage occurs in bovine tendon fascicles in the absence of changes in mechanics at either the gross or micro‐structural level. J Mech Behav Biomed Mater 38:163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Legerlotz K, Jones GC, Screen HRC, et al. 2013. Cyclic loading of tendon fascicles using a novel fatigue loading system increases interleukin‐6 expression by tenocytes. Scand J Med Sci Sports 23:31–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Borzacchiello G, Ambrosio V, Galati P, et al. 2003. Cyclooxygenase‐1 and ‐2 expression in urothelial carcinomas of the urinary bladder in cows. Vet Pathol 40:455–459. [DOI] [PubMed] [Google Scholar]
- 18. Cho MK, Cho YH, Lee GH, et al. 2004. Induction of cyclooxygenase‐2 by bovine type I collagen in macrophages via c/ebp and creb activation by multiple cell signaling pathways. Biochem Pharmacol 67:2239–2250. [DOI] [PubMed] [Google Scholar]
- 19. Devy J, Duca L, Cantarelli B, et al. 2010. Elastin‐derived peptides enhance melanoma growth in vivo by upregulating the activation of mcol‐a (MMP‐1) collagenase. Br J Cancer 103:1562–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hosseini SM, Veldink MB, Ito K, et al. 2013. Is collagen fiber damage the cause of early softening in articular cartilage? Osteoarthritis Cartilage 21:136–143. [DOI] [PubMed] [Google Scholar]
- 21. Billinghurst RC, Wu W, Ionescu M, et al. 2000. Comparison of the degradation of type II collagen and proteoglycan in nasal and articular cartilages induced by interleukin‐1 and the selective inhibition of type II collagen cleavage by collagenase. Arthritis Rheum 43:664–672. [DOI] [PubMed] [Google Scholar]
- 22. Lin PM, Chen C‐TC, Torzilli PA. 2004. Increased stromelysin‐1 (MMP‐3), proteoglycan degradation (3b3‐ and 7d4) and collagen damage in cyclically load‐injured articular cartilage. Osteoarthritis Cartilage 12:485–496. [DOI] [PubMed] [Google Scholar]
- 23. Danfelter M, Onnerfjord P, Heinegard D. 2007. Fragmentation of proteins in cartilage treated with interleukin‐1: specific cleavage of type IX collagen by matrix metalloproteinase 13 releases the nc4 domain. J Biol Chem 282:36933–36941. [DOI] [PubMed] [Google Scholar]
- 24. Langberg H, Skovgaard D, Karamouzis M, et al. 1999. Metabolism and inflammatory mediators in the peritendinous space measured by microdialysis during intermittent isometric exercise in humans. J Physiol 515:919–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gotoh M, Hamada K, Yamakawa H, et al. 2002. Interleukin‐1‐induced glenohumeral synovitis and shoulder pain in rotator cuff diseases. J Orthop Res 20:1365–1371. [DOI] [PubMed] [Google Scholar]
- 26. Behzad H, Sharma A, Mousavizadeh R, et al. 2013. Mast cells exert pro‐inflammatory effects of relevance to the pathophyisology of tendinopathy. Arthritis Res Ther 15:R184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fu SC, Wang W, Pau HM, et al. 2002. Increased expression of transforming growth factor‐beta1 in patellar tendinosis. Clin Orthop Relat Res 400:174–183. [DOI] [PubMed] [Google Scholar]
- 28. Nagase H, Suzuki K, Morodomi T, et al. 1992. Activation mechanisms of the precursors of matrix metalloproteinases1, 2 and 3. Matrix Suppl 1:237–244. [PubMed] [Google Scholar]
- 29. de Mos M, van El B, DeGroot J, et al. 2007. Achilles tendinosis: changes in biochemical composition and collagen turnover rate. Am J Sports Med 35:1549–1556. [DOI] [PubMed] [Google Scholar]
- 30. Burrage PS, Mix KS, Constance E, et al. 2006. Matrix metalloproteinases: role in arthritis. Front Biosci 11:529–543. [DOI] [PubMed] [Google Scholar]
- 31. Suzuki K, Enghild JJ, Morodomi T, et al. 1990. Mechanisms of activation of tissue procollagenase by matrix metalloproteinase 3 (stromelysin). Biochemistry 29:10261–10270. [DOI] [PubMed] [Google Scholar]
- 32. Woessner JF. 1991. Matrix metalloproteinases and their inhibitors in connective tissue remodeling. FASEB J 5:2145–2154. [PubMed] [Google Scholar]
- 33. Gotoh M, Mitsui Y, Shibata H, et al. 2013. Increased matrix metalloprotease‐3 gene expression in ruptured rotator cuff tendons is associated with postoperative tendon retear. Knee Surg Sports Traumatol Arthrosc 21:1807–1812. [DOI] [PubMed] [Google Scholar]
- 34. Billinghurst RC, Dahlberg L, Ionescu M, et al. 1997. Enhanced cleavage of type II collagen by collagenases in osteoarthritic articular cartilage. J Clin Invest 99:1534–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Svoboda SJ, Harvey TM, Owens BD, et al. 2013. Changes in serum biomarkers of cartilage turnover after anterior cruciate ligament injury. Am J Sports Med 41:2108–2116. [DOI] [PubMed] [Google Scholar]
- 36. Kim T‐H, Stone M, Payne U, et al. 2005. Cartilage biomarkers in ankylosing spondylitis: relationship to clinical variables and treatment response. Arthritis Rheum 52:885–891. [DOI] [PubMed] [Google Scholar]
- 37. Frisbie DD, Mc Ilwraith CW, Arthur RM, et al. 2010. Serum biomarker levels for musculoskeletal disease in two‐ and three‐year‐old racing thoroughbred horses: A prospective study of 130 horses. Equine Vet J 42:643–651. [DOI] [PubMed] [Google Scholar]
- 38. Rizzardi AE, Johnson AT, Vogel RI, et al. 2012. Quantitative comparison of immunohistochemical staining measured by digital image analysis versus pathologist visual scoring. Diagn Pathol 7:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mousavizadeh R, Khosravi S, Behzad H, et al. 2014. Cyclic strain alters the expression and release of angiogenic factors by human tendon cells. PLoS ONE 9:e97356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ahmed AS, Li J, Schizas N, et al. 2014. Expressional changes in growth and inflammatory mediators during achilles tendon repair in diabetic rats: new insights into a possible basis for compromised healing. Cell Tissue Res 357:109–117. [DOI] [PubMed] [Google Scholar]
- 41. Manning CN, Havlioglu N, Knutsen E, et al. 2014. The early inflammatory response after flexor tendon healing: a gene expression and histological analysis. J Orthop Res 32:645–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schubert TEO, Weidler C, Lerch K, et al. 2005. Achilles tendinosis is associated with sprouting of substance p positive nerve fibres. Ann Rheum Dis 64:1083–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Attia M, Huet E, Gossard C, et al. 2013. Early events of overused supraspinatus tendons involve matrix metalloproteinases and emmprin/cd147 in the absence of inflammation. Am J Sports Med 41:908–917. [DOI] [PubMed] [Google Scholar]
- 44. Baroneza JE, Godoy‐Santos A, Ferreira Massa B, et al. 2014. Leme Godoy dos Santos. MMP‐1 promoter genotype and haplotype association with posterior tibial tendinopathy. Gene 547:334–337. [DOI] [PubMed] [Google Scholar]
- 45. Castagna A, Cesari E, Gigante A, et al. 2013. Metalloproteases and their inhibitors are altered in both torn and intact rotator cuff tendons. Musculoskelet Surg 97:39–47. [DOI] [PubMed] [Google Scholar]
- 46. Riley GP, Curry V, DeGroot J, et al. 2002. Matrix metalloproteinase activities and their relationship with collagen remodelling in tendon pathology. Matrix Biol 21:185–195. [DOI] [PubMed] [Google Scholar]
- 47. Thorpe CT, Chaudhry S, Lei I, et al. 2015. Tendon overload results in alterations in cell shape and increased markers of inflammation and matrix degradation. Scand J Med Sci Sports 01/2015; DOI: 10.1111/sms.12333 [DOI] [PubMed] [Google Scholar]
- 48. Gardner K, Arnoczky SP, Caballero O, et al. 2008. The effect of stress‐deprivation and cyclic loading on the timp/mmp ratio in tendon cells: an in vitro experimental study. Disabil Rehabil 30:1523–1529. [DOI] [PubMed] [Google Scholar]
- 49. Nomura M, Hosaka Y, Kasashima Y, et al. 2007. Active expression of matrix metalloproteinase‐13 mRNA in the granulation tissue of equine superficial digital flexor tendinitis. J Vet Med Sci 69:637–639. [DOI] [PubMed] [Google Scholar]
- 50. Sun HB, Li Y, Fung DT, et al. 2008. Coordinate regulation of IL‐1beta and MMP‐13 in rat tendons following subrupture fatigue damage. Clin Orthop Relat Res 466:1555–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]