

Abstract
Background
Cow’s milk allergy (CMA) is a common disease in infancy. Early environmental factors are likely to contribute to CMA. It is known that epigenetic gene regulation can be altered by environmental factors. We have set up a proof of concept study, aiming to detect epigenetic associations specific with CMA.
Methods
We studied children from the Dutch EuroPrevall birth cohort study (N = 20 CMA, N = 23 controls, N = 10 tolerant boys), age and gender matched. CMA was challenge proven. Bisulfite converted DNA (blood) was analyzed using the 450K infinium DNA-methylation array. Four groups (combined, girls, boys and tolerant boys) were analysed between CMA and controls. Statistical analysis and pathway-analysis were performed in “R” using IMA, Minfi and the global-test package. Differentially methylated regions in DHX58, ZNF281, EIF42A and HTRA2 genes were validated by quantitative amplicon sequencing (ROCHE 454®).
Results
General hypermethylation was found in the CMA group compared to control children, while this effect was absent in the tolerant group. Methylation differences were, among others, found in regions of DHX58, ZNF281, EIF42A and HTRA2 genes. Several of these genes are known to be involved in immunological pathways and associated with other allergies.
Conclusion
We show that epigenetic associations are involved in CMA. Although, the statistical power of our study is limited and our sample was based on whole blood, we were still able to detect feasible loci and pathways. Therefore our findings might contribute to future diagnostic or therapeutic interventions for specific CMA. Further studies have to confirm the findings of our study.
Electronic supplementary material
The online version of this article (doi:10.1186/s13601-016-0105-z) contains supplementary material, which is available to authorized users.
Keywords: Cow’s milk allergy, Epigenetics, Food allergy, Gender differences, Tolerant
Background
Cow’s milk allergy
Cow’s milk allergy (CMA) is a common food allergy in young children. Accurate data on the worldwide incidences on CMA is still lacking because of discrepancies between self-reported and proper diagnosed allergy. A recent European study describes an incidence of CMA of 0.74 % [1–3]. CMA has a heterogeneous clinical presentation, with a low to moderate estimated heritability of 15 % compared to other (food) allergies [4, 5]. Accumulating evidence in the development of allergic diseases suggest involvement of different pre- and postnatal environmental factors, like gut-microbiota, maturation of the immune-system and epicutaneous allergen sensitization, but also a parent-of-origin factor [4, 6–8]. Young children are likely to develop tolerance for cow’s milk protein (CMP) within a few years [4, 9]. However, infants who suffered from CMA in their early childhood have an increased risk to develop other allergic diseases like asthma later in life, the so-called allergic march [9–13].
(Epi)genetic component of food allergy
Complex diseases are characterized by a considerable heterogenic phenotype, genetic aberrations but also include gene-environment interactions. Reports on heritability estimations for food allergy (FA) range from 15 % for milk to 82 % for peanut allergy [5, 14]. Some of these reported heritability estimates differ largely between study populations. Whether this is due to study design, geographic or ethnic background or involvement of different environmental factors is not clear [7, 14]. Several environmental components like maternal lifestyle, diet, stress and hygiene, have been suggested to be involved in food allergies [14–18]. Furthermore, accumulating evidence suggest the prevalence of FA in well-developed countries has been increasing over the last decades. Whether this increase is truly occurring is still under debate due to limited amount of comparative data. [19, 20] Genetic selection or genetic drift both are relative slow processes and generally occur in isolated populations, therefore it is highly unlikely that the suggested increase of FA is caused by genetic changes or drift within these populations. These observations favour the possibility that an epigenetic component is involved in the expression of FA and CMA.
Allergy with respect to gender
The role of gender and sex hormones in relation to the expression of allergy has been studied in epidemiological surveys. The distribution between genders is greatly skewed with regard to the expression of asthma before and after puberty [21, 22]. In general, allergy is more prevalent in boys before puberty, while after puberty allergies are more prevalent in girls. Reports on differences between genders suffering from specific food allergies are however inconsistent [6]. Especially, the studies based on self-reported allergy questionnaires and studies based on a food challenge (the gold standard) show different outcomes [6]. For CMA, differences in prevalence between genders are still under debate [23, 24].
In this pilot case–control study, we investigated if (1) epigenetic differences can be shown between children with proven CMA and healthy controls, (2) if epigenetic differences alter upon obtaining cow’s milk protein tolerance and (3) whether epigenetic differences are reflected by phenotypic differences with regard to gender in the CMA population.
Methods
Dutch EuroPrevall birth cohort study
All children were participating in the Dutch EuroPrevall birth cohort study. The EuroPrevall study has been described in detail previously [25, 26]. In summary, children were included around birth and standardized measurements were performed by questionnaires until 2.5 years of age. All children with symptoms suggestive of CMA underwent, among others, serum specific IgE (sIgE) measurement (Phadia Diagnostics, Uppsala, Sweden) and a double blind placebo controlled food challenge (DBPCFC). sIgE-values <0.35 kU/L, were considered negative. Healthy control children were selected from the entire Dutch cohort. Controls had no symptoms suggestive of any food allergy nor suffered from atopic dermatitis. In children with CMA and healthy controls, an attempt was made to obtain one full blood sample (EDTA KE 2.6 mL Monovette, Sarstedt BV, Etten-Leur, Netherlands) for DNA-isolation, besides the blood used for sIgE measurement. The Medical Ethics Committee of the Academic Medical Hospital (METC 06/005) approved the Dutch EuroPrevall Birth Cohort Study, including (epi) genetic studies. Written informed consent, both for the study and for the epigenetic study, were obtained from both parents of each child, unless only one of them had parental rights.
Patient characteristics were analysed with t test (Mann–Whitney U test when data was not normally distributed) for continuous parameters. Chi square test was used for categorical variables and Chi square test for trend for multiple categories categorical variables. P values <0.05 were considered significant. SPSS version 20 was used (IBM SPSS Statistics for Windows, Armonk, NY).
CMA diagnosis
DBPCFC is the gold standard for diagnosing CMA and the challenge of choice according to the study protocol [25–27]. The DBPCFC has been described in detail previously [28]. In summary, the child receives cow’s milk protein (CMP) free formula with added CMP (active day) or the same CMP free formula without the added CMP (placebo day) in increasing doses, double blinded, in a random order, on two different days. All children suspected of CMA were challenged with a DBPCFC. DBPCFC was repeated annually until the child was tolerant for CMP [25]. Meaning, the DBPCFC was negative and/or the child was consuming CMP regularly without symptoms.
Samples selected for epigenetic analysis
DNA samples of 20 children with proven and active CMA at the time of blood drawing were selected. Samples of healthy controls (N = 20) were gender matched with the 20 CMA-samples. Also ten DNA samples were selected of “tolerant boys”. These boys had proven CMA, but who were already CMP-tolerant at DNA-sampling. For the tolerant-children only boys were included, due to the limited availability of samples of tolerant girls. Samples of these children have also been studied in a different study on genetic aberrations regarding the Filaggrin-gene and SNPs known in allergic diseases [29]. A flowchart of the selected samples can be found in Fig. 1. To our knowledge no epigenome wide specific power calculator is available. Although not fully appropriate, we estimated the statistical power of our sample using a power calculator webtool, originally designed for expression array experiments (http://bioinformatics.mdanderson.org/MicroarraySampleSize). The following thresholds were used: Number of genes (25,000); Acceptable number of false positives (1); desired fold change (1.3); desired power (0.8); standard deviation (0.2). Using these parameters a sample size of 14 would be sufficient to detect an alpha of 0.00004 per gene.
Fig. 1.
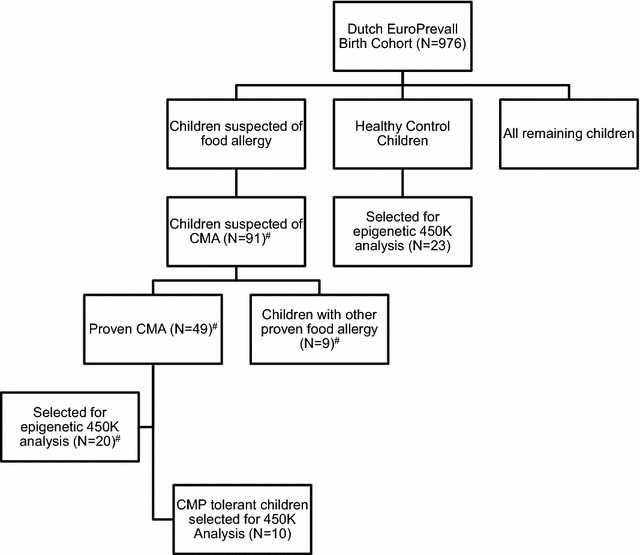
Flowchart of samples selected for our epigenetic and phenotypic studies. CMA cow’s milk allergy, N number, #selected for phenotypic factor analysis
Epigenetic analysis of selected samples
DNA was isolated from whole blood. Bisulfite treatment (ZYMO®) was applied on genomic DNA. DNA-methylation profiles for each sample were obtained using the 450K infinium array of Illumina. Raw data was processed using standard “genome studio” software of Illumina. This processed data was analyzed with statistical analysis scripted in “R” and “Perl”. “R” packages IMA, Minfi, wateRmelon, lumi and globaltest were used. We corrected for the two types of probes present on the Illumina array using BMIQ normalization. The presence of technical and/or biological batch effects was estimated and evaluated by means of principal component analysis (PCA). QQ-plots were generated, deflation or inflation factor ranging between 0.97 and 1.3 was assumed to be acceptable. Relative blood cell distribution was evaluated using the Minfi package according the method of Houseman et al. [30]. Standard output of the relative blood cell distribution comprises only CD8+ and CD4+ T cells, Natural killer cells, B cells, Monocytes and Granulocytes. Since CMA is an immunological disorder, blood cell distribution is related to our factor of interest. Therefore adjustment for blood cell distribution was assumed inappropriate. Statistical analysis on individual probe differentially methylated positions (DMPs) and on specific annotated differentially methylated regions (DMR) within a gene was performed using IMA package. For association analysis a linear model (limma; default settings) was used. All models were adjusted for age and where necessary the model was adjusted for gender. In the combined analysis probes representing chromosome X and Y were removed from the dataset. Analyses were conducted for children with CMA versus control children (combined analysis), girls with CMA versus control girls (girls), boys with CMA versus control boys (boys), and tolerant boys versus control boys (tolerant). In order to explore our CMA datasets we evaluated general methylation differences. For exclusion of the majority of non-informative probes we used solely probes with an unadjusted p < 0.05 and an absolute methylation difference >5 % in these analyses. Recent published information on the presence of common single nucleotide polymorphism (SNPs) beneath the Illumina 450K DNA-methylation probe and specificity of the probe was post hoc added to the output using in-house developed “Perl” scripts [31]. Common SNPs are especially effective confounding in DMPs. For the hypothesis free analysis approach we evaluated DMPs and DMRs, for the hypothesis driven analysis approach we extracted all individual probes within the candidate gene. Selection of candidate genes is described below. Post-hoc SNP evaluation was preferred over prior excluding common SNPs from the analysis dataset. DMPs were evaluated according the adjusted p values (Benjamini–Hochberg), an adjusted p value of <0.05 was assumed significant. The annotation of methylation regions used for DMR analysis in the IMA package is predefined and thus not based on biological/statistical correlation between CpGs. Moreover, IMA’s adjusted p values are calculated per region. Unfortunately, many of those regions overlap, which results seemingly in more independent tests and thus a false inflation of the multiple test penalty. In other words, we hypothesized that such annotation definition implicated more false negative findings per definition. Therefore our multiple test correction was based on a Bonferonni correction using p value of 0.05 divided by the maximum amount of genes (i.e. maximum number of genes found was 19,500) found in association analyses of this study. As such, DMRs were considered significant with p < 2.6 × 10−6.
Pathway analysis was performed in the combined, girls and boys groups using a global, untargeted, test method and the “Gene Ontology” (GO) databases [32]. Probes were filtered on the presence of entrez ID annotation. Entrez ID covered by less than 3 probes were excluded from the analysis. In our enrichment studies we used an arbitrary threshold of p < 0.01 in order to evaluate only the upper top and unique gene sets. Although enrichment or pathways that are plausible connected to the phenotype of interest is favorable, conclusions drawn from such analysis have to be taken with much greater caution compared to conclusions dawned from results like DMP and DMR analyses.
Illumina 450K DNA-methylation array profiles of all samples are available on request.
Epigenetic validation procedure
Promising aberrant methylated regions were verified using Roche ® 454 Next Generation Sequencing technology. The selection of these regions was based on the epigenome wide association results (top hits per group), feasible biological relevancy to CMA of the candidate and availability of DNA. Regions in the following genes were evaluated using multiple bisulphite primers sets: DHX58, ZNF281, EIF42A and HTRA2. Bisulphite primers were designed using Methprimer [33]. Primer information is described in Additional file 1: Table S1a. Cases and controls were random divided over 15 ID sequences, i.e. mids (Additional file 1: Table S1b). Concentration of polymerase chain reactions (PCR) fragments was obtained using “Lab on a Chip”. Subsequently PCR products were normalized and pooled. Sequence reactions were performed conform the Roche 454 protocol. Raw sequence data was mapped and aligned using the AvA software of Roche (Roche, Branford, United States). In-house developed scripts (PERL) were used to obtain methylation index (β) values for each CG position. QC of Roche 454 data was implemented in the script and was based on the presence of heterogeneous (non CG or TG) polymorphic sites (50 % distribution). Since bisulfite pyro sequencing is prone to a T-stretch bias, which may result in position shifts of the CG site, careful evaluation of flanking positions in the Roche 454 data was implemented in the script. Statistical analysis of the methylation index was performed in SPSS v20 using a Students T test. Pearsons correlation was used to evaluated the CG set within every amplicon. P values of correlated CG sites were combined using Fishers method combined p value method, p values <0.05 were assumed significant.
Phenotypic differences between several subgroups of allergic children
To detect phenotypic differences between genders: factor analysis was performed. Out of the group of children suspected of CMA (N = 91), there were 49 children with CMA and 33 children without food allergies. The 20 children with CMA who were selected for epigenetic analysis, were part of the group of 49 children with proven CMA (Fig. 1). Phenotypic counts on skin, gastrointestinal, and respiratory symptoms and number of allergies in all subsets of patients were investigated. Symptoms included in each category have been described previously [28]. Number of allergy was defined as more than 1 challenge proven food allergy.
Factor analysis for phenotypic differences in all subsets of patients were performed by univariate analysis. Factor regression coefficients and gender were dependent variables and age was a covariate. P values <0.05 were considered significant. SPSS version 20 was used (IBM SPSS Statistics for Windows, Armonk, NY).
Hypothesis driven (candidate gene) epigenetic association
Candidate genes were selected from a recent review of Bønnelykke et al. on allergy, sensitization, and related atopic disorders [34]. This review discusses previously reported genetic associations; mainly GWAS reports, with atopic disorders and/or sensitization mechanisms of approximately the last decade. We selected in total 66 genes previously associated with atopic disorders from large genome wide studies (GWAS) and 38 genes were associated with monogenic allergy related syndromes of which 22 genes were located at 22q11.1. 8 selected genes were reported for monogenic allergy related syndromes as well as GWAS (Additional file 1: Table S2). Individual probes were evaluated according the p value (hypothesis free) and adjusted p values based on p value of 0.05 divided by the maximum amount of genes. Since replication reports from which we obtained candidates followed not the same design and/or platform, we decided to evaluate these three reports independently, to overcome a too strict multiple test penalty when combining all three sets of hits. Therefore we used three separate levels of significance. Berni Canani et al. reported about 4 genes, Martino et al. reported about 49 genes and the review of Bønnelykke et al. reported about 66 genes, yielding significance thresholds of p < 1.25 × 10−2, p < 1.02 × 10−3 and p < 7.58 × 10−4 respectively.
Results
In Table 1 the patient characteristics of the children selected for epigenetic analyses are shown. Controls and children with CMA were well matched except for age at DNA-sampling, in which children with CMA were significantly younger (p = 0.008) compared to controls. Our CMA group consists of both IgE as well as non-IgE mediated CMA, with slightly more IgE positive children (specific IgE > 0.35 kU/L, p = 0.044) in the CMA group compared to controls. Comparison of the 10 tolerant boys with 10 control boys, shows no significant difference in age at DNA-sampling (p = 0.71) nor IgE status (p = 0.56), but did show a significant difference in maternal folic acid use during pregnancy (p = 0.012), with more folic acid use in the control group.
Table 1.
Characteristics of Dutch allergy study population; cow’s milk allergy versus controls
| CMA | Controls | p | Tolerant | Tolerant control boys | p | |
|---|---|---|---|---|---|---|
| N (♀)a | 20 (8) | 20 (10) | 0.342 | 10 (0) | 13 (0) | NA |
| Age diagnosis CMA ± SDb | 6.5 ± 2.5 | NA | NA | 5.8 ± 2.2 | NA | NA |
| Age at sampling ± SDb | 11.8 ± 4.8 | 17.2 ± 7.1 | 0.008 | 18.0 ± 5.5 | 17.2 ± 4.3 | 0.714 |
| N of children with CMP sIgE > 0.35 kU/Lc | 7 | 1 | 0.044 | 2 | 1 | 0.560 |
| Mean CMP sIgE value ± SD (kU/L) | 1.15 ± 3.6 | 0.16 ± 0.64 | 0.260 | 0.19 ± 0.39 | 0.23 ± 0.79 | 0.693 |
| Range CMP sIgE value (kU/L) | 16.4 | 2.9 | NA | 1.0 | 2.9 | NA |
| Maternal smokingd | 0.659 | 0.483 | ||||
| Pregnancy | 2 | 3 | 1 | 2 | ||
| Before pregnancy | 5 | 5 | 2 | 4 | ||
| Never | 13 | 12 | 7 | 7 | ||
| Maternal vitamin Dd | 0.317 | NA | ||||
| Regular | 0 | 0 | 0 | 0 | ||
| Period only | 0 | 0 | 0 | 0 | ||
| Irregular | 1 | 0 | 0 | 0 | ||
| None | 19 | 20 | 10 | 13 | ||
| Maternal multivitamind | 0.836 | 0.883 | ||||
| Regular | 12 | 11 | 1 | 5 | ||
| Period only | 2 | 2 | 4 | 3 | ||
| Irregular | 1 | 1 | 0 | 0 | ||
| None | 5 | 6 | 5 | 5 | ||
| Maternal folic acidd | 0.656 | 0.012 | ||||
| Regular | 8 | 9 | 0 | 5 | ||
| Period only | 9 | 9 | 10 | 6 | ||
| Irregular | 1 | 0 | 0 | 0 | ||
| None | 2 | 2 | 0 | 2 |
Significant p-values <0.05 are in italic
NA not applicable, CMA cow’s milk allergy, p p value, SD standard deviation, N number, CMP cow’s milk protein
aPearson Chi square test
bAge in months ANOVA
cFisher’s exact test, 2-sided
dPearson Chi square test for trend
Epigenetic 450K analysis of selected samples
All 450K analysed samples passed quality control. PCA showed no evidence of confounding technical batch effects according array or array-position (data not shown).General DNA-methylation analysis showed a hypermethylation in the CMA group. While in the tolerant boys versus controls we observed no deviation of the expected ratio hyper/hypomethylation (i.e. 50/50 %) (Fig. 2). A good concordance between CMA-children and controls with regard to the mean relative distribution of different types of, only the most abundant, blood cells: CD8+ and CD4+ T cells, natural Killer, B lymphocytes, monocytes and granulocytes was found (Additional file 2: Figure S1). Furthermore, the variation among blood cell type between groups (expressed as standard deviation) was not different. Relative cell distribution was not included as covariate in further analysis.
Fig. 2.
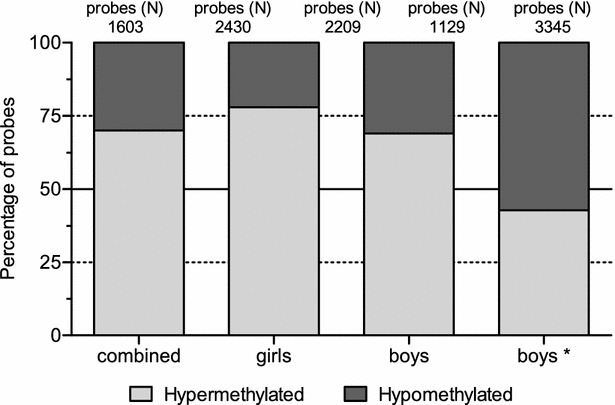
Percentage hyper and hypomethylated probes in cow’s milk allergy (CMA) patients versus controls. Filtering was based on a p value <0.05 and an absolute DNA-methylation difference (delta β values) >5 %; boy*: group boys who had CMA, but are now tolerant
Top 5 hits for DMP’s between CMA and controls for all analysed groups are presented in Table 2. None of individual probes reached genome wide significance. Table 3 shows DMR’s that were found to be associated in more than one group (combined, girls and boys) with a delta methylation difference >0.01. In total ten regions, with their respective gene, were found. Additional file 1: Table S3a–c describes all DMR top 5 hits in each analysed group regardless of the methylation difference (delta).
Table 2.
Top 5 differential methylated positions (DMPs) in combined, girls and boys analysis
| Probe | p | Adj. p | Delta | Gene | Region |
|---|---|---|---|---|---|
| Combined | |||||
| cg07687119 | 6.4E−06 | 0.942 | 0.009 | HOXC4/5/6 | 5UTR/body/body |
| cg01599189 | 1.1E−05 | 0.942 | 0.020 | NBPF1 | 5′UTR |
| cg09789536 | 2.8E−05 | 0.942 | 0.020 | KLHL17 | Body |
| cg11433629 | 2.9E−05 | 0.942 | −0.012 | IGR | NA |
| cg23339709 | 2.9E−05 | 0.942 | 0.009 | OR8U8/OR5M8 | Body/exon1 |
| Girls | |||||
| cg23876832a | 1.7E−07 | 0.081 | 0.214 | IGR | NA |
| cg13181291 | 1.8E−06 | 0.427 | −0.016 | GABPB2 | Body |
| cg13700939 | 1.3E−05 | 0.999 | 0.065 | IGR | NA |
| cg09573389 | 1.3E−05 | 0.999 | 0.039 | C10orf58 | TSS1500 |
| cg24691332 | 3.0E−05 | 0.999 | 0.024 | PARD3 | Body |
| Boys | |||||
| cg06644124 | 1.6E−06 | 0.773 | 0.034 | ZNF281 | 1stExon/5UTR |
| cg11126313 | 1.2E−05 | 0.997 | 0.009 | CCR3 | 5UTR |
| cg09357684 | 1.4E−05 | 0.997 | 0.064 | THAP3 | TSS1500 |
| cg09175834 | 1.5E−05 | 0.997 | 0.018 | NAV3 | 1stExon |
| cg21255128 | 2.1E−05 | 0.997 | 0.018 | C7orf50 | Body |
p p value, adj. p adjusted p value (BH), Delta expressed as β difference, IGR inter genic region, 5′-UTR 5′ untranslated region, Body genebody, TSS1500 transcription start site 1500 bp upstream
aHigh frequency (CEU MAF > 0.05) SNP located at position 34 of probe. Adjusted p values <0.05 were assumed significant
Table 3.
Selection of most significant differential methylated regions found in >1 group with delta >0.01
| Genea | Chromosome | Delta | p | Overlap | Function obtained from genecards | Valb |
|---|---|---|---|---|---|---|
| OR5M8 | 11 | 0.010 0.012 |
7.7E−06 1.6E−04 |
Exon1 (combined) Exon1 (girl) |
Codes for olfactory receptor located in the nose. Is involved in many neurotransmitter and hormone receptors and are responsible for the recognition and transduction of odorant signals | No |
| ZNF281 | 1 | 0.024 0.034 |
1.2E−04 1.3E−06 |
Exon1 (combined) Exon1 (boy) |
Transcription repressor. Represses the transcription of a number of genes including GAST, ODC1 and VIM | Yes |
| KIAA1324L | 7 | 0.012 0.016 |
4.8E−04 3.2E−04 |
Exon1 (combined) Exon1 (boy) |
Estrogen-Induced protein-coding gene | No |
| EIF4E2 | 2 | 0.027 0.036 |
2.6E−05 1.0E−04 |
Gene body (combined) Gene body (girl) |
Diseases associated with EIF4E2 include lung cancer, and among its related super-pathways are Interferon Signaling and Transcription Receptor-mediated HIF regulation | Yes |
| HTR2A | 13 | 0.051 0.069 |
6.4E−05 8.1E−05 |
Gene body (combined) Gene body (girl) |
Receptors for serotonin, a hormone that functions as a neurotransmitter and a mitogen. This receptor is involved in tracheal smooth muscle contraction, bronchoconstriction, and control of aldosterone production | Yes |
| ZNF366 | 5 | 0.022 0.033 |
2.2E−05 1.6E−04 |
TSS1500 (combined) TSS1500 (girl) |
Transcription repressor. Associated diseases include breast cancer, and prostatitis | No |
| DHX58 | 17 | −0.061 −0.061 −0.085 −0.085 |
3.1E−04 3.1E−04 3.3E−04 3.4E−04 |
3′-UTR (combined) SSHELF (combined) 3′-UTR (boy) SSHELF (boy) |
Involved in the innate immune response to various RNA viruses and some DNA viruses | Yes |
| PRB4 | 12 | −0.012 −0.015 |
6.3E−04 5.2E−04 |
3′-UTR (combined) 3′-UTR (boy) |
The protein encoded by this gene is a proline-rich salivary protein | No |
| DNM1 | 9 130996210–130996443 |
−0.022 −0.028 |
7.2E−05 2.4E−04 |
ISLAND (combined) ISLAND (boy) |
The encoded protein possesses unique mechanochemical properties used to tubulate and sever membranes, and is involved in clathrin-mediated endocytosis and other vesicular trafficking processes | No |
| WNT10A | 2 219762987–219763537 |
0.038 0.039 |
9.0E−05 4.1E−04 |
SSHELF (combined) SSHELF (boy) |
These proteins have been implicated in oncogenesis and in several developmental processes, including regulation of cell fate and patterning during embryogenesis | No |
aNearest gene; for genomic regions concerning ISLAND, NSHORE, SSHORE, NSHELF and SSHELF, UCSC browser was used to determine the nearest gene
b Val validated using Roche 454 sequencing
We observed no overlap in loci between boys and girls. The locus ZNF281 was found to be hypermethylated significantly in the boys analysis (p = 1.3 × 10−6, delta = 0.034). The remaining 9 loci showed nominal significant methylation differences and included OR5M8, KIAA1324L, EIF4E2, HTR2A, ZNF366, DHX58, PRB4, DNM1 and WNT10A (Table 3). Although these loci were good candidates for validation procedures, the availability of sufficient DNA was a strong limiting factor. Therefore only four candidate loci were selected for validation. The following four genes were selected for validation: (1) ZNF281; involved in the transcriptional regulation of several interesting genes like GAST, (2) EIF4E2; involved in the interferon pathway, (3) HTR2A; involved in smooth muscle contraction and aldosterone production and (4) DHX58; involved in the innate immune response to RNA and DNA (viruses).
Pathway evaluation of 450K data of selected samples
Biological Process pathways, i.e. GO terms, with a nominal p value <0.01 were evaluated in the combined, girls and boys analysis according to a global, untargeted, test method. Entrez IDs covered by less than 3 probes were excluded from the analysis. We selected enrichment of GO terms representing a unique set of genes in combination with the number of contributing probe sets within that GO annotation. This way similar defined pathways but with different GO annotation were removed from the datasets. We hypothesized that GO terms involved in immunological, epithelial and respiratory classes are feasible to be involved in CMA. Therefore, we applied such class annotation to the detected GO terms. Figure 3 illustrates the relative percentage of the three classes within the filtered global test analysis in the combined, girls and boys groups. Specific GO term annotations are described in Additional file 1: Table S4.
Fig. 3.
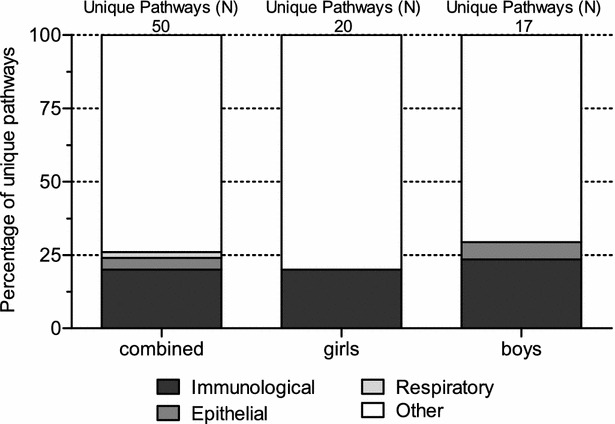
Global test and gene set enrichment analysis in CMA patients versus controls according GO terms. Y-axis contribution (%) of CMA related pathways, i.e. immunological, respiratory and epithelium related
The majority of detected pathways in the combined group involved immunological pathways (20 %), epithelial (4 %) and respiratory (2 %) pathways. In girls, only immunological pathways were detected (20 %). Boys on the other hand, showed in addition to immunological GO terms (23.5 %), epithelial related GO terms (5.9 %) as well. Interestingly, GO terms showed virtual no overlap between combined and girls or between girls and boys, while immunological terms between combined and boys suggests overlap in GO terms involved in epithelial cell polarity and immunological pathways.
Validation of 450K results
Validation for the following candidate regions was performed: ZNF281, EIF4E2, HTR2A and DHX58. Association values of the DMRs of these four genes are described in Table 3. Bisulphite primer sets were designed overlapping or near the CpG extension site of (nominal) significant found 450K probes. In total we designed 12 different amplicons for the four loci. For ZNF281 we were able to successfully optimize two amplicons, for EIF4E2 and HTR2A one amplicon and for DHX58 three amplicons. The exact chromosomal position (HG19) of covered CpGs as well as successful primer sequences are described in Additional file 1: Table S1a. PCR optimization was performed using a standard annealing temperature gradient protocol. 454 Sequencing preparation of the library was conform the Roche manual. Indices are presented in Additional file 1: Table S1b. The 450K DNA-methylation array data showed for several candidates methylation levels close to 10 or 90 %. The latter implies a lower reliability of the quantitative level of DNA methylation in case of a next generation sequencing (NGS) read count below 500. Therefore we aimed for an average depth of 1000 reads. Amplicon 2 and 3 of the ZNF281 gene covered the 450K cg26678649 and cg09255666 probes and the flanking cg24347950 and cg0644124 probes. Unfortunately the average methylation index of the amplicon CpGs did not correspond with the 450K array data, indicating a failed validation for the 450K array ZNF281 region data (Additional file 3: Figure S2A). The second amplicon for the EIF4E2 region flanked the 450K cg10364945 probe. Although the methylation pattern of the CpGs present in this amplicon were concordant to the 450K data we did not observe a similar methylation variation between girl CMA cases and girl controls (Additional file 3: Figure S2B). For the HTR2A region one amplicon was successfully optimized, covering in total 6 CpG positions of which 5 covered the 450K probes cg20102280 (47470793), cg15894389 (47470857), cg02250787 (47470989),cg06476131 (47471052) and cg06476131 (47471090). The first CpG sites located at position 47470760, 47470793, 47470857 showed high correlation and showed a significant hypermethylation (p = 5.1E−4) in CMA patients (Additional file 3: Figure S2C). The first DHX58 amplicon, showed a concordant DNA-methylation pattern with the upstream flanking the probe cg08577293 and the downstream flanking probe cg01405890 covered in total 4 CpG sites. In line with the 450K data, the NGS data showed significant hypomethylation in CMA boys compared to controls (p = 1.1E−3). In CMA girls hypomethylation at this position was absent. The second DHX58 amplicon covered in total 22 CpG sites covering and flanking the 450K probe cg02450064 (40260053). Although, the NGS obtained methylation index of this CpG site showed moderate replication with regard to the 450K data, an interesting and highly significant hypomethylation was observed upstream of cg02450064 (40260053) in CMA boys (p = 9.8E−06) and CMA girls (p = 2.8E−06) compared to controls (Additional file 3: Figure S2D). The third DHX58 amplicon showed insignificant but concordant methylation indices (0–5 %) compared to the 450K data (Additional file 3: Figure S2D).
Gender differences
Epigenetic results show clear differences between CMA boys and girls with respect to their controls with hardly any overlap in obtained loci between the groups, as shown in Tables 2, 3, Fig. 3, and Additional file 1: Table S3. Although such gender difference, even with exclusion of the X and Y-chromosomes is a common phenomenon in epigenetics, we evaluated whether such difference is also phenotypically present. A factor-analysis on phenotypic CMA characteristics of all 91 children suspected of CMA, showed a difference in the univariate analysis with gender as independent factor (p = 0.05). The same factor-analysis model in the children selected for epigenetic analyses showed no difference in the univariate analysis with gender as independent factor (Tables 4, 5). Furthermore, there were more boys compared to girls with proven CMA (Table 4).
Table 4.
Characteristics Subsets Dutch allergy study population
| Subset | N (%♀) | Mean age ± SD (months) | Phenotype counts (%) | Allergies (>1) | ||
|---|---|---|---|---|---|---|
| Skin | GI | Resp | ||||
| Suspicion of CMA allergy | 91 (37.4) | 36.2 ± 10.6 | 68 (55.7) | 47 (38.5) | 7 (5.7) | 14 |
| CMA | 49 (36.7) | 28.9 ± 13.7 | 35 (53.0) | 27 (40.9) | 4 (6.1) | 7 |
| CMA-subset | 20 (40.0) | 28.6 ± 11.4 | 15 (51.7) | 11 (37.9) | 3 (10.3) | 7 |
| FA | 58 (34.5) | 30.7 ± 14.2 | 44 (56.4) | 29 (37.2) | 5 (6.4) | 14 |
| No FA | 33 (42.4) | 45.9 ± 26.2 | 24 (54.5) | 18 (40.9) | 2 (4.5) | 0 |
SD standard deviation, FA food allergy, CMA cow’s milk allergy, CMA-subset 20 children selected for epigenetic analysis from the total group of children with proven CMA, GI gastrointestinal, Resp respiratory
Table 5.
Association of gender with factor analysis regression coefficients
| Subset componentsa | Skin | GI | Resp | FAb | p valuec |
|---|---|---|---|---|---|
| Allergy (C1 of 2) | 0.85 | −0.81 | 0.37 | −0.02 | 0.051 |
| Allergy (C2 of 2) | 0.07 | −0.18 | −0.52 | 0.85 | 0.702 |
| No FA (C1 of 2) | −0.73 | 0.85 | 0.20 | 0.49 | 0.794 |
| No FA (C2 of 2) | 0.51 | 0.15 | 0.91 | 0.12 | 0.292 |
| FA (C1 of 2) | 0.81 | −0.82 | 0.47 | 0.26 | 0.284 |
| FA (C2 of 2) | 0.12 | −0.02 | −0.54 | 0.85 | 0.249 |
| CMA (C1 of 2) | 0.81 | −0.81 | 0.52 | −0.20 | 0.324 |
| CMA (C2 of 2) | 0.22 | −0.20 | −0.29 | 0.91 | 0.107 |
| CMA-DNA (C1 of 2) | 0.83 | −0.86 | 0.30 | 0.51 | 0.843 |
| CMA-DNA (C2 of 2) | 0.16 | 0.02 | 0.85 | −0.72 | 0.213 |
FA food allergy, CMA cow’s milk allergy, GI gastrointestinal, Resp respiratory
aFactor analysis, extraction of components: eigenvalue >1
bUnivariate model: independent: gender, dependent: Regression coefficient factor analysis, covariate: age
cReported p value for gender
Hypothesis driven epigenetic association
For the candidate gene association studies we evaluated findings of three recent published reports with our dataset. First was the report of Berni Canani et al. on DNA methylation differences of IL-4, IL-5, IL-10, and INF-γ in IgE mediated CMA patients [35]. We did not detect any replication in any of the reported loci (data not shown) applying a multiple test significance level of p < 1.25 × 10−2 (Bonferonni, 0.05/4 independent tests). Second was the report of Martino et al. epigenome wide association in IgE mediated food allergy [36]. In total 2281 probes were extracted from our dataset. The six lowest p values (p < 0.005) covered 1 probe at GALNTL4, 2 probes at HDAC4 locus, 1 probe at KCNN3 locus and 2 probes at the RPS6KA2 locus (data not shown). However, we cannot claim replication for any of the probes annotated for the 49 reported genes (p < 1.02 × 10−3, Bonferonni, 0.05/49 independent tests). The final hypothesis driven evaluation was based on a review on genetic rather than epigenetic association studies of atopic diseases by Bønnelykke et al. [34]. In total we selected 66 genes covered by approximately 1650 individual probes (Additional file 1: Table S2a). We found one significant replication applying a multiple test significance level of p < 7.58 × 10−4 (Bonferonni, 0.05/66 independent tests). The probe cg03990811, located in the MICA gene showed significant association (p = 3.9 × 10−4) in the combined analysis (Additional file 1: Table S4b). This finding is strengthened by the fact that we found a second probe (cg14462939, p = 3.9 × 10−3) present in the top5 hits and located in the MICA gene (Additional file 1: Table S2b).
Discussion
In this pilot study we aimed to detect DNA-methylation differences between a group of mixed gender CMA and healthy controls. Several differential methylated loci were found, suggesting aberrant DNA-methylation is involved in the expression of CMA. Additional DNA-methylation analysis between a group of CMA tolerant boys and controls showed no DNA-methylation differences at all. We validate three loci of the top 10 of the 450K array results using bisulphite amplicon NGS. We found no evidence of confounding effects of inoculation or differences of blood cell distribution between cases and controls. Epigenetic gender differences were not reflected in a phenotypic symptom pattern in the CMA group but were present in the better-powered total allergy cohort.
Epigenetic results
Although it is a topic of high interest, we are the second to describe an epigenetic survey in children with CMA [14, 35]. General hypermethylation was found in children with CMA, which disappeared after developing tolerance. In addition a difference in methylation between boys with CMA and control boys was found in this study. Hypermethylation in general is associated with repressed gene transcription [37]. In case a repressed gene codes for a transcription factor, several of its target genes might show an excess of expression. In CMA the immune system is overactive, suggesting immunological suppressive genes should be hypermethylated in general, compared to non-CMA children [38]. In CMA tolerant children this hypermethylation effect completely disappeared. Therefore, hypermethylation can only serve as biomarker during the allergic state.
DMR analysis showed several feasible genes to be involved in CMA. ZNF281 was significantly hypermethylated in boys versus controls. This gene is a transcriptional regulator, known to repress several genes, for example GAST (located on chromosome 17) [39]. GAST is known to stimulate the stomach mucosa, production of digestive enzymes, smooth muscle contraction and releases histamines. Therefore hypermethylation of ZNF281 might be related to the gastro-intestinal symptoms in children with CMA. However, since the NGS amplicon analysis showed disconcordant and different methylation levels in CMA patients and controls, the claim of significance of the 450K results for the ZNF281 locus has to be taken with caution.
EIF4E2 is primarily involved in the initiation of protein synthesis [39]. Melnik et al. reported about the importance of breastmilk in early development and the mTORC1 signaling pathway. The EIF4 protein family plays a key role in this pathway that stimulates (post natal) growth and can be regulated by maternal excreted exosomal miRNAs [40]. Our finding of hypomethylation of EIF4E2 might be the result of absence of these maternal miRNAs due to CMP free diet. Dysregulation of the mTORC1 pathway might be related to the methylation status of EIF4E2.
Hypermethylation of HTR2A is likely related to less expressed protein and might on turn be related to gastro-intestinal symptoms in CMA patients via altered intestinal muscle contraction [39]. The methylation pattern of the bsr amplicon NGS was similar to the 450K although we observed a consistent lower methylation index in the NGS data.
Although, not significantly associated according to the applied multiple test threshold, DHX58 showed robust variability in all analyses including NGS bsr amplicon validation. The protein encoded by this gene plays a crucial step at the beginning of the cascade of antigen recognition in the innate immune response to various RNA viruses and some DNA viruses. Hypomethylation of DHX58 might lead to over expression of this gene, which on turn might favour viral nucleoprotein recognition by DDX58/RIG-I and IFIH1/MDA5 complexes [39]. Deretic et al. proposed that nutritional signals (mTORC pathway) and immune signalling (RIG-I/MDA5 pathway) might stimulate similar cascades. This suggestion might indicated that our findings on EIF4E2 and DHX58 underlie similar and maybe shared cascades [41]. Future studies should therefore investigate whether cow’s milk proteins are capable of mimicking epitopes, confirming a role of DHX58 in CMA.
Other genes in the top 5 findings are interesting as well. Unfortunately, the availability of DNA in these CMA samples was limited which disabled validation of further loci. Feasible biological relevance and their possible involvement in CMA have to be studied further.
Pathway evaluation of 450K data of selected samples
Gene set enrichment analyses based on a global test method, suggest that specific immunological mechanisms are involved in CMA. Interestingly, no overlap of these pathways between combined and girls or girls and boys were observed, suggesting involvement of different immunological biological processes between boys and girls with CMA (Additional file 1: Table S4a; Fig. 3). However, limited statistical power cannot be excluded as cause of this difference between genders. We did observe gender differences with regard to other, non-immunological, biological processes. A limited number of biological processes suggests involvement of epithelium development in combined CMA and boys CMA only. Furthermore, biological processes involved in the respiratory mechanism, was solely observed in boys. We can state that our results strongly suggest involvement of gender specific immunological processes and that other processes prone to be involved in CMA, are likely to be different between genders as well.
Gender difference
In this study we observed a clear difference, or in other words no overlap, of aberrant methylation loci, between boys and girls. These results were found in the differences in top-hits in the epigenetic results, but also in the number of boys with CMA and in the factor-analysis of all children suspected of CMA. This implies that different biological mechanisms underlie CMA in boys and girls. However we were not able to confirm this difference in the factor-analysis in our subgroup of 20 CMA-children and 20 controls. This is probably due to very small subgroups in a very heterogeneous disease. Furthermore symptom descriptions, obtained through questionnaires answered by parents are very subjective, compared to epigenetic results. It is very likely that these effects are smaller in larger sample sizes. However, differences between boys and girls are not unexpected. It has been described both, in allergy studies, as well as in epigenetic studies [21–24].
Allergic march
The epigenetic results presented in this study, show methylation differences in children suffering from CMA relative to controls, which disappears in the children who developed tolerance to CMA.
So far, it has been considered that children who suffer from FA, are at risk of developing other allergic diseases later in life [42]. However, since the methylation differences found in our study disappear upon developing CMP tolerance, there is no indication that the studied loci are involved in development of the allergic march in children with CMA, despite some (top hit) loci have been described in childhood asthma as well [39]. A recent review discussed the short-term dynamics of epigenetic programming within human immunological responses. They stated that epigenetic regulation plays a key role in proliferation and differentiation of specific immune systems. Immune cells share for example many specific transcription factors, but remodelling of the epigenome is thought to be essential regulating specific gene expression profiles for different lineages [43]. Since our data suggest the absence of CMA “epigenetic memory” or evidence of certain enrichment of specific immune cells, it may disprove the hypothesis arguing for an epigenetic basis underlying the “allergic march”. Literature on the development of the allergic march is contradicting, with studies reporting on the development of the allergic march, as well as studies describing a co-manifestation instead of an allergic march [9, 42, 44, 45]. An epigenetic risk profile might also be a contributing factor. Since methylation can change back and forth more than once in life, certain people might be more at risk for expressing diseases involved by epigenetic changes. However, our results need to be interpreted with great caution, with a very little sample size in a heterogeneous disease. Furthermore, these results are based on CMA and CMP-tolerance development only, clinical data regarding development of other allergic diseases in life are not yet available, due to the prospective character of this study. It would be interesting to evaluate these children in the future in case of the development of other allergic diseases.
Replication other reports involving CMA
Although no significant replication was found with the review of Bønnelykke et al., according the q value based on the number of selected probes, applying a multiple test correction based on the number of genes, showed significant replication for the MICA gene [34]. In addition, this finding is strengthened by the fact that another probe located in this gene was present in the DMP top 5 hits. According UCSC public databases, both probes are located in the first intron of the MICA gene which, also represents a strong H3K27AC mark that is associated with an active regulatory element [46]. MICA is closely positioned to HLA-B and C on chromosome 6, which both are thought to play an important role in MHC interactions and immune related disorders. MICA itself codes for a protein that functions as a stress-induced antigen. Interestingly, MICA is broadly recognized by intestinal epithelial gamma delta T cells, favouring an association with FA.
We were not able to replicate the results of two other studies on epigenetics in food allergy studies [34, 35]. Differences in the design between our study and the studies of others are likely to be responsible for the absence of replication. First of all, we used whole blood sample analyses, while both other studies used subsets of blood cells, namely peripheral blood mononuclear cells and CD4+ T cell respectively. Second, we studied children with IgE and non-IgE mediated CMA, while Berni Canani et al. studied children with CMA with high levels of IgE only and Martino et al. studied children with IgE-mediated FA, and included only one child with CMA. Furthermore the study of Berni Canani et al. used a candidate gene approach. The study of Martino et al. was based on a pooled sample strategy and analysis was based on a mixed gender design [34, 35]. Our analysis was untargeted and we have followed both a combined and gender stratified strategy. This emphasizes the importance and the great bias potential of epigenetic, tissue related, heterogeneity which is very likely to block positive replication between studies.
Strength and weaknesses of the study
The main strength of the studied samples is that CMA is diagnosed according to the current available gold standard [27]. Furthermore, clinical data are well documented due to the setup of the study with regular questionnaires [25].
Since our sample size is small, we used strict thresholds for evaluation of our results. For p values this threshold was based on bonferonni and for effect size we applied an absolute difference of >5 %. Despite these thresholds results need to be interpreted with great caution, until they are replicated in an independent study.
Our sample involves IgE and non-IgE mediated CMA cases. Additional analysis of epigenetic profiles of CMA patients with relative high levels of IgE (N = 12 combined genders, IgE ranging between 0.01 and 16.4, three CMA cases IgE > 0.5) showed no additional significant hits with regard to the total group of CMA children (data not shown). In general the IgE plasma level is not an essential parameter in CMA diagnosis. Moreover, IgE mediated and non-IgE mediated CMA might origin from an overlapping or similar sensitization mechanism [43]. Our study is thus limited in the sense that we solely are able to detect general CMA associated epigenetic loci. Future research using a larger CMA sample should include IgE and non-IgE stratified analyses. Ideally these IgE measurements should be measured several times during the course of disease.
Unfortunately, there was only one DNA-sample available per child. Therefore, comparisons over time within the same child were not possible. Also, it was not possible to obtain DNA-samples in all children. Furthermore the amount of blood drawing for research purposes in infants is in the Netherlands limited to 2.5 mL. The latter also implies that a sufficient DNA yield in DNA extraction of a specific blood cell is virtual impossible.
Proper meta-analysis nor replication of our findings in an independent well characterized CMA cohort unfortunately appeared not to be feasible. Previous reports generally are based on specific immune cells while our study is based on DNA obtained from whole blood. Validation our dataset was therefore solely based on replicating findings of others in our sample.
In general, discrepancy between the NGS and 450K methylation index might be the result of a failing normalization procedure (BMIQ) to overcome the probe bias of the array. A suboptimal primerset, in case of amplicon NGS, cannot be excluded as another cause of the discrepancy.
The national Dutch vaccination program is extensive and starts already at young age. Since vaccination can have a major impact on the blood cell distribution and/or on methylation status of particular loci we preselected our sample and evaluated post hoc the period between vaccination and blood drawing (Additional file 4: Figure S3). We excluded all children who received a vaccination up to 4 weeks prior to DNA-sampling [47]. Since no significant differences between CMA cases and controls and their period (>4 weeks) between vaccination and blood drawing were found, we are confident vaccination plays an insignificant role with regard to the epigenetic aberrations we detected in this study. Finally there is a statistical significant difference in the folic acid consumption of mothers during pregnancy between the tolerant boys and control boys. It is known that folic acid has the ability to influence epigenetics. Since our sample size is very limited, folic acid is widely used by pregnant women and this difference was only found between already tolerant boys and controls (and not in children still suffering from CMA), we consider the relevance of this difference limited [48–50].
Future studies
The present study should be classified as pilot study or proof of principle study. Moreover, CMA can be characterized as a complex disease involving multiple loci and a heterogeneous phenotype. Thus, in combination with the fact we analysed only relative small groups, further studies in larger cohorts are essential in order to replicate or validate our findings. Ideally, sample size in this study would be large enough to investigate boys and girls as well as IgE and non-IgE mediated CMA separately or to perform subgroup-analysis.
To investigate whether there is a evidence for the allergic march, it would be interesting to compare epigenetic analyses during infancy, school-age and later life. Boys and girls should be separately analysed to also investigate a gender effect.
Since infants develop CMA at a very young age. Diet is known to be involved in the development of certain diseases, probably through epigenetic alterations, in the next generations [51]. And milk is known to influence the infants immune system [40, 52]. It would be very interesting to investigate the role of (breast)milk in a future epigenetic study in children with cow’s milk allergy, especially seen our findings in EIF4E2.
Conclusion
Current studies indicate the involvement of epigenetic mechanisms underlying the expression of CMA. In particular DHX58, involved in DDX58/RIG-I and IFIH1/MDA5 immunological pathways, are involved in CMA in young boys, while in CMA girls EIF4E2, HTR2A seems to play a role. Our data strongly suggests a different mechanism for girls and boys underlying CMA. We have no indication DNA methylation aberrations play a crucial role in the allergic march, since all effects disappeared upon developing CMA tolerance in boys. However, due to small sample size further studies are necessary before definite conclusions can be drawn.
Authors’ contributions
NP: acquisition, analysis and interpretation of data; drafting the manuscript. PH: supervision of laboratory experiments; analysis and interpretation of data; drafting the manuscript. AV: performing laboratory experiments; critically revising the manuscript. AM: performing laboratory experiments; critically revising the manuscript. FS: performing laboratory experiments; critically revising the manuscript. MH: performing laboratory experiments; critically revising the manuscript. OM: performing laboratory experiments; critically revising the manuscript. RH: interpretation of the data; critically revising the manuscript. AS: supervising the clinical studies; critically revising the manuscript. MM: supervising the laboratory studies; critically revising the manuscript. All authors read and approved the final manuscript.
Acknowledgements
The authors would like to thank all children and their parents for participating in the Dutch EuroPrevall Birth Cohort Study. Furthermore the authors would like to thank everyone involved in collecting clinical data. The employees of the department of Parasitology, Leiden University Medical Center, for storing blood samples.
Competing interests
This study was supported by an unrestricted grant from the “Stichting Steun Emma” Academic Medical Hospital Amsterdam. The European EuroPrevall Birth Cohort study has been funded by the European Commission (FOOD-CT_2005-514000). The Dutch cohort received unrestricted grants from Nutricia Netherlands, Nutricia Advanced Medical Nutrition Netherlands, AstraZeneca Netherlands, TEVA Netherlands and GlaxoSmithKline Netherlands. These companies were not involved in the design of the study, collecting the data or writing the manuscript. The authors declare that they have no competing interests.
Abbreviations
- CMA
cow’s milk allergy
- CMP
cow’s milk protein
- DBPCFC
double blind placebo controlled food challenge
- DMP
differentially methylated positions
- DMR
differential methylated regions
- DNA
deoxyribonucleic acid
- EWAS
epigenome-wide association studies
- FA
food allergy
- GO
gene ontology
- GWAS
genome wide association studies
- IgE
immunoglobuline E
- NGS
next generation sequencing
- PCA
principal component analysis
- PCR
Polymerase chain reaction
- RNA
ribonucleic acid
- sIgE
serum specific IgE
- SNP
single nucleotide polymorphism
Additional files
10.1186/s13601-016-0105-z Relative blood cell type distribution estimate in CMA patients and controls. CD8T: CD8+ T cells; CD4T: CD4+ T cells; NK: natural killer cells; Bcell: B cells; Mono: monocytes; Gran: granulocytes.
10.1186/s13601-016-0105-z Average methylation index. A: ZNF281 amplicon 2 and 3 in CMA boys and control boys. B: EIF4E2 amplicon in CMA girls and control girls. C: HTR2A amplicon in CMA girls and control girls. D: DHX58 amplicon 1–3 in CMA boys and control boys. Y-axis: Methylation index expressed in (normalized) β-values; X-axis: ranked chromosomal probe position according hg19.
10.1186/s13601-016-0105-z Categorical time span between vaccination and blood drawing (DNA) of CMA cases and controls.
Footnotes
Nicole C. M. Petrus and Peter Henneman contributed equally to this work
Aline B. Sprikkelman and Marcel Mannens contributed equally to this work
Contributor Information
Nicole C. M. Petrus, Phone: 0031 20 5662851, Email: n.c.petrus@amc.nl
Peter Henneman, Email: p.henneman@amc.nl.
Andrea Venema, Email: a.venema@amc.nl.
Adri Mul, Email: a.n.mul@amc.nl.
Femke van Sinderen, Email: femke.vsinderen@gmail.com.
Martin Haagmans, Email: m.a.haagmans@amc.nl.
Olaf Mook, Email: o.r.mook@amc.nl.
Raoul C. Hennekam, Email: r.c.hennekam@amc.nl
Aline B. Sprikkelman, Email: a.b.sprikkelman@amc.nl
Marcel Mannens, Email: m.a.mannens@amc.nl.
References
- 1.Chafen JJ, Newberry SJ, Riedl MA, Bravata DM, Maglione M, Suttorp MJ, et al. Diagnosing and managing common food allergies: a systematic review. JAMA. 2010;303(18):1848–1856. doi: 10.1001/jama.2010.582. [DOI] [PubMed] [Google Scholar]
- 2.Rona RJ, Keil T, Summers C, Gislason D, Zuidmeer L, Sodergren E, et al. The prevalence of food allergy: a meta-analysis. J Allergy Clin Immunol. 2007;120(3):638–646. doi: 10.1016/j.jaci.2007.05.026. [DOI] [PubMed] [Google Scholar]
- 3.Schoemaker AA, Sprikkelman AB, Grimshaw KE, Roberts G, Grabenhenrich L, Rosenfeld L, et al. Incidence and natural history of challenge-proven cow’s milk allergy in European children–EuroPrevall birth cohort. Allergy. 2015;70(8):963–972. doi: 10.1111/all.12630. [DOI] [PubMed] [Google Scholar]
- 4.Benhamou AH, Schappi Tempia MG, Belli DC, Eigenmann PA. An overview of cow’s milk allergy in children. Swiss Med Wkly. 2009;139(21–22):300–307. doi: 10.4414/smw.2009.12258. [DOI] [PubMed] [Google Scholar]
- 5.Tsai HJ, Kumar R, Pongracic J, Liu X, Story R, Yu Y, et al. Familial aggregation of food allergy and sensitization to food allergens: a family-based study. Clin Exp Allergy. 2009;39(1):101–109. doi: 10.1111/j.1365-2222.2008.03111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen W, Mempel M, Schober W, Behrendt H, Ring J. Gender difference, sex hormones, and immediate type hypersensitivity reactions. Allergy. 2008;63(11):1418–1427. doi: 10.1111/j.1398-9995.2008.01880.x. [DOI] [PubMed] [Google Scholar]
- 7.Matsumoto K, Saito H. epicutaneous immunity and onset of allergic diseases-per-”eczema” tous sensitization drives the allergy march. Allergol Int. 2013;62(3):291–296. doi: 10.2332/allergolint.13-RAI-0603. [DOI] [PubMed] [Google Scholar]
- 8.Noverr MC, Huffnagle GB. The ‘microflora hypothesis’ of allergic diseases. Clin Exp Allergy. 2005;35(12):1511–1520. doi: 10.1111/j.1365-2222.2005.02379.x. [DOI] [PubMed] [Google Scholar]
- 9.Host A, Halken S, Jacobsen HP, Christensen AE, Herskind AM, Plesner K. Clinical course of cow’s milk protein allergy/intolerance and atopic diseases in childhood. Pediatr Allergy Immunol. 2002;13(Suppl 15):23–28. doi: 10.1034/j.1399-3038.13.s.15.7.x. [DOI] [PubMed] [Google Scholar]
- 10.Saarinen KM, Pelkonen AS, Makela MJ, Savilahti E. Clinical course and prognosis of cow’s milk allergy are dependent on milk-specific IgE status. J Allergy Clin Immunol. 2005;116(4):869–875. doi: 10.1016/j.jaci.2005.06.018. [DOI] [PubMed] [Google Scholar]
- 11.Host A, Halken S. A prospective study of cow milk allergy in Danish infants during the first 3 years of life. Clinical course in relation to clinical and immunological type of hypersensitivity reaction. Allergy. 1990;45(8):587–596. doi: 10.1111/j.1398-9995.1990.tb00944.x. [DOI] [PubMed] [Google Scholar]
- 12.Longo G, Berti I, Burks AW, Krauss B, Barbi E. IgE-mediated food allergy in children. Lancet. 2013;382(9905):1656–1664. doi: 10.1016/S0140-6736(13)60309-8. [DOI] [PubMed] [Google Scholar]
- 13.Morita H, Nomura I, Matsuda A, Saito H, Matsumoto K. Gastrointestinal food allergy in infants. Allergol Int. 2013;62(3):297–307. doi: 10.2332/allergolint.13-RA-0542. [DOI] [PubMed] [Google Scholar]
- 14.Tan TH, Ellis JA, Saffery R, Allen KJ. The role of genetics and environment in the rise of childhood food allergy. Clin Exp Allergy. 2012;42(1):20–29. doi: 10.1111/j.1365-2222.2011.03823.x. [DOI] [PubMed] [Google Scholar]
- 15.Bird A. Perceptions of epigenetics. Nature. 2007;447(7143):396–398. doi: 10.1038/nature05913. [DOI] [PubMed] [Google Scholar]
- 16.Hong X, Wang X. Early life precursors, epigenetics, and the development of food allergy. Semin Immunopathol. 2012;34(5):655–669. doi: 10.1007/s00281-012-0323-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anacker C, O’Donnell KJ, Meaney MJ. Early life adversity and the epigenetic programming of hypothalamic-pituitary-adrenal function. Dialogues Clin Neurosci. 2014;16(3):321–333. doi: 10.31887/DCNS.2014.16.3/canacker. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Andersson NW, Hansen MV, Larsen AD, Hougaard KS, Kolstad HA, Schlunssen V. Prenatal maternal stress and atopic diseases in the child: a systematic review of observational human studies. Allergy. 2016;71(1):15–26. doi: 10.1111/all.12762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Platts-Mills TA. The allergy epidemics: 1870–2010. J Allergy Clin Immunol. 2015;136(1):3–13. doi: 10.1016/j.jaci.2015.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prescott SL, Pawankar R, Allen KJ, Campbell DE, Sinn JK, Fiocchi A, et al. A global survey of changing patterns of food allergy burden in children. World Allergy Organ J. 2013;6(1):21. doi: 10.1186/1939-4551-6-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Naumova AK, Al TA, Morin A, Vaillancourt VT, Madore AM, Berlivet S, et al. Sex- and age-dependent DNA methylation at the 17q12-q21 locus associated with childhood asthma. Hum Genet. 2013;132(7):811–822. doi: 10.1007/s00439-013-1298-z. [DOI] [PubMed] [Google Scholar]
- 22.Vink NM, Postma DS, Schouten JP, Rosmalen JG, Boezen HM. Gender differences in asthma development and remission during transition through puberty: the TRacking Adolescents’ Individual Lives Survey (TRAILS) study. J Allergy Clin Immunol. 2010;126(3):498–504. doi: 10.1016/j.jaci.2010.06.018. [DOI] [PubMed] [Google Scholar]
- 23.DunnGalvin A, Hourihane JO, Frewer L, Knibb RC, Oude Elberink JN, Klinge I. Incorporating a gender dimension in food allergy research: a review. Allergy. 2006;61(11):1336–1343. doi: 10.1111/j.1398-9995.2006.01181.x. [DOI] [PubMed] [Google Scholar]
- 24.Kelly C, Gangur V. Sex disparity in food allergy: evidence from the PubMed database. J Allergy (Cairo) 2009;2009:159845. doi: 10.1155/2009/159845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Keil T, McBride D, Grimshaw K, Niggemann B, Xepapadaki P, Zannikos K, et al. The multinational birth cohort of EuroPrevall: background, aims and methods. Allergy. 2010;65(4):482–490. doi: 10.1111/j.1398-9995.2009.02171.x. [DOI] [PubMed] [Google Scholar]
- 26.McBride D, Keil T, Grabenhenrich L, Dubakiene R, Drasutiene G, Fiocchi A, et al. The EuroPrevall birth cohort study on food allergy: baseline characteristics of 12,000 newborns and their families from nine European countries. Pediatr Allergy Immunol. 2012;23(3):230–239. doi: 10.1111/j.1399-3038.2011.01254.x. [DOI] [PubMed] [Google Scholar]
- 27.Boyce JA, Assa’ad A, Burks AW, Jones SM, Sampson HA, Wood RA, et al. Guidelines for the diagnosis and management of food allergy in the United States: summary of the NIAID-sponsored expert panel report. J Allergy Clin Immunol. 2010;126(6):1105–1118. doi: 10.1016/j.jaci.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Petrus NC, Schoemaker AF, van Hoek MW, Jansen L, Jansen-van der Weide MC, van Aalderen WM, Sprikkelman AB. Remaining symptoms in half the children treated for milk allergy. Eur J Pediatr. 2015;174(6):759–765. doi: 10.1007/s00431-014-2456-6. [DOI] [PubMed] [Google Scholar]
- 29.Henneman P, Petrus NC, Venema A, van SF, van der Lip K, Hennekam RC, et al. Genetic susceptibility for cow’s milk allergy in Dutch children: the start of the allergic march? Clin Transl Allergy. 2015;6:7. doi: 10.1186/s13601-016-0096-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Houseman EA, Kelsey KT, Wiencke JK, Marsit CJ. Cell-composition effects in the analysis of DNA methylation array data: a mathematical perspective. BMC Bioinform. 2015;16:95. doi: 10.1186/s12859-015-0527-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen YA, Lemire M, Choufani S, Butcher DT, Grafodatskaya D, Zanke BW, et al. Discovery of cross-reactive probes and polymorphic CpGs in the illumina infinium humanmethylation450 microarray. Epigenetics. 2013;8(2):203–209. doi: 10.4161/epi.23470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goeman JJ, van de Geer SA, de KF, van Houwelingen HC. A global test for groups of genes: testing association with a clinical outcome. Bioinformatics. 2004;20(1):93–99. doi: 10.1093/bioinformatics/btg382. [DOI] [PubMed] [Google Scholar]
- 33.Methprimer. www.urogene.org/cgi-bin/methprimer/methprimer.cgi. Accessed 1-5-2014.
- 34.Bonnelykke K, Sparks R, Waage J, Milner JD. Genetics of allergy and allergic sensitization: common variants, rare mutations. Curr Opin Immunol. 2015;36:115–126. doi: 10.1016/j.coi.2015.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berni CR, Paparo L, Nocerino R, Cosenza L, Pezzella V, Di CM, et al. Differences in DNA methylation profile of Th1 and Th2 cytokine genes are associated with tolerance acquisition in children with IgE-mediated cow’s milk allergy. Clin Epigenetics. 2015;7(1):38. doi: 10.1186/s13148-015-0070-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martino D, Joo JE, Sexton-Oates A, Dang T, Allen K, Saffery R, Prescott S. Epigenome-wide association study reveals longitudinally stable DNA methylation differences in CD4+ T cells from children with IgE-mediated food allergy. Epigenetics. 2014;9(7):998–1006. doi: 10.4161/epi.28945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.He XJ, Chen T, Zhu JK. Regulation and function of DNA methylation in plants and animals. Cell Res. 2011;21(3):442–465. doi: 10.1038/cr.2011.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Walker-Smith J. Cow’s milk allergy: a new understanding from immunology. Ann Allergy Asthma Immunol. 2003;90(6 Suppl 3):81–83. doi: 10.1016/S1081-1206(10)61666-3. [DOI] [PubMed] [Google Scholar]
- 39.Genecards. www.genecards.org. Accessed 1-5-2014.
- 40.Melnik BC, John SM, Schmitz G. Milk is not just food but most likely a genetic transfection system activating mTORC1 signaling for postnatal growth. Nutr J. 2013;12:103. doi: 10.1186/1475-2891-12-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013;13(10):722–737. doi: 10.1038/nri3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Spergel JM. From atopic dermatitis to asthma: the atopic march. Ann Allergy Asthma Immunol. 2010;105(2):99–106. doi: 10.1016/j.anai.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 43.Kondilis-Mangum HD, Wade PA. Epigenetics and the adaptive immune response. Mol Aspects Med. 2013;34(4):813–825. doi: 10.1016/j.mam.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Illi S, Voon ME, Lau S, Nickel R, Gruber C, Niggemann B, Wahn U. The natural course of atopic dermatitis from birth to age 7 years and the association with asthma. J Allergy Clin Immunol. 2004;113(5):925–931. doi: 10.1016/j.jaci.2004.01.778. [DOI] [PubMed] [Google Scholar]
- 45.Nissen SP, Kjaer HF, Host A, Nielsen J, Halken S. The natural course of sensitization and allergic diseases from childhood to adulthood. Pediatr Allergy Immunol. 2013;24(6):549–555. doi: 10.1111/pai.12108. [DOI] [PubMed] [Google Scholar]
- 46.UCSC Public databases. https://genome.ucsc.edu. Accessed 1-5-2014.
- 47.Amanna IJ, Slifka MK. Mechanisms that determine plasma cell lifespan and the duration of humoral immunity. Immunol Rev. 2010;236:125–138. doi: 10.1111/j.1600-065X.2010.00912.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Amarasekera M, Prescott SL, Palmer DJ. Nutrition in early life, immune-programming and allergies: the role of epigenetics. Asian Pac J Allergy Immunol. 2013;31(3):175–182. [PubMed] [Google Scholar]
- 49.De-Regil LM, Fernandez-Gaxiola AC, Dowswell T, Pena-Rosas JP. Effects and safety of periconceptional folate supplementation for preventing birth defects. Cochrane Database Syst Rev 2010;(10):CD007950. doi:10.1002/14651858.CD007950.pub2 [DOI] [PMC free article] [PubMed]
- 50.Harb H, Renz H. Update on epigenetics in allergic disease. J Allergy Clin Immunol. 2015;135(1):15–24. doi: 10.1016/j.jaci.2014.11.009. [DOI] [PubMed] [Google Scholar]
- 51.Kaati G, Bygren LO, Edvinsson S. Cardiovascular and diabetes mortality determined by nutrition during parents’ and grandparents’ slow growth period. Eur J Hum Genet. 2002;10(11):682–688. doi: 10.1038/sj.ejhg.5200859. [DOI] [PubMed] [Google Scholar]
- 52.Tooley KL, El-Merhibi A, Cummins AG, Grose RH, Lymn KA, DeNichilo M, Penttila IA. Maternal milk, but not formula, regulates the immune response to beta-lactoglobulin in allergy-prone rat pups. J Nutr. 2009;139(11):2145–2151. doi: 10.3945/jn.109.108845. [DOI] [PubMed] [Google Scholar]