

Abstract
Aicardi–Goutières syndrome (AGS) provides a monogenic model of nucleic acid‐mediated inflammation relevant to the pathogenesis of systemic autoimmunity. Mutations that impair ribonuclease (RNase) H2 enzyme function are the most frequent cause of this autoinflammatory disorder of childhood and are also associated with systemic lupus erythematosus. Reduced processing of either RNA:DNA hybrid or genome‐embedded ribonucleotide substrates is thought to lead to activation of a yet undefined nucleic acid‐sensing pathway. Here, we establish Rnaseh2b A174T/A174T knock‐in mice as a subclinical model of disease, identifying significant interferon‐stimulated gene (ISG) transcript upregulation that recapitulates the ISG signature seen in AGS patients. The inflammatory response is dependent on the nucleic acid sensor cyclic GMP‐AMP synthase (cGAS) and its adaptor STING and is associated with reduced cellular ribonucleotide excision repair activity and increased DNA damage. This suggests that cGAS/STING is a key nucleic acid‐sensing pathway relevant to AGS, providing additional insight into disease pathogenesis relevant to the development of therapeutics for this childhood‐onset interferonopathy and adult systemic autoimmune disorders.
Keywords: Aicardi–Goutières syndrome, autoinflammation, cGAS‐STING, ribonuclease H2
Subject Categories: Immunology
Introduction
Nucleic acid sensing is a key element of antimicrobial immunity (Wu & Chen, 2014). Diverse nucleic acid sensors act as pattern recognition receptors (PRRs), initiating innate immune responses necessary to protect against pathogens containing a diverse range of nucleic acid species (Wu & Chen, 2014). Such innate immune activation, arising from sensing of cellular nucleic acids, is also implicated in autoimmune disease, most notably systemic lupus erythematosus (SLE) (Wahren‐Herlenius & Dorner, 2013).
The autoinflammatory disorder Aicardi–Goutières syndrome (AGS) has provided important insights into mechanisms underlying nucleic acid‐mediated inflammation (Crow & Manel, 2015). AGS typically presents in infancy following a period of normal development, with sterile pyrexia, irritability, seizures and loss of developmental milestones (Crow & Livingston, 2008). Persisting severe physical and intellectual disability is frequent, and extra‐neurological features can include vasculitic skin lesions (Crow & Livingston, 2008; Crow et al, 2015). Increased type 1 interferon activity is most reliably detected during the early stages of the disease (Crow et al, 2015). However, upregulated interferon‐stimulated gene (ISG) transcripts in blood have proven to be the most robust diagnostic biomarker in AGS patients, and an “ISG signature” often persists long after the initial stages of disease (Rice et al, 2013; Crow et al, 2015).
Although AGS is a monogenic disorder, it is genetically heterogeneous, with seven genes implicated to date, encoding several nucleic acid processing enzymes and a cytosolic nucleic acid sensor. These comprise the RNASEH2A, RNASEH2B and RNASEH2C proteins of the RNase H2 endonuclease complex (Crow et al, 2006b) as well as TREX1, SAMHD1, ADAR and IFIH1 (Crow et al, 2006a; Rice et al, 2009, 2012, 2014). Heterozygous mutations in the three RNase H2 genes (Günther et al, 2015) and TREX1 (Lee‐Kirsch et al, 2007) are also associated with systemic lupus erythematosus.
Partial loss‐of‐function biallelic mutations in the RNase H2 genes are the major cause of AGS, accounting for over half of all cases (Crow et al, 2015). RNase H2 is ubiquitously expressed and functions alongside RNase H1 to degrade cellular RNA:DNA heteroduplexes. Although the exact in vivo substrates of these nucleases remain to be identified, they are thought to act on RNA:DNA hybrids such as those arising during nuclear DNA replication and R‐loop formation (Cerritelli & Crouch, 2009). Unlike RNase H1, RNase H2 also cleaves and initiates the removal of single ribonucleotides embedded in DNA (Eder et al, 1993; Rydberg & Game, 2002), a process known as ribonucleotide excision repair (RER) (Sparks et al, 2012). RNase H2 is essential for mammalian genome stability, with complete loss of RNase H2 resulting in embryonic lethality (Reijns et al, 2012; Hiller et al, 2012).
Intracellular accumulation of aberrant nucleic acid species is believed to trigger intrinsic nucleic acid sensors initiating autoimmunity in AGS (Crow & Manel, 2015). Trex1 −/− and Adar1 −/− mouse models have, respectively, implicated the cGAS‐STING (Ablasser et al, 2014; Ahn et al, 2014a; Gray et al, 2015; Gao et al, 2015) and MDA5/IFIH1 (Liddicoat et al, 2015) pathways in driving immune activation. However, to date, an immune phenotype in mice has not been described for mutations in any of the RNase H2 genes. It therefore remains to be determined whether a similar pathophysiological mechanism underlies RNase H2 deficiency, and consequently, the nature of the activated immune pathway in the majority of AGS patients is yet to be defined.
Here, we identify a sub‐clinical phenotype of ISG induction in a mouse model of the RNASEH2B‐A177T mutation, the single most common missense mutation found in AGS patients. We demonstrate that this is dependent on the cGAS/STING pathway, consistent with PRR sensing of cell‐intrinsic nucleic acids in RNase H2‐deficient cells.
Results
A hypomorphic RNase H2 mouse model for Aicardi–Goutières syndrome
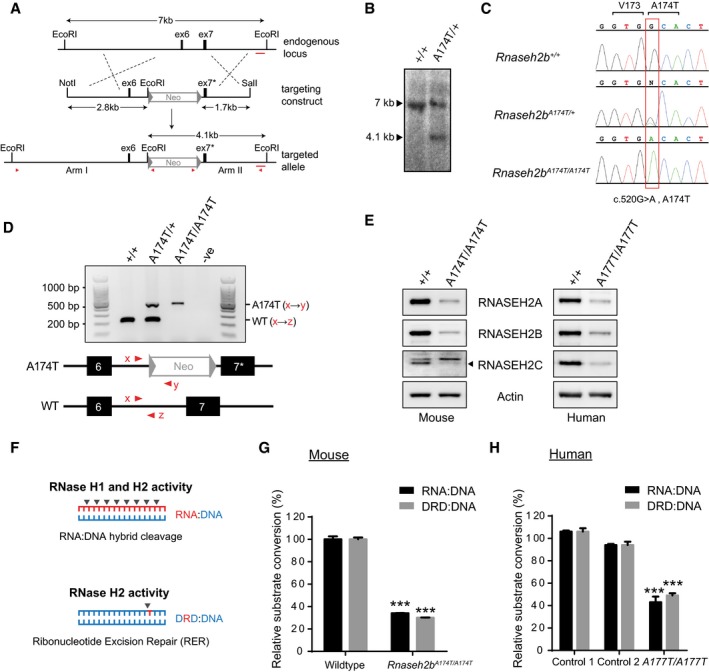
A mouse model was created by targeted knock‐in of the A174T missense mutation (c.520G>A) into exon 7 of Rnaseh2b in mouse embryonic stem cells using homologous recombination (Fig 1A–D and Appendix Fig S1). A C57BL/6J congenic Rnaseh2b A174T/A174T mouse line was established using these cells, orthologous to the most common pathogenic mutation identified in patients with AGS, RNASEH2B‐A177T. This mutation resulted in reduced cellular levels of all three RNase H2 subunits in Rnaseh2b A174T/A174T mouse embryonic fibroblasts (MEFs) and RNASEH2B A177T/A177T AGS patient lymphoblastoid cells (LCLs) (Fig 1E). This is consistent with reduced RNase H2 complex stability predicted from structural and biochemical studies that showed that the RNASEH2B‐RNASEH2C interaction interface is disrupted by the A177T substitution (Figiel et al, 2011; Reijns et al, 2011). Cellular RNase H2 activity was also significantly reduced to 30 ± 2% activity in Rnaseh2b A174T/A174T MEFs (Fig 1F and G) and 49 ± 3% activity in RNASEH2B A177T/A177T LCLs (Fig 1H), assessed against an embedded ribonucleotide substrate. More pronounced reduction in the mouse cells may be explained by the presence of a neomycin selection cassette between exon 6 and 7, causing reduced Rnaseh2b transcript levels (~60% of wild type; data not shown).
Figure 1. RNase H2 complex levels and enzymatic activity are reduced in Rnaseh2b A174T/A174T mouse and RNASEH2 BA 177T/A177T AGS patient cells.
-
ATargeted mutagenesis of the Rnaseh2b gene. Top: A 7‐kb region of the Rnaseh2b genomic locus; black boxes, exons 6 (ex6) and 7 (ex7). Middle: NotI/SalI restriction fragment of the targeting construct, comprising 4.5 kb of genomic DNA and a neomycin selection cassette (Neo) flanked by loxP sites (triangles). (Bottom) Targeted locus containing exon 7 with the c.520G>A mutation (ex7*). Red arrowheads, primers used to confirm correct targeting. Red bar, 400‐bp probe for Southern blotting.
-
BSouthern blotting confirms successful targeting. Introduction of an additional EcoRI site results in a 4.1‐kb restriction fragment detectable by Southern for targeted ES cells (A174T/+) but not for parental DNA (+/+).
-
CCapillary sequencing for Rnaseh2b +/+, Rnaseh2b A174T/+ and Rnaseh2b A174T/A174T DNA confirmed the presence of the introduced missense mutation.
-
DMouse genotyping by multiplex PCR. Top: A 221‐bp PCR product is present in wild‐type mice (+/+); the Rnaseh2b A174T allele (also) gives a 460‐bp product. Bottom: Position of forward (x) and reverse primers (y, z).
-
EImmunoblotting demonstrates depletion of all three RNase H2 protein subunits in Rnaseh2b A174T/A174T MEFs and RNASEH2B A177T/A177T LCLs. Representative of three independent experiments.
-
FSchematic showing enzyme activities attributed to RNase H1 and RNase H2 (DNA blue, RNA red).
-
G, HRNase H2 enzyme activity is reduced in mouse and patient cells. (G) Enzyme activity for Rnaseh2b A174T/A174T MEFs and passage‐matched Rnaseh2b +/+ controls, against RNase H substrate (RNA:DNA heteroduplex) and RNase H2‐specific substrate, double‐stranded DNA with a single‐embedded ribonucleotide (DRD:DNA). Mean activity for three independent cell lines, error bars represent SEM. Enzymatic activity expressed relative to the average value of control MEFs. ***P < 0.001, two‐tailed t‐test (n = 3 Rnaseh2b A174T/A174T and n = 3 Rnaseh2b +/+ control MEF lines). (H) RNase H2 activity in LCLs from two independent healthy controls and an AGS patient homozygous for the RNASEH2B‐A177T mutation. Enzyme activity normalised to average activity of control lines. Three independent experiments, error bars represent SEM. ***P < 0.001 versus either control, two‐tailed t‐test.
Source data are available online for this figure.
Despite marked impairment of RNase H2 activity, Rnaseh2b A174T/A174T mice had no overt phenotype and remained healthy when aged. Full pathological examination of brain, liver, heart, lungs, thymus, spleen, gastrointestinal tract, kidneys, skin and tongue from mice (n = 9) did not detect histological features of inflammation, infection or neoplasia at 1 year (data not shown). In particular, there was no evidence of intracranial calcification, leukodystrophy, chilblain vasculitis or cardiomyopathy, clinical features associated with AGS in humans (Crow et al, 2015). Notably, histopathological signs of inflammation are also not evident in Samhd1 −/− mice, although activation of innate immune signalling does occur, with ISG upregulation evident (Behrendt et al, 2013; Rehwinkel et al, 2013). We therefore investigated whether this was also the case in Rnaseh2b A174T/A174T mice.
ISG upregulation is present in tissues from Rnaseh2b A174T/A174T mice
Since an ISG transcriptional response is the most robust biomarker of inflammation in human patients (Rice et al, 2013), we performed RT–qPCR for a panel of AGS‐relevant ISGs on RNA extracted from adult Rnaseh2b A174T/A174T mouse tissues. A broad upregulation of ISGs was detected in heart (Fig 2A) along with significant induction of a subset of ISGs in the kidney (Fig 2B), but without any ISG response evident in brain (Fig 2C). The twofold to fourfold induction of ISGs we observed in Rnaseh2b A174T/A174T heart tissue is comparable to the fourfold to sevenfold induction seen in Samhd1 −/− mouse tissue (Rehwinkel et al, 2013). While an overt inflammatory phenotype is seen in Trex1 −/− mice (Morita et al, 2004; Gall et al, 2012), pathological signs of neuroinflammation are not evident, although ISG upregulation in brain tissue can be detected (Pereira‐Lopes et al, 2013). Given that the autoinflammatory process appears to initiate in the Trex1 −/− heart (Gall et al, 2012), there are similarities between the pattern of inflammation in Trex1 −/− mice and the ISG tissue expression pattern in Rnaseh2b A174T/A174T mice.
Figure 2. Increased ISG expression in tissues from Rnaseh2b A174T/A174T mice.
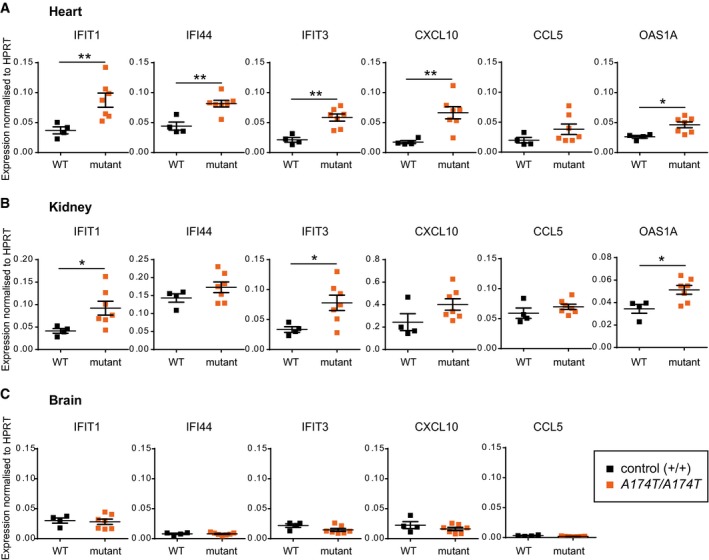
- Transcript levels of multiple ISGs are significantly elevated in heart.
- Transcript levels of a subset of ISGs are significantly increased in kidney.
- No ISG upregulation is evident in the brain.
A proinflammatory response in Rnaseh2b −/− MEFs
We next investigated whether the observed ISG induction was directly attributable to reduced RNase H2 activity. To address this, we examined cells from a second independent mouse line carrying a null allele of Rnaseh2b (Rnaseh2b tm1d), derived from the EUCOMM knockout‐first Rnaseh2b tm1a allele, from which exon 5 of Rnaseh2b had been excised by Cre recombinase. Given the early embryonic lethality of Rnaseh2b −/− mice (Reijns et al, 2012), mouse embryonic fibroblasts (MEFs) were generated on a p53 −/− background. Hereafter, we will refer to the resulting Rnaseh2b −/− p53−/− and Rnaseh2b +/+ p53−/− cells simply as Rnaseh2b −/− and Rnaseh2b +/+, respectively. Loss of RNase H2 activity in Rnaseh2b −/− cells was confirmed (Fig 3A).
Figure 3. A proinflammatory response is detectable in vitro in Rnaseh2b −/− MEFs.
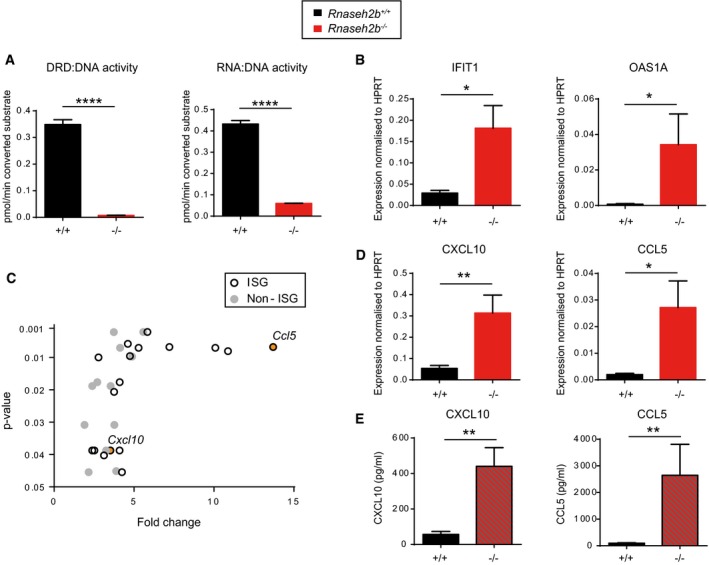
- Validation of Rnaseh2b −/− MEF lines. RER activity (DRD:DNA) is undetectable in a Rnaseh2b −/− line consistent with complete inactivation of the Rnaseh2b gene (Reijns et al, 2012). Mean of three independent experiments; error bars, SEM, ****P < 0.0001, two‐tailed t‐test. Rnaseh2b −/− and Rnaseh2b +/+ control MEFs on a C57BL/6 p53 −/− background.
- ISG transcript levels are increased in Rnaseh2b −/− MEFs.
- BeadArray transcript analysis (Illumina) detected induction of multiple ISGs, including the cytokines CXCL10 and CCL5, but not other cytokine transcripts in Rnaseh2b −/− MEFs. Plotted, average fold enrichment versus P‐value of significantly upregulated transcripts (P < 0.05, after multiple testing correction) comparing two Rnaseh2b −/− MEF lines versus 4 Rnaseh2b +/+ MEF lines. Seventeen out of 29 transcripts are ISGs.
- Cxcl10 and Ccl5 transcripts (detected by RT–qPCR) are significantly elevated.
- Increased CXCL10 and CCL5 protein (detected by ELISA) is secreted by Rnaseh2b −/− MEFs.
Significant upregulation of ISG transcripts was again apparent, on comparing six independent Rnaseh2b −/− MEF lines with four Rnaseh2b +/+ control MEF lines (Fig 3B and Appendix Fig S2A). To further characterise the nature of the response, two of the Rnaseh2b −/− MEF lines with stronger ISG upregulation were compared to control lines by gene expression microarray (Fig 3C and Appendix Table S1). Of the 29 upregulated genes achieving genomewide significance, 17 were documented ISGs and activation of non‐ISG immune response pathways was not evident. In particular, there was significant induction of two ISG genes encoding proinflammatory cytokines: Ccl5 (P = 0.0067) and Cxcl10 (P = 0.039). Upregulation of both transcripts was confirmed by RT–qPCR, and ELISA of culture supernatants from all six Rnaseh2b −/− lines demonstrated significantly increased CCL5 and CXCL10 secretion (Fig 3D and E, and Appendix Fig S2B). Notably, these cytokines have previously been implicated in AGS neuroinflammation in humans (van Heteren et al, 2008; Takanohashi et al, 2013; Cuadrado et al, 2015). We therefore concluded that impaired RNase H2 activity in vitro and in vivo results in a similar inflammatory response to that observed in patients with AGS, and like for Trex1 deficiency (Gall et al, 2012) is present in non‐immune cells.
ISG activation is dependent on the cGAS‐STING nucleic acid‐sensing pathway
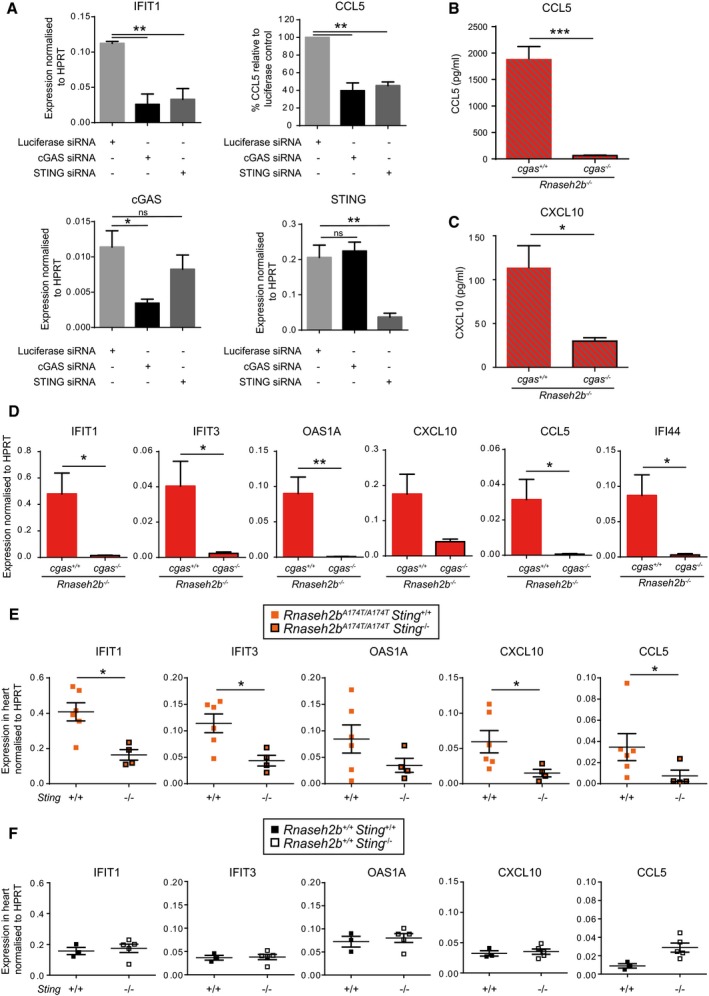
We next sought to determine the nucleic acid‐sensing pathway responsible for ISG induction in Rnaseh2b −/− cells. Three pathways have recently been implicated in the sensing of RNA:DNA hybrids: TLR9 (Rigby et al, 2014), cGAS‐STING (Mankan et al, 2014) and the NLRP3 inflammasome (Kailasan Vanaja et al, 2014). We therefore postulated that one of these nucleic acid sensors would be responsible for the ISG signature, given that RNase H2 is the major enzyme degrading RNA:DNA hybrids in mammalian cells (Büsen, 1980; Reijns et al, 2012). However, as IL‐1β and IL‐18 were both undetectable in culture supernatants from Rnaseh2b −/− cells (data not shown), an inflammasome‐mediated response was unlikely. While the observed ISG induction would be consistent with TLR9 activation, signalling is impaired by deficient proteolytic processing of the receptor in MEFs (Ewald et al, 2008), and we therefore prioritised assessment of the cGAS‐STING pathway. We performed siRNA depletion of cGAS in Rnaseh2b −/− MEFs and found that cGAS depletion significantly abrogated the ISG response and CCL5 production (Fig 4A). siRNA depletion of STING, the adaptor associated with cGAS sensing, also significantly reduced ISG induction and CCL5 secretion (Fig 4A), implicating the cGAS/STING‐sensing pathway in innate immune activation in Rnaseh2b −/− cells. The siRNA knock‐down was specific to cGAS and STING, respectively (Fig 4A), and cells remained fully responsive to poly(I:C) (Appendix Fig S3).
Figure 4. ISG induction in RNase H2 deficiency is dependent on the cGAS‐STING nucleic acid‐sensing pathway.
-
AISG activation and cytokine secretion in Rnaseh2b −/− MEFs is markedly impaired by cGAS or STING siRNA depletion. Upper left and lower panels: RT–qPCR of Ifit1, cGas and Sting transcripts, 48 h after siRNA targeting luciferase (control), cGAS or STING. Upper right panel: CCL5 is significantly reduced in culture supernatants 48 h after cGAS or STING depletion. Concentration of CCL5 (ELISA), normalised to luciferase siRNA control levels in each experiment. Mean from three independent experiments using one Rnaseh2b −/− MEF line; error bars, SEM. *P < 0.05, **P < 0.01, two‐tailed t‐test for RT–qPCR, one‐sample t‐test for CCL5 ELISA.
-
B–DISG induction and cytokine secretion is abolished in Rnaseh2b −/− cGas −/− MEFs. cGas was targeted by CRISPR/Cas9 genome editing of a Rnaseh2b −/− MEF line to inactivate cGAS/STING signalling. In addition to sequence validation, functional inactivation of cGAS was confirmed in Rnaseh2b −/− cGas −/− CRISPR lines by the absence of CCL5 secretion in response to dsDNA (Appendix Fig S4). CCL5 (B) and CXCL10 (C) production, as well as ISG expression (D), was abrogated in Rnaseh2b −/− cGas −/− clones, assessed by ELISA and RT–qPCR, respectively. Four independent experiments, n = 2 Rnaseh2b −/− cGas −/− clones, n = 4 Rnaseh2b −/− cGas +/+ clones, error bars, SEM of each experiment; *P < 0.05, **P < 0.01, ***P < 0.001, two‐tailed t‐test.
-
EISG induction in Rnaseh2b A174T/A174T mice is STING dependent. RT–qPCR of RNA extracted from hearts from Sting +/+ Rnaseh2b A174T/A174T (n = 4) and Sting −/− Rnaseh2b A174T/A174T (n = 6) 3‐month‐old mice. Each data point represents the mean of technical triplicates from one mouse. Horizontal line, mean; error bars, SEM. *P < 0.05, Mann–Whitney U‐test.
-
FAbsence of STING does not significantly decrease basal ISG expression. RT–qPCR of RNA extracted from hearts from Sting +/+ (n = 3) and Sting −/− (n = 5) three‐month‐old mice. Each data point represents the mean of technical triplicates from one mouse. Horizontal line, mean; error bars, SEM. *P < 0.05, Mann–Whitney U‐test.
To substantiate that cGAS is required for ISG induction, CRISPR/Cas9 genome editing was performed to knock out the cGAS gene in Rnaseh2b −/− MEFs. Rnaseh2b −/− MEF clones in which cGAS deletion had not occurred were also identified, to act as experimental controls. While ISG transcript upregulation and cytokine production (CCL5 and CXCL10) were still observed in such control cells, both were abrogated in CRISPR/Cas9 targeted Rnaseh2b −/− cGas −/− cells (Fig 4B–D), establishing that the innate immune activation found in Rnaseh2b −/− MEFs is dependent on cGAS.
To determine whether ISG induction was dependent on the cGAS‐STING‐sensing pathway in vivo, we intercrossed Rnaseh2b A174T/A174T and Sting −/− mice. We found that ISG transcript levels from Rnaseh2b A174T/A174T Sting −/− heart tissue were significantly reduced compared to Rnaseh2b A174T/A174T Sting +/+ controls (Fig 4E), while no significant reduction was observed when comparing Rnaseh2b +/+ Sting −/− and Rnaseh2b +/+ Sting +/+ controls (Fig 4F). Hence, a STING‐dependent ISG response also occurs in vivo in Rnaseh2b A174T/A174T mice, implicating the cGAS‐STING pathway in the ISG induction observed in RNase H2 AGS patients.
Loss of RNase H2‐specific activity results in ISG induction
Finally, the fact that both dsDNA (Sun et al, 2013; Gao et al, 2013) and RNA:DNA hybrids (Mankan et al, 2014) can bind and activate the cytoplasmic nucleic acid sensor cGAS prompted us to consider the origin of the cGAS ligand present in RNase H2‐deficient cells. Abrogated RNase H2 function could give rise to cytosolic accumulation of RNA:DNA heteroduplexes as a consequence of reduced RNA:DNA degradation of structures such as R‐loops or active retroelements/endogenous retroviruses (Chon et al, 2013; Rigby et al, 2014; Moelling & Broecker, 2015). Alternatively, cytoplasmic DNA with embedded ribonucleotides may accumulate as a consequence of impaired RER that resulted in genome instability (Reijns et al, 2012; Hiller et al, 2012). To address these possibilities, we performed complementation experiments in RNase H2 null cells, to establish which enzymatic activity was associated with the pro‐inflammatory response.
Rnaseh2b −/− cells were complemented by retroviral transduction of either Rnaseh1 or Rnaseh2b, respectively, to reconstitute cellular RNase H activity or RNase H plus RER activity. Overexpression of RNase H1 in Rnaseh2b −/− cells restored cellular enzyme activity against RNA:DNA hybrids to 81 ± 10% of the level seen in Rnaseh2b +/+ cells (Fig 5A), but did not alleviate DNA damage (Fig 5B and C). Complementation with Rnaseh2b reconstituted activity against both types of substrates (Fig 5A) and also returned DNA damage to levels seen in Rnaseh2b +/+ cells (Fig 5B and C). Reconstitution with Rnaseh2b was able to reduce cytokine and ISG responses close to wild‐type levels (Fig 5D–F), while Rnaseh1 complementation did not (Fig 5D–F). While some reduction was seen in CXCL10, ISG expression in Rnaseh1 complemented cells was otherwise the same, if not greater than, in parental Rnaseh2b −/− cells. cGAS‐dependent ISG induction in Rnaseh2b −/− cells is therefore associated with DNA damage and loss of RNase H2‐specific activity, rather than an overall reduction in cellular activity against RNA:DNA hybrids.
Figure 5. Cellular RER and not enzyme activity against RNA:DNA hybrids correlates with DNA damage and proinflammatory response.
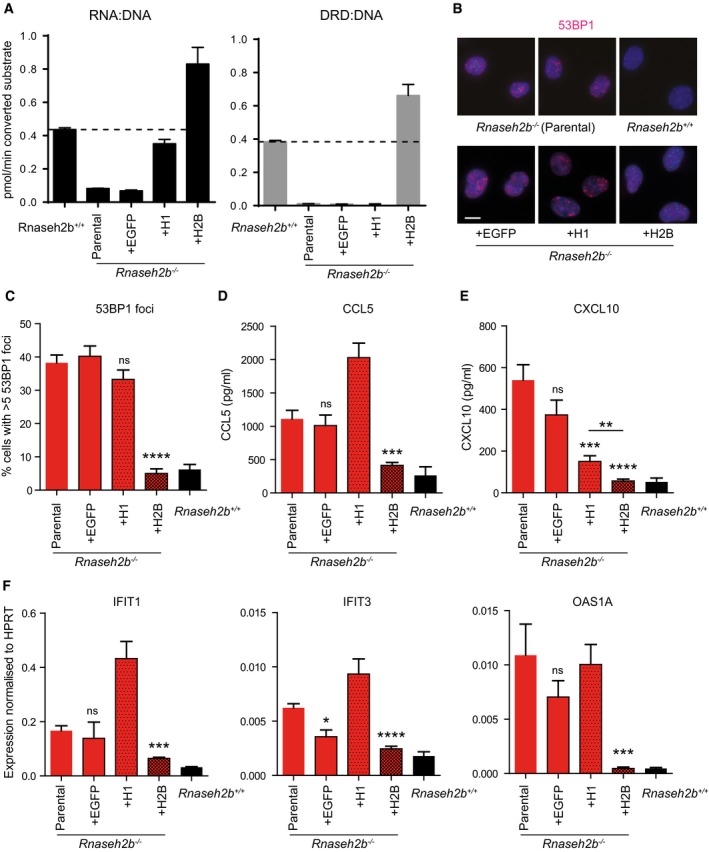
-
AOverexpression of RNase H1 in Rnaseh2b −/− cells restores RNase H activity against RNA:DNA hybrids to 81 ± 10% of wild‐type levels, while overexpression of RNASEH2B restores cellular enzyme activity for cleavage of both RNA:DNA and DRD:DNA substrates (RER). Rnaseh2b −/− MEFs were complemented with Rnaseh1 (+H1), Rnaseh2b (+H2B) or EGFP by retroviral infection. Mean of n = 3 independent experiments ± SEM.
-
B, CDNA damage is reduced to wild‐type levels by complementation with Rnaseh2b but not Rnaseh1, measured by 53BP1 foci formation in detergent‐extracted fixed cells. (B) Representative images (scale bar, 10 μm). (C) At least 150 cells were counted for each cell line in three independent experiments. Mean ± SEM, ****P < 0.0001 two‐tailed t‐test.
-
D–FCCL5 (D) and CXCL10 production (E), as well as ISG induction (F) in Rnaseh2b −/− MEFs are reduced close to wild‐type levels (Rnaseh2b +/+), by complementation with Rnaseh2b but not Rnaseh1. Mean of n = 6 independent experiments ± SEM for complemented cells; n = 3 independent experiments for Rnaseh2b −/− parental and Rnaseh2b +/+ controls cells. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 two‐tailed t‐test indicates significantly reduced expression compared to Rnaseh2b −/− parental cells.
Discussion
Here, we establish that RNase H2 deficiency leads to a proinflammatory response which is dependent upon the cGAS/STING pathway. The resulting ISG transcriptional response and induction of pro‐inflammatory cytokines are consistent with cell‐intrinsic innate immune activation. ISG activation varied between tissues, which may explain why such transcriptional changes were not reported previously in Rnaseh2b −/− or Rnaseh2b tm1a/tm1a embryos (Reijns et al, 2012; Hiller et al, 2012). The precise factors determining tissue‐specific cGAS activation are currently unclear, but could include the sensitivity of the cGAS/STING pathway in particular cell types, their level of cell proliferation, the rate of accumulation of immunogenic nucleic acids and the counteracting influences of cellular processes degrading such nucleic acids.
Despite tissue‐specific ISG upregulation, Rnaseh2b A174T/A174T mice have a subclinical phenotype without any overt inflammatory pathology, and so, like all other AGS mouse models do not recapitulate the neuroinflammation seen in human AGS patients (Rabe, 2013; Behrendt & Roers, 2014). It will therefore be important to confirm that the innate immune pathways implicated in Trex1, Adar1 and now RNase H2 deficiency, through the use of mouse models, are also relevant to the human autoinflammatory phenotype in AGS patients in whom these genes are affected. Samhd1 −/− mice, like Rnaseh2b A174T/A174T mice, display an ISG response in the absence of detectable pathology (Behrendt et al, 2013; Rehwinkel et al, 2013). In contrast a strong ISG response in Adar1 null or editing‐deficient mice is associated with embryonic lethality (Mannion et al, 2014; Liddicoat et al, 2015; Pestal et al, 2015), and with autoinflammatory cardiomyopathy and multi‐tissue involvement in Trex1 −/− mice (Morita et al, 2004; Stetson et al, 2008; Gall et al, 2012). The variation in severity between different AGS gene mouse models remains unexplained, although it may be meaningful that mutations in human RNASEH2B are associated with the least severe disease course, with AGS onset generally in infancy, in contrast to the prenatal/neonatal onset more commonly seen in TREX1 patients (Crow et al, 2015). Also, additional triggers, such as viral infection, have been proposed to be relevant to the pathogenesis of AGS (Crow & Manel, 2015). Reports of marked phenotypic variability in sibling pairs with identical RNASEH2B or RNASEH2C mutations (Vogt et al, 2013; Tüngler et al, 2014) are consistent with the possibility of such environmental factors impacting on disease severity. The Rnaseh2b A174T/A174T mouse and other models provide an opportunity for future investigation of these aspects.
Identification of the precise source of the immunogenic nucleic acids responsible for the inflammatory phenotype remains central to providing further mechanistic insight. Dependence of the ISG response in RNase H2‐ and Trex1‐deficient mouse cells on the cGAS/STING pathway suggests that accumulation of cytoplasmic DNA is common to both TREX1 and RNase H2 AGS, given that DNA is the canonical ligand for cGAS (Sun et al, 2013; Gao et al, 2013). Our complementation experiments would favour this possibility, as normalising cellular enzymatic activity against RNA:DNA hybrids does not rescue the proinflammatory response. However, immunogenic RNA:DNA hybrids cannot be completely excluded, given that some hybrids might be specifically degraded by the RNase H2 enzyme (Chon et al, 2013), although the mechanism for such differential substrate specificity for RNase H1 and H2 enzymes is currently unclear.
So far, we have been unable to ascertain whether cytoplasmic double‐stranded nucleic acids accumulate in RNase H2‐deficient cells. However, given that impaired RER causes reduced genome stability, DNA fragments resulting from DNA strand breaks are a potential origin of immunogenic cytoplasmic DNA (Ahn et al, 2014b; Hartlova et al, 2015; Shen et al, 2015), while an alternative source could be reverse‐transcribed retroelements, which are known to be activated upon DNA damage (Farkash & Luning Prak, 2006). Irrespective of the chemical nature of the ligand, our observation that cGAS plays a central role in the ISG response in RNase H2 deficiency provides further impetus for defining the source of the immunogenic nucleic acids. While cytoplasmic DNA was first detected in 2007 in Trex1 −/− cells, even here the precise origin still remains elusive, with conflicting evidence supporting either endogenous retroviruses/retroelements (Stetson et al, 2008) or genome instability (Yang et al, 2007). Purification of the responsible immunostimulatory nucleic acids could provide the solution; however, the specific isolation of such low‐abundant cytoplasmic nucleic acids remains a formidable technical challenge. This has precluded a definitive answer to date, but may be aided by implicating cGAS as the nucleic acid sensor binding immunogenic nucleic acids in both Trex1‐ and RNase H2‐deficient cells, informing potential future biochemical strategies.
In summary, our findings implicate the cGAS‐STING pathway in RNase H2 AGS and, together with the previously attributed role in Trex1 deficiency, suggest it is the most common signalling pathway driving inflammation in AGS, relevant also to the pathogenesis of SLE (Lee‐Kirsch et al, 2007; Günther et al, 2015). Targeting this pathway therefore represents a relevant therapeutic strategy for the treatment of this childhood interferonopathy and related adult‐onset autoimmune conditions.
Materials and Methods
Generation of Rnaseh2b‐A174T ES cells
Gap repair was used to retrieve a genomic fragment of 4.5 kb from BAC bMQ454F14 (Source Bioscience Lifesciences), which included exon 6 and 7 of the mouse Rnaseh2b locus (nucleotides 62977197‐62981727 of Chr14, Ensembl release 64). Subsequent bacterial recombination was used to insert a neomycin cassette flanked by loxP sites between exon 6 and 7, generating a targeting vector with two external homology arms of 2.8 kb and 1.7 kb. A point mutation c.520G>A (A174T) was inserted into exon 7 of mouse Rnaseh2b by site‐directed‐mutagenesis (Quikchange, Agilent Technologies). After linearisation and vector backbone removal by digestion with NotI and SalI, and electroelution/purification by Elutrap, the targeting cassette was electroporated into 129/Ola E14Tg2AIV embryonic stem cells. Southern blotting and long‐range PCR were used to identify correctly targeted clones. Capillary sequencing was performed to ensure the presence of the c.520G>A mutation and absence of other coding changes. The ES cell clone was karyotyped before injection into C57BL/6J mouse blastocysts.
Mice
Rnaseh2b A174T
Male chimeras resulting from blastocyst injection with Rnaseh2b A174T/+ ES cells were crossed to C57BL/6J females, giving rise to heterozygous Rnaseh2b knock‐in mice carrying the A174T mutation (Rnaseh2b tm2‐hgu‐A174T, elsewhere referred to as Rnaseh2b A174T). These were backcrossed to F11 on the C57BL/6J background to establish congenicity and subsequently maintained as a homozygous mutant line. C57Bl/6J control mice were bought in at 4–6 weeks of age from the same source as those used in backcrossing, to ensure genetic matching to a level of > 99.97% identity. All mice were housed in the same facility in the same room in conventional cages and fed the same water and food until they were analysed. Murine data were analysed unblinded to genotype. Sample sizes of 3–7 mice per group were similar to previous studies.
Rnaseh2b tm1d
Knockout‐first Rnaseh2b mice were generated by blastocyst injection of the Rnaseh2b tm1a(EUCOMM)Wtsi C57BL/6N ES cell clone EPD0087_4_A02 (EUCOMM ID: 24441) and crossing of male chimeras to C57BL/6J females. The Rnaseh2b tm1d allele, in which exon 5 is deleted, was generated by crossing of Rnaseh2b tm1a/+ mice to mice with ubiquitous expression of FLPe recombinase to delete the genetrap cassette. Rnaseh2b tm1c/+ offspring were subsequently crossed to Cre745 mice (a kind gift from DJ Kleinjan, University of Edinburgh), containing a CAGGS‐Cre construct in which Cre recombinase is under control of a chicken β‐actin promoter (Araki et al, 1995; Kleinjan et al, 2006) to excise Rnaseh2b exon 5. The resulting Rnaseh2b tm1d/+ (Rnaseh2b +/−) mice were maintained on the C57BL/6J background. The phenotype of Rnaseh2b tm1d/tm1d embryos was indistinguishable from that previously described for Rnaseh2b E202X/E202X embryos (Reijns et al, 2012).
Other strains
Trp53 tm1Tyj /J mice on the C57BL/6J background have been described previously (Jacks et al, 1994) and were kindly provided by Andrew Wood, University of Edinburgh. Interbreeding with Rnasehb tm1d/+ mice was used to generate Rnaseh2b +/+ p53 −/− and Rnaseh2b −/− p53 −/− MEFs.
Sting/Mpys −/− mice on the C57BL/6 background have been described previously (Jin et al, 2011) and were kindly provided by Jan Rehwinkel, University of Oxford, with the consent of John Cambier, University of Colorado SOM and National Jewish Health. Interbreeding with Rnaseh2b A174T/A174T mice generated Rnaseh2b A174T/A174T Sting −/− mice.
Mb21d1/cGas −/− MEFs derived from C57BL/6NTac‐Mb21d1tm1a(EUCOMM)Hmgu/IcsOrl mice were a kind gift from Jan Rehwinkel, Oxford University. These mice were originally obtained from the Institute Clinique de la Souris through the European Mouse Mutant Archive.
All mouse studies were conducted according to UK Home Office regulations under a UK Home Office project licence.
Genotyping
DNA was extracted from earclips (boiled in 50 μl 25 mM NaOH, 0.2 mM EDTA for 30 min at 95°C, cooled and then neutralised with 50 μl 40 mM Tris base) or embryos (tail tips treated with DirectPCR Lysis Reagent (Viagen) according to the manufacturer's instructions). All genotyping PCRs were performed using a multiplex three‐primer strategy and Taq ReddyMix PCR Master Mix (Thermo Scientific), as previously described for Rnaseh2b E202X/E202X (Reijns et al, 2012) and p53 −/− (Jacks et al, 1994). For Rnaseh2b tm1d/+, Sting/Mpys +/− as well as all other primers and product sizes, see Appendix Table S2.
Generation of murine embryonic fibroblast lines and assessment of innate immune activation
Independent Rnaseh2b A174T/A174T and Rnaseh2b +/+ MEF lines were generated from individual E13.5 embryos. After removing the head and internal organs, the embryos were mechanically dissociated in growth medium (DMEM, 10% FCS, 50 U/ml penicillin and 50 μg/ml streptomycin, 0.1 mM β‐mercaptoethanol). Resulting suspensions were grown at 37°C, 5% CO2 and 3% O2, and non‐adherent cells removed after 24 h. MEFs were subsequently maintained and passaged under the same conditions. Independent Rnaseh2b −/− p53 −/− and Rnaseh2b +/+ p53 −/− MEF lines were similarly generated from individual whole E10.5 embryos. For assessment of ISG upregulation and cytokine production, MEFs were plated at 2 × 105 cells per well in 12‐well plates. The following day culture medium was replaced with 800 μl of fresh medium. After a further 20 h, the culture medium was removed for assessment by ELISA to determine CXCL10 and CCL5 concentrations (R&D Systems). RNA was extracted from adherent cells using the RNeasy kit (Qiagen) as per manufacturer's instructions and included DNase I treatment. Extracted RNA was stored at −80°C until analysis.
AGS patient cells
A lymphoblastoid cell line from an AGS patient with the RNASEH2B‐A177T mutation (kindly donated by Professor Yanick Crow, University of Manchester) and non‐affected controls were generated from peripheral blood samples by EBV transformation using standard methods. Lymphoblastoid cell lines (LCLs) were maintained in RPMI 1640 supplemented with 15% foetal bovine serum, L‐glutamine, 50 U/ml penicillin and 50 μg/ml streptomycin at 37°C, 5% CO2 and normoxic conditions. The A177T/A177T mutation in the patient cell line was validated using Sanger sequencing.
Immunoblotting
Whole‐cell lysates were prepared by lysing cells in 50 mM Tris (pH 8), 280 mM NaCl, 0.5% NP‐40, 0.2 mM EDTA, 0.2 mM EGTA, 10% glycerol (vol/vol), 1 mM DTT and 1 mM PMSF for 10 min at 4°C. Lysed cells were then diluted 1:1 with 20 mM HEPES (pH 7.9), 10 mM KCl, 1 mM EDTA, 10% glycerol (vol/vol), 1 mM DTT and 1 mM PMSF for an additional 10 min, and extracts were cleared by centrifugation (17,000 g, 5 min, 4°C). Equal amounts of proteins from supernatants (concentrations determined using the Bradford method) were separated by SDS–PAGE on NuPAGE Novex Bis‐Tris 4–12% protein gels (Thermo Fisher Scientific) and transferred to PVDF membrane. Membranes were blocked in 5% milk in TBS with 0.2% Tween and probed with antibodies raised against mouse or human recombinant RNase H2, as previously described (Reijns et al, 2012), RNASEH2A (TA306706, Origene) or actin (A2066, Sigma).
RNase H enzyme assays
Enzyme activity assays were performed using a FRET‐based fluorescent substrate release assay, as previously described (Reijns et al, 2011). Briefly, 10 mM of fluorescein‐labelled oligonucleotides (GATCTGAGCCTGGGaGCT for RNase H2 specific activity, DRD:DNA, or gaucugagccugggagcu for total RNase H activity, RNA:DNA; upper case DNA, lower case RNA) was annealed to a complementary DABCYL‐labelled DNA oligonucleotide (Eurogentec) in 60 mM KCl, 50 mM Tris–HCl pH 8. Activity against double‐stranded DNA substrate of the same sequence was measured and used to correct for non‐RNase H2 activity against DRD:DNA substrate. Reactions were performed in 100 μl of buffer (60 mM KCl, 50 mM Tris–HCl pH 8, 10 mM MgCl2, 0.01% BSA, 0.01% Triton X‐100) with 250 nM substrate in 96‐well flat‐bottomed plates at 24 ± 2°C. Whole‐cell lysates were prepared as described above, and the final protein concentration used per reaction was 100 ng/μl (for DRD:DNA substrate), 50 ng/μl (for RNA:DNA substrate) for MEFs, and 32 or 16 ng/μl, respectively, for LCLs. Fluorescence was read for 100 ms using a VICTOR2 1420 multilabel counter (Perkin Elmer), with a 480‐nm excitation filter and a 535‐nm emission filter.
RT–qPCR
Murine tissues were snap‐frozen in liquid nitrogen and stored at −80°C until RNA extraction via homogenisation and using the RNeasy kit (Qiagen) as per manufacturer's instructions. cDNA was prepared from RNA from murine tissues or MEFs using AMV RT (Roche) and random oligomer primers (Thermo Fisher). Brilliant II SyBR Green qPCR Master Mix (Stratagene) was used to conduct RT–qPCR on the ABI Prism HT7900 Sequence Detection System (Applied Biosciences). The expression of target genes was normalised to the housekeeping gene HPRT using the formula (). Appendix Table S3 shows the primers used.
Illumina microarray transcriptome analysis
RNA was extracted from MEFs as described above. The Illumina TotalPrep RNA amplification kit (Ambion) was used to generate cRNA and whole‐genome gene expression analysis performed for two independent Rnaseh2b −/− p53 −/− MEFs and four Rnaseh2b +/+ p53 −/− lines using MouseWG‐6 v2.0 Expression BeadChips (Illumina). Microarray data were analysed with R 3.1.0, using the beadarray and Limma v.3.24.15 packages. Raw, non‐normalised bead‐summary values were imported from the Illumina BeadStudio software into R using the beadarray package. The limma function neqc was used to perform background correction and quantile normalisation of the raw probe signals. Prior to gene level differential analysis, probes that were not detected on any arrays were removed (detection P‐value < 0.01), and genes with multiple probes were replaced with their average. A linear model was applied to the expression data for each gene. To determine statistically differentially expressed genes, the results of the linear model were summarised and a Bayes moderated t‐test applied. To control for multiple testing, a Benjamini and Hochberg false discovery rate value of < 0.05 was used. Gene Ontology enrichment analysis was performed using the R package clusterProfiler. Genes in WT MEFs significantly upregulated against Rnaseh2b knockout (q‐value < 0.05 and log2FC > 0) were tested for functional enrichment against the background of genes detected on the array. Microarray data were deposited at the Gene Expression Omnibus (GEO)—accession number GSE76942.
siRNA knock‐down
MEFs were plated overnight at 2 × 104 cells per well in a 24‐well plate. The following day cells were transfected with short‐interfering RNA (siRNA) oligonucleotides targeting cGAS, STING or designed against Luciferase, using Dharmafect 1 in Opti‐MEM reduced serum medium (Thermo Fisher Scientific). Appendix Table S4 lists the oligonucleotide sequences. Transfection medium was replaced with 800 μl complete medium after 6 h, and culture medium was removed 48 h after transfection and used for ELISA analysis. Total RNA was extracted from cells as described above. To assess responsiveness to poly(I:C) following siRNA treatment, cells were further transfected with a final concentration of 10 μg/ml of high molecular weight poly(I:C) (InvivoGen) and culture supernatants assessed 22 h later for CCL5 using ELISA.
CRISPR/Cas9 genome editing
Rnaseh2b −/− p53 −/− MEFs were electroporated using the neon transfection system (Invitrogen) with vectors based on pSpCas9n(BB)‐2A‐GFP, a gift from Feng Zhang (Addgene plasmid # 48140), expressing two guide RNAs designed against the coding part of exon 1 of murine cGAS/Mb21d1 (NM_173386.5), using previously described methods (Ran et al, 2013). Rnaseh2b −/− p53 −/− cGAS −/− clones were selected by sizing of PCR products of the targeted region to detect clones in which deletions had occurred; these were subsequently confirmed by Sanger sequencing ensuring out of frame deletions in all alleles. Rnaseh2b −/− p53 −/− cGAS +/+ MEF clones were identified in parallel to act as controls. An additional functional assay was performed to confirm inactivation of cGAS by assessing cellular responsiveness to transfected dsDNA (1.33 μg/ml final concentration, ISD naked (InvivoGen)) to ensure the absence or presence of functional cGAS, and 2′3′‐cGAMP (2 μg/ml final concentration (InvivoGen)) to confirm the presence of functional STING. Innate immune activation of Rnaseh2b −/− p53 −/− cGAS +/+ and Rnaseh2b −/− p53 −/− cGAS −/− clones was assessed as described above.
Complementation of RNase H2 null MEFs
The Rnaseh2b −/− p53 −/− MEF line used for complementation was previously published and was derived from an E10.5 embryo, resulting from interbreeding Rnaseh2b tm1‐hgu‐A174T,E202X/+ p53 +/− mice (Reijns et al, 2012). These cells were infected in the presence of 4 μg/ml polybrene with retroviral supernatant from the Phoenix Ecotropic packaging cell line (Swift et al, 2001) transfected with pMSCVpuro, allowing expression of EGFP, RNASEH2B (NP_080277) or RNASEH1 (NP_001273794), and selected for stable integration with 2 μg/ml puromycin. Phoenix cells and pMSCVpuro were a kind gift from Dr. Irena Stancheva, University of Edinburgh.
Immunofluorescence
Parental Rnaseh2b −/− p53 −/− MEFs, matching Rnaseh2b +/+ p53 −/− controls (Reijns et al, 2012) and complemented Rnaseh2b −/− p53 −/− MEFs (5 × 104 cells per well), were plated on coverslips in 6‐well plates. After 24 h, non‐chromatin bound proteins were extracted using pre‐extraction buffer (25 mM HEPES pH 7.4, 50 mM NaCl, 1 mM EDTA, 3 mM MgCl2, 0.3 M sucrose, and 0.5% Triton X‐100) for 7 min on ice. Cells were fixed with 4% PFA for 14 min at room temperature (RT). After blocking with 3% FCS in PBS for 30 min at RT, 53BP1 antibody (NB100‐904, Novus Biologicals) was added for 1 h at RT. The next day, Alexa Fluor 568 goat anti‐rabbit secondary antibody (Life technologies) was applied and incubated for 1 h at RT. Coverslips were mounted using Vectashield antifade mounting medium with DAPI (Vector laboratories) and imaged at RT using a Coolsnap HQ CCD camera (Photometrics) and a Zeiss Axioplan II fluorescence microscope with Plan‐neofluar objectives (×40 and ×63) and acquired with iVision software (BioVision Technologies). Scoring was done under blinded conditions.
Statistical analysis
Data were analysed by unpaired t‐test for normally distributed quantitative data and Mann–Whitney U‐tests for nonparametric data as indicated in the text. One‐sample t‐tests were employed where parametric data were plotted in terms of percentage of control. Prism (GraphPad Software, Inc) was used throughout.
Author contributions
KJM, PC, LL, ZT, RER, BR, FK, AF, PSD, MAMR performed the experiments. KJM, PC, LL, ZT, EA, RER, BR, GG, MAMR and APJ analysed data. FD, AR and AF developed protocols and provided reagents. KJM, KR, RER, MAMR and APJ planned the project/supervised experiments. KJM, MAMR and APJ wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Review Process File
Source Data for Figure 1
Acknowledgements
We thank J. Dorin, D. Hunt, J. Rehwinkel, F. Semple and W. Bickmore for helpful discussions and Y. Crow, J. Cambier, J. Rehwinkel, J. Dorin, F. Semple, E. Hall, I. Stancheva and A. Wood for generous sharing of reagents. We thank the IGMM Transgenic Facility, E. Freyer and J. Ding for technical assistance. This work was funded by the MRC, Newlife Foundation for Disabled Children and The Wellcome Trust‐University of Edinburgh ISSF2.
The EMBO Journal (2016) 35: 831–844
See also: M Gentili & N Manel (April 2016)
References
- Ablasser A, Hemmerling I, Schmid‐Burgk JL, Behrendt R, Roers A, Hornung V (2014) TREX1 deficiency triggers cell‐autonomous immunity in a cGAS‐dependent manner. J Immunol 192: 5993–5997 [DOI] [PubMed] [Google Scholar]
- Ahn J, Ruiz P, Barber GN (2014a) Intrinsic self‐DNA triggers inflammatory disease dependent on STING. J Immunol 193: 4634–4642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn J, Xia T, Konno H, Konno K, Ruiz P, Barber GN (2014b) Inflammation‐driven carcinogenesis is mediated through STING. Nat Commun 5: 5166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki K, Araki M, Miyazaki J, Vassalli P (1995) Site‐specific recombination of a transgene in fertilized eggs by transient expression of Cre recombinase. Proc Natl Acad Sci U S A 92: 160–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrendt R, Schumann T, Gerbaulet A, Nguyen LA, Schubert N, Alexopoulou D, Berka U, Lienenklaus S, Peschke K, Gibbert K, Wittmann S, Lindemann D, Weiss S, Dahl A, Naumann R, Dittmer U, Kim B, Mueller W, Gramberg T, Roers A (2013) Mouse SAMHD1 has antiretroviral activity and suppresses a spontaneous cell‐intrinsic antiviral response. Cell Rep 4: 689–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrendt R, Roers A (2014) Mouse models for Aicardi‐Goutieres syndrome provide clues to the molecular pathogenesis of systemic autoimmunity. Clin Exp Immunol 175: 9–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Büsen W (1980) Purification, subunit structure, and serological analysis of calf thymus ribonuclease H I. J Biol Chem 255: 9434–9443. [PubMed] [Google Scholar]
- Cerritelli SM, Crouch RJ (2009) Ribonuclease H: the enzymes in eukaryotes. FEBS J 276: 1494–1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chon H, Sparks JL, Rychlik M, Nowotny M, Burgers PM, Crouch RJ, Cerritelli SM (2013) RNase H2 roles in genome integrity revealed by unlinking its activities. Nucleic Acids Res 41: 3130–3143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow YJ, Hayward BE, Parmar R, Robins P, Leitch A, Ali M, Black DN, van Bokhoven H, Brunner HG, Hamel BC, Corry PC, Cowan FM, Frints SG, Klepper J, Livingston JH, Lynch SA, Massey RF, Meritet JF, Michaud JL, Ponsot G, et al (2006a) Mutations in the gene encoding the 3′‐5′ DNA exonuclease TREX1 cause Aicardi‐Goutieres syndrome at the AGS1 locus. Nat Genet 38: 917–920 [DOI] [PubMed] [Google Scholar]
- Crow YJ, Leitch A, Hayward BE, Garner A, Parmar R, Griffith E, Ali M, Semple C, Aicardi J, Babul‐Hirji R, Baumann C, Baxter P, Bertini E, Chandler KE, Chitayat D, Cau D, Dery C, Fazzi E, Goizet C, King MD, et al (2006b) Mutations in genes encoding ribonuclease H2 subunits cause Aicardi‐Goutieres syndrome and mimic congenital viral brain infection. Nat Genet 38: 910–916 [DOI] [PubMed] [Google Scholar]
- Crow YJ, Livingston JH (2008) Aicardi‐Goutieres syndrome: an important Mendelian mimic of congenital infection. Dev Med Child Neurol 50: 410–416 [DOI] [PubMed] [Google Scholar]
- Crow YJ, Chase DS, Lowenstein Schmidt J, Szynkiewicz M, Forte GM, Gornall HL, Oojageer A, Anderson B, Pizzino A, Helman G, Abdel‐Hamid MS, Abdel‐Salam GM, Ackroyd S, Aeby A, Agosta G, Albin C, Allon‐Shalev S, Arellano M, Ariaudo G, Aswani V, et al (2015) Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am J Med Genet A 167a: 296–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow YJ, Manel N (2015) Aicardi‐Goutieres syndrome and the type I interferonopathies. Nat Rev Immunol 15: 429–440 [DOI] [PubMed] [Google Scholar]
- Cuadrado E, Michailidou I, van Bodegraven EJ, Jansen MH, Sluijs JA, Geerts D, Couraud PO, De Filippis L, Vescovi AL, Kuijpers TW, Hol EM (2015) Phenotypic variation in Aicardi‐Goutieres syndrome explained by cell‐specific IFN‐stimulated gene response and cytokine release. J Immunol 194: 3623–3633 [DOI] [PubMed] [Google Scholar]
- Eder PS, Walder RY, Walder JA (1993) Substrate specificity of human RNase H1 and its role in excision repair of ribose residues misincorporated in DNA. Biochimie 75: 123–126 [DOI] [PubMed] [Google Scholar]
- Ewald SE, Lee BL, Lau L, Wickliffe KE, Shi GP, Chapman HA, Barton GM (2008) The ectodomain of Toll‐like receptor 9 is cleaved to generate a functional receptor. Nature 456: 658–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farkash EA, Luning Prak ET (2006) DNA damage and L1 retrotransposition. J Biomed Biotechnol 2006: 37285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figiel M, Chon H, Cerritelli SM, Cybulska M, Crouch RJ, Nowotny M (2011) The structural and biochemical characterization of human RNase H2 complex reveals the molecular basis for substrate recognition and Aicardi‐Goutieres syndrome defects. J Biol Chem 286: 10540–10550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gall A, Treuting P, Elkon KB, Loo YM, Gale M Jr, Barber GN, Stetson DB (2012) Autoimmunity initiates in nonhematopoietic cells and progresses via lymphocytes in an interferon‐dependent autoimmune disease. Immunity 36: 120–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P, Ascano M, Wu Y, Barchet W, Gaffney BL, Zillinger T, Serganov AA, Liu Y, Jones RA, Hartmann G, Tuschl T, Patel DJ (2013) Cyclic [G(2′,5′)pA(3′,5′)p] is the metazoan second messenger produced by DNA‐activated cyclic GMP‐AMP synthase. Cell 153: 1094–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao D, Li T, Li XD, Chen X, Li QZ, Wight‐Carter M, Chen ZJ (2015) Activation of cyclic GMP‐AMP synthase by self‐DNA causes autoimmune diseases. Proc Natl Acad Sci USA 112: E5699–E5705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray EE, Treuting PM, Woodward JJ, Stetson DB (2015) Cutting edge: cGAS is required for lethal autoimmune disease in the Trex1‐deficient mouse model of aicardi‐goutieres syndrome. J Immunol 195: 1939–1943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Günther C, Kind B, Reijns MA, Berndt N, Martinez‐Bueno M, Wolf C, Tungler V, Chara O, Lee YA, Hubner N, Bicknell L, Blum S, Krug C, Schmidt F, Kretschmer S, Koss S, Astell KR, Ramantani G, Bauerfeind A, Morris DL, et al (2015) Defective removal of ribonucleotides from DNA promotes systemic autoimmunity. J Clin Invest 125: 413–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartlova A, Erttmann SF, Raffi FA, Schmalz AM, Resch U, Anugula S, Lienenklaus S, Nilsson LM, Kroger A, Nilsson JA, Ek T, Weiss S, Gekara NO (2015) DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti‐microbial innate immunity. Immunity 42: 332–343 [DOI] [PubMed] [Google Scholar]
- van Heteren JT, Rozenberg F, Aronica E, Troost D, Lebon P, Kuijpers TW (2008) Astrocytes produce interferon‐alpha and CXCL10, but not IL‐6 or CXCL8, in Aicardi‐Goutieres syndrome. Glia 56: 568–578 [DOI] [PubMed] [Google Scholar]
- Hiller B, Achleitner M, Glage S, Naumann R, Behrendt R, Roers A (2012) Mammalian RNase H2 removes ribonucleotides from DNA to maintain genome integrity. J Exp Med 209: 1419–1426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, Weinberg RA (1994) Tumor spectrum analysis in p53‐mutant mice. Curr Biol 4: 1–7 [DOI] [PubMed] [Google Scholar]
- Jin L, Hill KK, Filak H, Mogan J, Knowles H, Zhang B, Perraud AL, Cambier JC, Lenz LL (2011) MPYS is required for IFN response factor 3 activation and type I IFN production in the response of cultured phagocytes to bacterial second messengers cyclic‐di‐AMP and cyclic‐di‐GMP. J Immunol 187: 2595–2601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kailasan Vanaja S, Rathinam VA, Atianand MK, Kalantari P, Skehan B, Fitzgerald KA, Leong JM (2014) Bacterial RNA:DNA hybrids are activators of the NLRP3 inflammasome. Proc Natl Acad Sci U S A 111: 7765–7770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinjan DA, Seawright A, Mella S, Carr CB, Tyas DA, Simpson TI, Mason JO, Price DJ, van Heyningen V (2006) Long‐range downstream enhancers are essential for Pax6 expression. Dev Biol 299: 563–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee‐Kirsch MA, Gong M, Chowdhury D, Senenko L, Engel K, Lee YA, de Silva U, Bailey SL, Witte T, Vyse TJ, Kere J, Pfeiffer C, Harvey S, Wong A, Koskenmies S, Hummel O, Rohde K, Schmidt RE, Dominiczak AF, Gahr M, et al (2007) Mutations in the gene encoding the 3′‐5′ DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat Genet 39: 1065–1067 [DOI] [PubMed] [Google Scholar]
- Liddicoat BJ, Piskol R, Chalk AM, Ramaswami G, Higuchi M, Hartner JC, Li JB, Seeburg PH, Walkley CR (2015) RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 349: 1115–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mankan AK, Schmidt T, Chauhan D, Goldeck M, Honing K, Gaidt M, Kubarenko AV, Andreeva L, Hopfner KP, Hornung V (2014) Cytosolic RNA:DNA hybrids activate the cGAS‐STING axis. EMBO J 33: 2937–2946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannion NM, Greenwood SM, Young R, Cox S, Brindle J, Read D, Nellaker C, Vesely C, Ponting CP, McLaughlin PJ, Jantsch MF, Dorin J, Adams IR, Scadden AD, Ohman M, Keegan LP, O'Connell MA (2014) The RNA‐editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep 9: 1482–1494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moelling K, Broecker F (2015) The reverse transcriptase‐RNase H: from viruses to antiviral defense. Ann N Y Acad Sci 1341: 126–135 [DOI] [PubMed] [Google Scholar]
- Morita M, Stamp G, Robins P, Dulic A, Rosewell I, Hrivnak G, Daly G, Lindahl T, Barnes DE (2004) Gene‐targeted mice lacking the Trex1 (DNase III) 3′–>5′ DNA exonuclease develop inflammatory myocarditis. Mol Cell Biol 24: 6719–6727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira‐Lopes S, Celhar T, Sans‐Fons G, Serra M, Fairhurst AM, Lloberas J, Celada A (2013) The exonuclease Trex1 restrains macrophage proinflammatory activation. J Immunol 191: 6128–6135 [DOI] [PubMed] [Google Scholar]
- Pestal K, Funk CC, Snyder JM, Price ND, Treuting PM, Stetson DB (2015) Isoforms of RNA‐editing enzyme ADAR1 independently control nucleic acid sensor MDA5‐driven autoimmunity and multi‐organ development. Immunity 43: 933–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabe B (2013) Aicardi‐Goutieres syndrome: clues from the RNase H2 knock‐out mouse. J Mol Med (Berl) 91: 1235–1240 [DOI] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F (2013) Genome engineering using the CRISPR‐Cas9 system. Nat Protoc 8: 2281–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehwinkel J, Maelfait J, Bridgeman A, Rigby R, Hayward B, Liberatore RA, Bieniasz PD, Towers GJ, Moita LF, Crow YJ, Bonthron DT, Reis e Sousa C (2013) SAMHD1‐dependent retroviral control and escape in mice. EMBO J 32: 2454–2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reijns MA, Bubeck D, Gibson LC, Graham SC, Baillie GS, Jones EY, Jackson AP (2011) The structure of the human RNase H2 complex defines key interaction interfaces relevant to enzyme function and human disease. J Biol Chem 286: 10530–10539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reijns MA, Rabe B, Rigby RE, Mill P, Astell KR, Lettice LA, Boyle S, Leitch A, Keighren M, Kilanowski F, Devenney PS, Sexton D, Grimes G, Holt IJ, Hill RE, Taylor MS, Lawson KA, Dorin JR, Jackson AP (2012) Enzymatic removal of ribonucleotides from DNA is essential for mammalian genome integrity and development. Cell 149: 1008–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice GI, Bond J, Asipu A, Brunette RL, Manfield IW, Carr IM, Fuller JC, Jackson RM, Lamb T, Briggs TA, Ali M, Gornall H, Couthard LR, Aeby A, Attard‐Montalto SP, Bertini E, Bodemer C, Brockmann K, Brueton LA, Corry PC, et al (2009) Mutations involved in Aicardi‐Goutieres syndrome implicate SAMHD1 as regulator of the innate immune response. Nat Genet 41: 829–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice GI, Kasher PR, Forte GM, Mannion NM, Greenwood SM, Szynkiewicz M, Dickerson JE, Bhaskar SS, Zampini M, Briggs TA, Jenkinson EM, Bacino CA, Battini R, Bertini E, Brogan PA, Brueton LA, Carpanelli M, De Laet C, de Lonlay P, del Toro M, et al (2012) Mutations in ADAR1 cause Aicardi‐Goutieres syndrome associated with a type I interferon signature. Nat Genet 44: 1243–1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice GI, Forte GM, Szynkiewicz M, Chase DS, Aeby A, Abdel‐Hamid MS, Ackroyd S, Allcock R, Bailey KM, Balottin U, Barnerias C, Bernard G, Bodemer C, Botella MP, Cereda C, Chandler KE, Dabydeen L, Dale RC, De Laet C, De Goede CG, et al (2013) Assessment of interferon‐related biomarkers in Aicardi‐Goutieres syndrome associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and ADAR: a case‐control study. Lancet Neurol 12: 1159–1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice GI, del Toro Duany Y, Jenkinson EM, Forte GM, Anderson BH, Ariaudo G, Bader‐Meunier B, Baildam EM, Battini R, Beresford MW, Casarano M, Chouchane M, Cimaz R, Collins AE, Cordeiro NJ, Dale RC, Davidson JE, De Waele L, Desguerre I, Faivre L, et al (2014) Gain‐of‐function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nat Genet 46: 503–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigby RE, Webb LM, Mackenzie KJ, Li Y, Leitch A, Reijns MA, Lundie RJ, Revuelta A, Davidson DJ, Diebold S, Modis Y, MacDonald AS, Jackson AP (2014) RNA:DNA hybrids are a novel molecular pattern sensed by TLR9. EMBO J 33: 542–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rydberg B, Game J (2002) Excision of misincorporated ribonucleotides in DNA by RNase H (type 2) and FEN‐1 in cell‐free extracts. Proc Natl Acad Sci U S A 99: 16654–16659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen YJ, Le Bert N, Chitre AA, Koo CX, Nga XH, Ho SS, Khatoo M, Tan NY, Ishii KJ, Gasser S (2015) Genome‐derived cytosolic DNA mediates type I interferon‐dependent rejection of B cell lymphoma cells. Cell Rep 11: 460–473 [DOI] [PubMed] [Google Scholar]
- Sparks JL, Chon H, Cerritelli SM, Kunkel TA, Johansson E, Crouch RJ, Burgers PM (2012) RNase H2‐initiated ribonucleotide excision repair. Mol Cell 47: 980–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stetson DB, Ko JS, Heidmann T, Medzhitov R (2008) Trex1 prevents cell‐intrinsic initiation of autoimmunity. Cell 134: 587–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Wu J, Du F, Chen X, Chen ZJ (2013) Cyclic GMP‐AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339: 786–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swift S, Lorens J, Achacoso P, Nolan GP (2001) Rapid production of retroviruses for efficient gene delivery to mammalian cells using 293T cell‐based systems. Curr Protoc Immunol Chapter 10: Unit 10 17C [DOI] [PubMed] [Google Scholar]
- Takanohashi A, Prust M, Wang J, Gordish‐Dressman H, Bloom M, Rice GI, Schmidt JL, Crow YJ, Lebon P, Kuijpers TW, Nagaraju K, Vanderver A (2013) Elevation of proinflammatory cytokines in patients with Aicardi‐Goutieres syndrome. Neurology 80: 997–1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tüngler V, Schmidt F, Hieronimus S, Reyes‐Velasco C, Lee‐Kirsch MA (2014) Phenotypic variability in a family with aicardi‐goutières syndrome due to the common A177T RNASEH2B mutation. Case Rep Clin Med 3: 153–156 [Google Scholar]
- Vogt J, Agrawal S, Ibrahim Z, Southwood TR, Philip S, Macpherson L, Bhole MV, Crow YJ, Oley C (2013) Striking intrafamilial phenotypic variability in Aicardi‐Goutieres syndrome associated with the recurrent Asian founder mutation in RNASEH2C. Am J Med Genet A 161a: 338–342 [DOI] [PubMed] [Google Scholar]
- Wahren‐Herlenius M, Dorner T (2013) Immunopathogenic mechanisms of systemic autoimmune disease. Lancet 382: 819–831. [DOI] [PubMed] [Google Scholar]
- Wu J, Chen ZJ (2014) Innate immune sensing and signaling of cytosolic nucleic acids. Annu Rev Immunol 32: 461–488 [DOI] [PubMed] [Google Scholar]
- Yang Y, Lindahl T, Barnes D (2007) Trex1 exonuclease degrades ssDNA to prevent chronic checkpoint activation and autoimmune disease. Cell 131: 873–886 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Review Process File
Source Data for Figure 1