

Abstract
Purpose
To examine whether Nrf2-regulated antioxidant defense and p53 are activated in human corneal endothelial cells (CEnCs) by environmental levels of ultraviolet A (UV-A), a known stimulator of oxidative stress.
Methods
Immortalized human CEnCs (HCEnCi) were exposed to UV-A fluences of 2.5, 5, 10, or 25 J/cm2, then allowed to recover for 3 to 24 hours. Control HCEnCi did not receive UV-A. Reactive oxygen species (ROS) were measured using H2DCFDA. Cell cytotoxicity was evaluated by lactate dehydrogenase (LDH) release. Levels of Nrf2, HO-1, NQO-1, p53, and caspase3 were detected by immunnoblotting or real-time PCR. Activated caspase3 was measured by immunoblotting and a fluorescence assay.
Results
Exposure of HCEnCi to 5, 10, and 25 J/cm2 UV-A increased ROS levels compared with controls. Nrf2, HO-1, and NQO-1 mRNA increased 1.7- to 3.2-fold at 3 and 6 hours after irradiation with 2.5 and 5 J/cm2 UV-A. At 6 hours post irradiation, UV-A (5 J/cm2) enhanced nuclear Nrf2 translocation. At 24 hours post treatment, UV-A (5, 10, and 25 J/cm2) produced a 1.8- to 2.8-fold increase in phospho-p53 and a 2.6- to 6.0-fold increase in activated caspase3 compared with controls, resulting in 20% to 42% cell death.
Conclusions
Lower fluences of UV-A induce Nrf2-regulated antioxidant defense and higher fluences activate p53 and caspase3, indicating that even near-environmental levels of UV-A may affect normal CEnCs. This data suggest that UV-A may especially damage cells deficient in antioxidant defense, and thus may be involved in the etiology of Fuchs' endothelial corneal dystrophy (FECD).
Keywords: corneal endothelial cells, ultraviolet A, Nrf2, p53, oxidative stress
The cornea is exposed daily to solar ultraviolet (UV) radiation, which is known to induce damage to DNA as well as initiate oxidative stress.1 In a natural environment, UV-A with the wavelengths of 320 to 400 nm constitutes approximately 95% of the UV radiation that reaches the earth's surface. Different from UV-B (280–320 nm), which can be absorbed by the bases in DNA directly, UV-A wavelengths are not well absorbed by native DNA and induce reactive oxygen species (ROS) causing oxidative damage.1 Based on the observations of UV-A intensity of different areas around the world, it is estimated that 10 J/cm2 of UV-A equates to approximately 2 hours of exposure of sunlight at noon time during the summer.2 Ultraviolet A penetrates all corneal layers including endothelium.3 Human corneal endothelial cells (CEnCs) are arrested in the post mitotic state and have limited proliferative and repair capacity in vivo. Therefore, CEnCs damaged by aging or disease do not have the capacity to regenerate. The adverse effect of UV-A on CEnCs remains unclear. Limited safety studies of corneal collagen crosslinking using UV-A showed that high doses of UV-A irradiation caused rabbit CEnC cell apoptosis and necrosis.4,5 Given that CEnCs are particularly susceptible to oxidative damage due to their high aerobic metabolic activity and lifelong exposure to sunlight6 studying the effect of UV-A, the natural physical oxidative stressor on CEnCs would provide the basis for the role of UV-A in corneal endothelial degeneration.
Fuchs endothelial corneal dystrophy (FECD) is a leading cause of endogenous corneal endothelial degeneration. Previous studies showed that oxidant–antioxidant imbalance plays a major role in the chronic degenerative process of corneal endothelium (CE) and apoptosis seen in FECD.7 Corneal endotheliums exhibited an increased level of oxidative DNA damage and apoptosis in FECD as compared with that seen in normal cells.7,8 Moreover, decreased levels of different antioxidants, such as glutathione S-transferase (GST), aldehyde dehydrogenase 3A1 (ALDH3A1), peroxiredoxins (Prdx), superoxide dismutase 2 (SOD2), thioredoxin reductase 1 (TXNRD1), and heme oxygenase 1(HO-1) were detected in FECD.7,9–11 Proximal promoter sequence analysis of downregulated antioxidants revealed a consensus sequence called antioxidant responsive element (ARE). Importantly, nuclear factor erythroid 2–related factor 2 (Nrf2), which is a master regulator of ARE-mediated antioxidants, showed decreased protein levels in FECD as well.7
Transcription factors Nrf2 and p53 play key roles in the stress response. Under oxidative stress conditions, Nrf2 is known to cause a coordinated upregulation of antioxidant defense.12–15 p53 may coordinate many biological processes including cell-cycle arrest, apoptosis, senescence, and DNA repair.16 p53 phosphoprotein acts as a guardian of the cell by either repairing DNA damage or by inducing cell apoptosis.17–19 When ROS-induced damage is irreparable, p53 promotes cell apoptosis.20,21 At present, the interaction of Nrf2 and p53 under UV-A–induced oxidative damage remains unclear.22–31
Our previous studies showed the deficiency of DJ-1, Nrf2, and Nrf2-regulated antioxidants, such as HO-1 in FECD.7,32 Moreover, elevated levels of p53 along with heightened oxidative damage and apoptosis were found in FECD.8 Importantly, a downregulation of DJ-1, the main regulator of Nrf2, leads to CEnC susceptibility to UV-A–induced damage.33 Nevertheless, the effect of UV-A, the main source of physiological oxidative stress, has not yet been explored in relation to Nrf2 and p53 pathways in human CEnCs. Because these pathways are affected by FECD, the study herein has specific relevance to FECD pathogenesis. We aimed to determine the responses of Nrf2 and p53 under UV-A–induced oxidative stress in CEnCs. Using the fluences similar to those the CE are exposed to in daily life, we evaluated ROS production, Nrf2-regulated antioxidant defense, p53, and cell apoptosis in human CEnCs exposed to varying UV-A fluences along with cells allowed to recover following UV-A treatment. Our findings provided not only the cellular response of CEnCs to UV-A–induced oxidative stress, but also an in vitro model for studying CEnC pathogenesis under natural environmental conditions.
Materials and Methods
Human Corneal Endothelial Cell Culture
Normal human corneal endothelial cells (HCEnCi), immortalized by infection with an amphotropic recombinant retrovirus containing human papilloma virus type 16 genes E6 and E7, were a generous gift from May Griffith, PhD, (Ottawa Hospital Research Institute, Ottawa, Ontario, Canada).34 Cells were grown in tissue culture flasks or petri dishes in Chen's medium35 at 37°C with 5% CO2.
UV-A Irradiation
A fluorescent UV-A broadband lamp (The Southern New England Ultraviolet Co., Branford, CT, USA) was used for irradiation. Twenty-four hours prior to the UV-A treatment, HCEnCi growth medium was replaced with Opti-MEM I reduced serum medium (Invitrogen, Carlsbad, CA, USA). Ultraviolet A treatment was performed in a biosafety laminar hood with the fluence of 2.5, 5, 10, or 25 J/cm2. Immortalized human CEnCs were washed with 37°C Hank's balanced salt solution (HBSS) and then the cells were placed under UV-A light without a petri dish lid followed by recovery period in Opti-MEM I reduced serum medium at 37°C with 5% CO2 for 0, 3, 6, 18, or 24 hours. Controls were treated identically, except they were not exposed to UV-A.
ROS Production Assay
Immediately post UV-A treatment, the cells were washed with warm HBSS (Invitrogen) and then loaded with 25 μM 5-(and −6)-carboxy-2′,7′-dichlorodihydrofluorescein diacetate (carboxy-H2DCFDA; Invitrogen) for 30 minutes at 37°C. Cells were harvested with a scraper, washed with warm HBSS, and centrifuged at 500g for 5 minutes. Cells were then suspended in 110 μL of warm HBSS; 100 μL of the cell suspension was transferred to a flat-bottom black 96-well plate for the carboxy-H2DCFDA fluorescence assay and 10 μL was used for the cell viability assay. The cell number and viability immediately after UV-A irradiation were measured by Trypan blue staining using an automatic cell counter (Countess; Life Technologies, Carlsbad, CA, USA). The carboxy-H2DCFDA fluorescence (485/520 nm) was analyzed using a fluorescence microplate reader (BioTek-Synergy 2; BioTek Instruments, Inc., Winooski, VT, USA). Relative fluorescence units were normalized to the cell number.
Cytotoxicity Assay
Cytotoxicity was measured by evaluating lactate dehydrogenase (LDH) release according to the manufacturer's instructions (CytoTox 96 Non-radioactive Cytotoxicity Assay; Promega, Madison, WI, USA). Briefly, at 3, 6, and 24 hours post UV-A irradiation, 50 μL of cell culture medium was transferred to a 96-well plate after centrifugation, and 50 μL of substrate was added to each well and mixed. The mixture was incubated for 30 minutes at room temperature in the dark, followed by the addition of 50 μL of stop solution to each well. The plate was read at 490 nm using an absorbance microplate reader (SpectraMax 34; Molecular Devices, Silicon Valley, CA, USA). One hundred percent cell death was determined by rupturing all cells using cell lysis solution provided in the kit. The LDH release levels were normalized to the control of 100% cell death.
Real-Time RT-PCR
Total RNA from HCEnCi was extracted using RNeasy Mini Kit (Qiagen, Valencia, CA, USA). Complementary cDNA was prepared by reverse transcription with a commercial kit (Promega) in a MyCycler Thermal cycler (Bio-Rad, Hercules, CA, USA), according to the manufacturer's protocol. TaqMan primers and probes for Nrf2, HO-1and NQO1 genes, as well as for the endogenous control β2-microglobulin (β2-MG), were obtained from Applied Biosystems (Foster City, CA, USA). Real-time RT PCR reactions were set up with the Probe Fast master mix (Kapa Biosystems, Woburn, MA, USA) and performed in Mastercycler Realplex2 (Eppendorf, Hamburg, Germany). Each gene was detected in duplicate and repeated three times. For data analysis, the comparative threshold cycle (CT) method was performed using β2-MG as the calibrator.
Subcellular Fractionation
Immortalized human CEnCi were pretreated with 5 mM of N-acetyl-L-cysteine (NAC; Sigma-Aldrich Corp., St. Louis, MO, USA) 1 hour prior to 5 J/cm2 UV-A irradiation. At 6 hours post UV-A, cytosolic and nuclear extracts were sequentially isolated using a nuclear/cytosol fractionation kit (BioVision, Milpitas, CA, USA) according to the manufacturer's instructions. Untreated HCEnCi served as controls.
Western Blot Analysis
Whole-cell extracts from HCEnCi were lysed with ×1 RIPA buffer (Cell Signaling Technology, Danvers, MA, USA) supplemented with a protease and phosphatase inhibitor cocktail (Pierce, Waltham, MA, USA), followed by brief sonication (Sonifier 250; Branson, Danbury, CT, USA). Protein concentrations were determined by BCA protein assay (Pierce). Proteins were separated in 10% or 12% Bis-Tris NuPAGE gels (Invitrogen) and electroblotted to polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, MA, USA). Membranes were blocked with 5% dry non-fat milk in TBS for 1 hour and then incubated overnight at 4°C with the following primary antibodies: mouse monoclonal anti-Nrf2 (1:400; R&D Systems); mouse monoclonal anti-phospho-p53 (Ser15; 1:1000; Cell Signaling Technology); rabbit polyclonal anti-p53 (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA, USA) rabbit monoclonal anti-cleaved Caspase 3 (1:1500; Cell Signaling Technology); rabbit polyclonal anti-GAPDH (1:4000; Santa Cruz Biotechnology) and mouse anti-β-actin (1:4000; Sigma-Aldrich Corp.). Blots were washed with TTBS (50 nM Tris, pH 7.5, 0.9% NaCl2 and 0.1% Tween-20) and exposed for 1 hour to horseradish peroxidase (HRP)-conjugated secondary antibody: goat anti-mouse IgG, 1:1000 for Nrf2 (R&D Systems), 1:2000 for phospho-p53 (Ser 15), 1:4000 for cleaved caspase3 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) blots. After washing in TTBS, antibody binding was detected with a chemiluminescent substrate (Thermo Scientific, Waltham, MA, USA). Densitometry was analyzed with ImageJ software (http://imagej.nih.gov/ij/; provided in the public domain by the National Institutes of Health, Bethesda, MD, USA).
Immunocytochemistry
Prior to UV-A irradiation, HCEnCi were pretreated with sulforaphane (1-isothiocyanato-[4R]-[methylsulfinyl]-butane, SFN, 2 μM) over night or with N-acetyl-L-cysteine (NAC, 5 mM) for 1 hour. Immunocytochemistry was performed at 6 hours post 5 J/cm2 UV-A. Nrf2 subcellular localization was detected using anti-Nrf2 antibody (H-300, 1:200; Santa Cruz Biotechnology). Untreated HCEnCi served as controls. Images were viewed by confocal microscopy (Leica DM6000S; Leica Microsystems, Mannheim, Germany).
Apoptosis Assay
Activated caspase3 was detected by Western blot analysis with anti-cleaved caspase3 (Asp175; 5A1) rabbit mAb (Cell Signaling Technology) and by the EnzChek Caspase3 Assay Kit #2 (Invitrogen; Molecular Probes) following the manufacturer's instructions. Briefly, after treatment, the cells were washed in cold PBS and lysed. Caspase 3 activity in the cell lysates was determined by the detection of fluorescence generated by the cleavage of the substrate Z-DEVD-rhodamine 110 using a fluorescence microplate reader (BioTek-Synergy 2) with excitation/emission at 496/520 nm. Procaspase3 was detected by western blot analysis with an anti-casapse 3 (8G10) rabbit mAb (Cell Signaling Technology).
Statistical Analysis
Statistical analysis was carried out using the Bonferroni posttest two-way ANOVA. P less than 0.05 was considered to be statistically different. Data were expressed as the mean ± SEM.
Results
UV-A Irradiation Causes Cytotoxicity
Post UV-A irradiation, we examined the HCEnCi morphology by imaging and evaluated the cell cytotoxicity by measuring the LDH levels. Phase-contrast microscopy showed that the cell confluence and morphology immediately after UV-A exposure were similar to controls. However, a small number of bright and rounded up cells that loosely adhered to the cell layer were observed 6 hours after 25 J/cm2 UV-A treatment, indicating cell death. This change was also observed 24 hours after 5, 10, and 25 J/cm2 UV-A treatments (Fig. 1A). Concurrently, the LDH based cytotoxicity assay showed that UV-A exposure caused elevated levels of LDH release with increased UV-A fluences, indicating cell death from 13% to 42% at 24 hours post 2.5, 5, 10, and 25 J/cm2 UV-A treatments (Fig. 1B). Two-way ANOVA indicated that after 24 hours recovery, 5, 10, and 25 J/cm2 UV-A treatments produced significant changes in cell death compared with no UV-A treatment (Fig. 1B, P < 0.05).
Figure 1.

Cell cytotoxicity and ROS production in UV-A irradiated HCEnCi. (A) Representative phase-contrast microcopy images of HCEnCi at 0, 6, and 24 hours post UV-A irradiation with the fluences of 2.5, 5, 10, and 25 J/cm2. Immortalized human CEnCi not exposed to UV-A irradiation were used as a baseline control. Bar represents 100 μm. (B) Lactate dehydrogenase–based cytotoxicity assay showed elevated LDH release with increased UV-A fluences, indicating cell death increases with higher fluences of UV-A irradiation. (C) DCFDA assay of HCEnCi post various UV-A irradiation. Results showed that ROS levels increased with increasing UV-A fluences. *P < 0.05.
UV-A Irradiation Induces ROS Production
It has been shown that UV-A stimulates the production of ROS in many cell types. Consequently, we determined whether ROS were formed using our UV-A treatment regimen. We measured the ROS production levels and cell viability immediately after UV-A irradiation. As Figure 1C shows, exposure of HCEnCi to 5, 10, and 25 J/cm2 of UV-A radiation generated elevated levels of ROS production compared with those produced in the untreated controls (P < 0.05). The same UV-A treatments did not alter cell viability in those samples (Supplementary Fig. S1, P > 0.05), suggesting that ROS were produced by viable HCEnCi with a fluence of 5, 10, or 25 J/cm2 UV-A.
UV-A Irradiation Activates Nrf2-Regulated Antioxidant Defense
To investigate whether Nrf2-regulated antioxidant defense plays a critical role in protection against UV-A irradiation in HCEnCi, we analyzed the transcriptional and translational levels of Nrf2 and its regulated antioxidants post UV-A exposure. Real-time RT-PCR showed the mRNA levels of Nrf2 increased greater than 2.0-fold at 3 hours after 2.5 and 5 J/cm2 UV-A and at 6 hours after 5 J/cm2 UV-A irradiation compared with controls (Fig. 2A, P < 0.05). The mRNA levels of the Nrf2-regulated gene HO-1 showed a significant 2.3-fold increase with the 2.5 J/cm2 UV-A and peaked with a 3.2-fold increase with 5 J/cm2 UV-A irradiated cells at 3-hours recovery (Fig. 2B, P < 0.05), which correlated with the Nrf2 mRNA increases. In addition, NQO-1, another Nrf2-regulated gene showed a 1.7-fold increase in mRNA levels 3 hours after 2.5 J/cm2 UV-A exposure (Fig. 2C, P < 0.05). Interestingly, the fluence and recovery time-dependent increases of Nrf2 and HO-1were not observed after the higher UV-A fluence exposures (Figs. 2A, 2B). Western blot analysis demonstrated that elevated protein levels of Nrf2 and HO-1occured in 2.5 and 5 J/cm2 UV-A–treated HCEnCi in a fluence and recovery time-dependent manner (Fig. 2D).
Figure 2.

The effect of UV-A irradiation on Nrf2 and its target genes. The mRNA levels of Nrf2 (A), HO-1 (B), and NQO 1 (C) were analyzed with real time RT-PCR post UV-A irradiation at different recovery time points. Results were expressed as fold changes and normalized to β2-microglobulin. The protein levels of Nrf2, HO-1 and NQO-1 were analyzed by Western blotting (D). Ultraviolet A upregulates Nrf2, HO-1, and NQO1 with 2.5 and 5 J/cm2 UV-A treatment. *P < 0.05.
UV-A Irradiation Promotes Nrf2 Nuclear Translocation
Knowing that UV-A treatments activate Nrf2 and its target genes, we postulated that UV-A enhances Nrf2 nuclear translocation, which is essential for the activation of Nrf2-regulated antioxidant defense. To test this hypothesis, we examined nuclear Nrf2 levels after 5 J/cm2 UV-A treatment combined with and without NAC or SFN pretreatments, respectively, at 6 hours post irradiation. NAC is an aminothiol and considered an important antioxidant. SFN is a well-studied chemoprotective compound inducing Nrf2-regulated antioxidant responses. The confocal images (Fig. 3A) showed diffuse cytosolic and nuclear staining of Nrf2 in cells without UV-A treatment (Fig. 3A, columns 1–3). However, increased Nrf2 staining was observed in nuclei 6 hours post UV-A treatment (Fig. 3A, column 4). Similarly, intense Nrf2 nuclear staining was seen in NAC pretreatment at 6 hours post UV-A (Fig. 3A, column 5). With SFN pretreatment, increased Nrf2 nuclear staining was observed in UV-A irradiated cells (Fig. 3A, column 6) as compared with that of UV-A treatment alone, suggesting that SFN enhances Nrf2 movement from the cytoplasm to the nucleus under UV-A oxidative stress. Meanwhile, Western blot analysis of subcellular Nrf2 showed that there was an increase in nuclear Nrf2 levels in UV-A–treated cells as compared with no UV-A treatment (Fig. 3B), suggesting the translocation of Nrf2 from the cytoplasm to nuclei in response to UV-A irradiation. With NAC pretreatments, both immunostaining and Western blotting showed similar nuclear Nrf2 levels as seen in UV-A irradiated cells (Fig. 3A, columns 4, 5; Fig. 3B), indicating that NAC along with UV-A irradiation did not significantly change Nrf2 nuclear translocation compared with UV-A treatment alone.
Figure 3.

Nuclear localization of Nrf2 in UV-A irradiated cells. (A) Representative confocal images of Nrf2 localization in cells with no treatment (column 1), NAC-treated (column 2), SFN-treated (column 3), UV-A irradiated (column 4), UV-A irradiated with NAC-pretreatment (column 5), and UV-A irradiated with SFN-pretreatment (column 6). Nrf2 (green) was initially diffused in cytoplasm and nuclei. After treatment with UV-A and SFN, Nrf2 accumulated primarily in the nuclei. DAPI was used for nuclei staining (blue). Bar represents 25 μm. (B) Western blot analysis of cytosolic and nuclear Nrf2 in UV-A or UV-A– and NAC-treated cells. GAPDH and histone H3 were used for normalization of cytosolic and nuclear protein loading, respectively. Cells were irradiated with 5 J/cm2 UV-A and collected at 6 hours post UV-A.
UV-A Irradiation Activates p53 and Induces Accumulation of Nuclear Phospho-p53
To determine the role of p53 in HCEnCi under UV-A–induced oxidative stress, the protein levels of p53 and phosphorylated p53 (phospho-p53) at serine 15 were analyzed. Figures 4A and 4B indicate that phospho-p53 showed a recovery time-dependent accumulation at all of the UV-A fluences. Fluences of 5, 10, and 25 J/cm2 UV-A yielded a significant 1.8- to 2.8-fold increase in phospho-p53 (P < 0.05, Fig. 4A) compared with no UV-A treatment at 24 hours post exposure, indicating that these fluences functionally activate p53 by a phosphorylation modification at serine 15 (Fig. 4B). It is worthy to point out that 5 J/cm2 UV-A showed the most robust accumulation of phospho-p53 as compared with the other fluences at 24 hours post UV-A treatment. Fractionation of cytosolic and nuclear protein showed that phospho-p53 is exclusively expressed in nuclei after 5 J/cm2 UV-A treatment at 6-hours recovery. Moreover, UV-A irradiation enhanced phospho-p53 accumulation in the nuclei. Meanwhile, total p53 levels showed a decrease in the cytosol of UV-A irradiated cells, suggesting UV-A treatment may facilitate the movement of total p53 from the cytosol to the nuclei (Fig. 4C). In addition, elevated phospho-p53 levels were detected in UV-A irradiated cells with NAC pretreatment (Fig. 4C).
Figure 4.
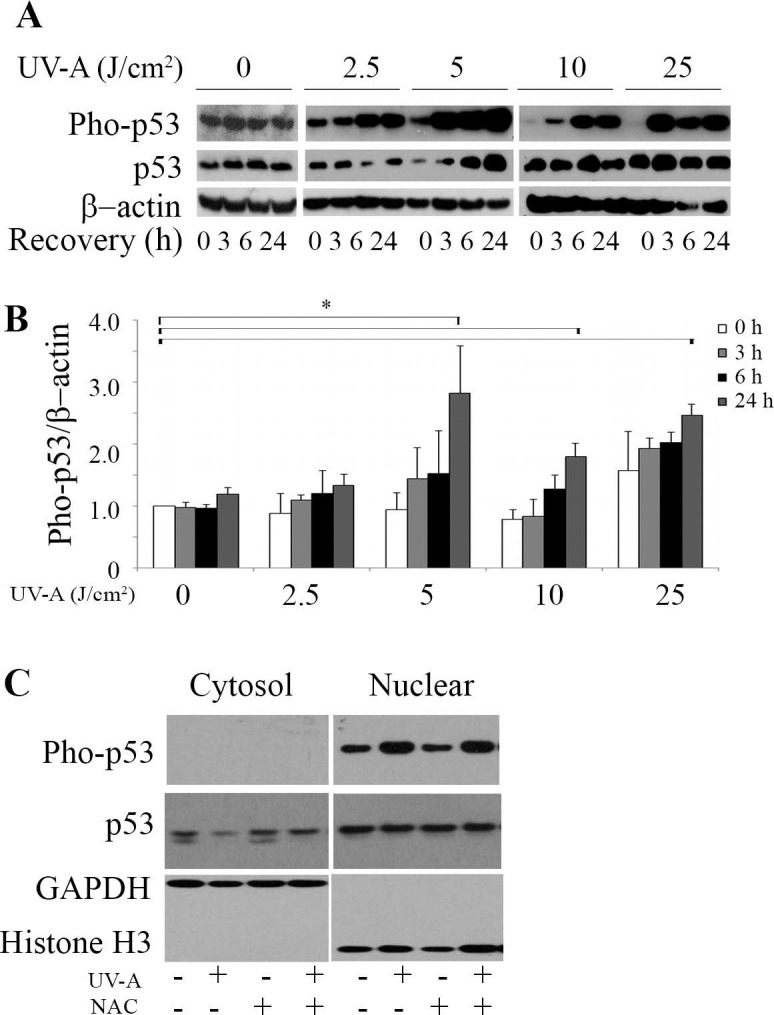
Ultraviolet-A irradiation activates p53 in HCEnCi. (A) Representative Western blot of phospho-p53 and total p53 protein levels and (B) densitometric analysis of phospho-p53 with normalization to β-actin. (C) Western blot analysis of cytosolic and nuclear phospho-p53 and total p53 in 5 J/cm2 UV-A– or NAC-pretreated cells followed by 5 J/cm2 UV-A at 6 hours recovery. Results showed that phospho-p53 increases at 24 hours post 5, 10, and 25 J/cm2 as compared with no UV-A treatment, and UV-A causes accumulation of nuclear phospho-p53.
UV-A Irradiation Activates Cell Apoptosis
Caspase3 plays a central role in the execution-phase of cell apoptosis. To assess the effect of UV-A on HCEnCi apoptosis, we examined the protein levels of activated caspase3 in UV-A irradiated cells. Western blot analysis (Fig. 5A) showed that cleaved caspase3 levels were elevated by the increased UV-A fluences and the recovery times that followed despite similar levels of pro-caspase3. Figure 5B shows the results from a rhodamine 110 fluorescence-based caspase3 activation assay. At 24 hours post treatment with 5, 10, and 25 J/cm2 UV-A, a 2.6- to 6.0-fold increase was produced in activated caspase3 as compared with no UV-A treatment (P < 0.05, Fig. 5B).
Figure 5.

UV-A irradiation activates caspase3 in HCEnCi. (A) Western blot analysis of cleaved caspase3 and pro-caspase3 in HCEnCi cells. (B) Z-DEVD-rhodamine 110 fluorescence-based casapase3 activity assay. Results showed that activated caspase3 increases at 24 hours post 5, 10, and 25 J/cm2 as compared with no UV-A treatment.
Discussion
There is growing evidence indicating that CE degeneration is associated with oxidative stress in aged and diseased corneal endothelium.7–9,32,33,36,37 Our previous studies on FECD specimens and FECD CEnCs showed that Nrf2 and p53 play critical roles in complex mechanisms regulating oxidative-stress–induced apoptosis in FECD.7,8,32 Ultraviolet A is the most abundant source of solar radiation and it induces ROS production. To investigate the cellular response of CEnC to UV-A, we examined the effect of UV-A on Nrf2-regulated antioxidant defense, p53 activation, and cell apoptosis. Using UV-A fluences similar to those the CE are exposed to in daily life, we found that UV-A irradiation causes fluence-dependent ROS production and cytotoxicity. Ultraviolet A facilitates Nrf2 nuclear translocation and induces Nrf2-regulated antioxidant defense. Meanwhile, UV-A activates p53 and caspase3. Collectively, these findings suggested that UV-A activates Nrf2-regulated antioxidant defense and induces apoptosis.
Nrf2 plays a critical role in antioxidant defense under oxidative stress conditions.12–15 It has been demonstrated that, in normal physiological condition, Nrf2 localizes in the cytoplasm before being ubiquitinated and degraded by Kelch-like ECH-associated protein (Keap1) and Cullin3. However, under oxidative stress conditions, the ROS and electrophiles bind to sulfhydryl residues of Keap1. As a result, the released Nrf2 translocates to the nucleus, where it binds to ARE in the promoter region of some anti-oxidative genes and transcriptionally facilitates expression of antioxidants, including HO-1, NAD(P)H: quinone oxidoreductase 1 (NQO1).12–15 In this study, we detected increased levels of Nrf2 and its target genes HO-1 and NQO-1 with the UV-A fluences of 2.5 and 5 J/cm2. Moreover, we observed increased levels of nuclear Nrf2 in UV-A treated HCEnCi, indicating that UV-A facilitate Nrf2 nuclear translocation. These results suggest that UV-A activates Nrf2 and its regulated antioxidant defense. This finding is supported by previous reports that UV-A irradiation with a fluence of 5 to 37.2 J/cm2 leads to the induction of Nrf2-regulated antioxidant HO-1 in dermal fibroblasts22–24 and keratinocytes,25,38 retinal pigment epithelial cells39 as well as mouse embryonic fibroblasts.40 Studies on skin fibroblasts demonstrated that UV-A irradiation resulted in an increase in Nrf2 and its regulated gene HO-1at both the mRNA22 and protein24 level. Downregulation of Nrf2 inhibits mRNA expression of ARE dependent genes,22,24 such as HO-1, but not other UV-A regulated genes, such as Cox2 and IL-6,22 indicating that UV-A selectively induces Nrf2-dependent antioxidant defense. However, analysis of keratinocytes from mice transgenic for ARE activation showed that 5 to 20 J/cm2 UV-A did not activate Nrf2 regulated antioxidants in vivo and in vitro although UV-A irradiation strongly increased intracellular ROS levels.26 These findings suggested that the induction of Nrf2-regulated antioxidant defense may be cell-type dependent.
P53 plays dual roles under oxidative stress, either by repairing DNA damage or by inducing cell apoptosis.17–19 In the present study, we found that UV-A irradiation functionally activates p53 by phosphorylation modification at serine 15 at the amino terminus in HCEnCi with UV-A fluences of 5, 10, and 25 J/cm2 at 24 hours post exposure. Additionally, Western blot analysis showed that 5 J/cm2 UV-A promotes phospho-p53 nuclear accumulation. Concurrently, increased caspase3 activation and cytotoxicity were detected following treatment with 5, 10, and 25 J/cm2 UV-A. These results provided the evidence that UV-A activates p53 and apoptosis. Similarly, it has been reported that UV-A irradiation activates p53 in skin fibroblasts27,28 and that p53 is essential for UV-A–induced apoptosis in mouse epidermal cells.29,41 In contrast, a recent study showed that p53 is not active in response to UV-A and that UV-A facilitates apoptosis via a p53-independent pathway.30 This study reported that there was no detectable increase of sequence-specific DNA-binding activity of p53 and no induction of Ser 15 phosphorylation of p53 in 10 to 30 J/cm2 UV-A–treated mouse embryonic fibroblasts at 6 to 24 hours post irradiation. Another time course study on human neonatal fibroblasts displayed that phospho-53 was detected at 2 hours but not 6 to 24 hours after 10 to 30 J/cm2 UV-A exposure.31 Our result showing that UV-A activates p53 fits well within the range of activation profiles previously reported.27,28 Performing p53 knockdown studies in the future will investigate whether blocking p53 action leads to decreased apoptosis indicating that UV-A induces p53-mediated apoptosis.
As transcription factors involved in oxidative stress, Nrf2 and p53 respond to ROS via different mechanisms. Nrf2-regulated antioxidant defense scavenges ROS, however, p53-induced apoptosis requires the accumulation ROS for its activation. It remains unclear whether the crosstalk between Nrf2 and p53 is synergistic, additive or suppressive. A carcinogenesis study on Nrf2 knockout and p53 heterozygotic mice showed that a Nrf2−/−::p53± mutation in mice increased the incidence of a chemical induced bladder carcinogenesis as compared with those carrying the Nrf2−/− or p53± mutation alone, indicating the cooperative and compensatory interaction between Nrf2 and p53.42 This observation, however, is challenged by more direct examination between Nrf2 and p53, which showed that overexpressed or endogenous p53 suppresses the Nrf2-dependent transactivation by interacting with and inhibiting the ARE- containing promoter of antioxidant genes, such as NQO-1, GST-α1.19 A recent study demonstrated that Nrf2-medidated antioxidant defense is regulated by p53 in two phases in a dose-dependent manner.43 Under low levels of ROS, p53 upregulates the expression of antioxidant defense genes, protecting cells from ROS-induced oxidative damage. However, at high levels of ROS, p53 expression increases and downregulates Nrf2 and its target genes.43 In our study, we found that UV-A irradiation with 5, 10, or 25 J/cm2 generates ROS product in a fluence-dependent manner. Nrf2-regulated antioxidants occur only with lower UV-A fluences of 2.5 and 5 J/cm2 at 3 to 6 hours post UV-A exposure, while increased phospho-p53 was observed at higher UV-A fluence of 5, 10, and 25 J/cm2 at 24 hour post treatment. It is reasonable to speculate that Nrf2-regulated antioxidants protect CEnCs from oxidative stress caused by UV-A at low levels of damage, however, high levels of oxidative damage activate p53. Additionally, the phenomenon we observed when only the lower UV-A fluences enhanced the expression of Nrf2 and its target genes is consistent with the concept of hormesis in which lower levels of a stimulator are more effective than higher ones.44
Interestingly, we observed that pretreatment of NAC prior to UV-A irradiation did not decrease Nrf2 nuclear translocation, as would be expected due to the ROS-scavenging effect of NAC in HCEnCi.45 By convention, NAC is known to scavenge ROS directly by acting as a cysteine donor and upregulating glutathione levels.46,47 However, recent studies have shown that Nrf2 expression was directly elevated after NAC administration in phosgene-stressed rat lung tissue48 and carbon tetrachloride (CCl4)-stressed rat liver tissue.49 We speculate that NAC directly causes Nrf2 nuclear translocation in addition to scavenging ROS during UV-A irradiation of corneal endothelium.
In summary, our findings demonstrate that UV-A causes ROS-induced cytotoxicity in CEnCs. Low fluences of UV-A activate Nrf2-regulated antioxidant defense by promoting Nrf2 nuclear translocation and upregulating its target genes HO-1 and NQO-1. High fluences of UV-A irradiation lead to the activation of p53 and apoptosis. Given that previous studies have shown the critical roles of Nrf2-mediated antioxidant defense and p53 in regulating oxidative stress induced apoptosis in FECD,7,8,32 this study yielded not only knowledge about the cellular responses of CEnC to oxidative stress, but it may also provide an in vitro oxidative stress model for investigating CEnC degeneration, especially, in FECD pathogenesis.
Supplementary Material
Acknowledgments
Supported by grants from National Institutes Health/National Eye Institutes R01 EY20581 (Bethesda, MD, USA) and Research to Prevent Blindness Award (UVJ; New York, NY, USA).
Disclosure: C. Liu, None; D. Vojnovic, None; I.E. Kochevar, None; U.V. Jurkunas, None
References
- 1. Cadet J,, Gentner NE,, Rozga B,, Paterson MC. Rapid quantitation of ultraviolet-induced thymine-containing dimers in human cell DNA by reversed-phase high-performance liquid chromatography. J Chromatogr. 1983; 280: 99–108. [DOI] [PubMed] [Google Scholar]
- 2. Xia Q,, Yin JJ,, Zhao Y,, et al. UVA photoirradiation of nitro-polycyclic aromatic hydrocarbons-induction of reactive oxygen species and formation of lipid peroxides. Int J Environ Res Public Health. 2013; 10: 1062–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Parrish J,, Anderson R,, Urbach F,, Pitts D. UV-A: Biological Effects of Ultraviolet Radiation with Emphasis on Human Responses to Longwave Ultraviolet. 1st ed. New York: Plenum Press; 1978. [Google Scholar]
- 4. Spoerl E,, Hoyer A,, Pillunat LE,, Raiskup F. Corneal cross-linking and safety issues. Open Ophthalmol J. 2011; 5: 14–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wollensak G,, Spoerl E,, Wilsch M,, Seiler T. Endothelial cell damage after riboflavin-ultraviolet-A treatment in the rabbit. J Cataract Refract Surg. 2003; 29: 1786–1790. [DOI] [PubMed] [Google Scholar]
- 6. Brightbill F,, McDonnell PJ,, McGhee CNJ,, Farjo AA,, Serdarevic O. Corneal Surgery: Theory Technique and Tissue. Maryland Heights: Mosby Elsevier; 2009. [Google Scholar]
- 7. Jurkunas UV,, Bitar MS,, Funaki T,, Azizi B. Evidence of oxidative stress in the pathogenesis of Fuchs endothelial corneal dystrophy. Am J Pathol. 2010; 177: 2278–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Azizi B,, Ziaei A,, Fuchsluger T,, Schmedt T,, Chen Y,, Jurkunas UV. p53-regulated increase in oxidative-stress–induced apoptosis in Fuchs endothelial corneal dystrophy: a native tissue model. Invest Ophthalmol Vis Sci. 2011; 52: 9291–9297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Buddi R,, Lin B,, Atilano SR,, Zorapapel NC,, Kenney MC,, Brown DJ. Evidence of oxidative stress in human corneal diseases. J Histochem Cytochem. 2002; 50: 341–351. [DOI] [PubMed] [Google Scholar]
- 10. Gottsch JD,, Bowers AL,, Margulies EH,, et al. Serial analysis of gene expression in the corneal endothelium of Fuchs' dystrophy. Invest Ophthalmol Vis Sci. 2003; 44: 594–599. [DOI] [PubMed] [Google Scholar]
- 11. Jurkunas UV,, Rawe I,, Bitar MS,, et al. Decreased expression of peroxiredoxins in Fuchs' endothelial dystrophy. Invest Ophthalmol Vis Sci. 2008; 49: 2956–2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xu Z,, Wei Y,, Gong J,, et al. NRF2 plays a protective role in diabetic retinopathy in mice. Diabetologia. 2014; 57: 204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Osburn WO,, Wakabayashi N,, Misra V,, et al. Nrf2 regulates an adaptive response protecting against oxidative damage following diquat-mediated formation of superoxide anion. Arch Biochem Biophys. 2006; 454: 7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kensler TW,, Wakabayashi N,, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007; 47: 89–116. [DOI] [PubMed] [Google Scholar]
- 15. Espinosa-Diez C,, Miguel V,, Mennerich D,, et al. Antioxidant responses and cellular adjustments to oxidative stress. Redox Biol. 2015; 6: 183–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Campbell C,, Quinn AG,, Angus B,, Farr PM,, Rees JL. Wavelength specific patterns of p53 induction in human skin following exposure to UV radiation. Cancer Res. 1993; 53: 2697–2699. [PubMed] [Google Scholar]
- 17. Vousden KH,, Prives C. Blinded by the light: the growing complexity of p53. Cell. 2009; 137: 413–431. [DOI] [PubMed] [Google Scholar]
- 18. Vousden KH,, Ryan KM. p53 and metabolism. Nat Rev Cancer. 2009; 9: 691–700. [DOI] [PubMed] [Google Scholar]
- 19. Faraonio R,, Vergara P,, Di Marzo D,, et al. p53 suppresses the Nrf2-dependent transcription of antioxidant response genes. J Biol Chem. 2006; 281: 39776–39784. [DOI] [PubMed] [Google Scholar]
- 20. Hall PA,, McKee PH,, Menage HD,, Dover R,, Lane DP. High levels of p53 protein in UV-irradiated normal human skin. Oncogene. 1993; 8: 203–207. [PubMed] [Google Scholar]
- 21. Vogelstein B,, Lane D,, Levine AJ. Surfing the p53 network. Nature. 2000; 408: 307–310. [DOI] [PubMed] [Google Scholar]
- 22. Gruber F,, Mayer H,, Lengauer B,, et al. NF-E2-related factor 2 regulates the stress response to UVA-1-oxidized phospholipids in skin cells. FASEB J. 2010; 24: 39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gruber F,, Ornelas CM,, Karner S,, et al. Nrf2 deficiency causes lipid oxidation, inflammation, and matrix-protease expression in DHA-supplemented and UVA-irradiated skin fibroblasts. Free Radic Biol Med. 2015; 88 (pt B): 439–451. [DOI] [PubMed] [Google Scholar]
- 24. Zhong JL,, Edwards GP,, Raval C,, Li H,, Tyrrell RM. The role of Nrf2 in ultraviolet A mediated heme oxygenase 1 induction in human skin fibroblasts. Photochem Photobiol Sci. 2010; 9: 18–24. [DOI] [PubMed] [Google Scholar]
- 25. Zhong JL,, Raval C,, Edwards GP,, Tyrrell RM. A role for Bach1 and HO-2 in suppression of basal and UVA-induced HO-1 expression in human keratinocytes. Free Radic Biol Med. 2010; 48: 196–206. [DOI] [PubMed] [Google Scholar]
- 26. Durchdewald M,, Beyer TA,, Johnson DA,, Johnson JA,, Werner S, auf dem Keller U. Electrophilic chemicals but not UV irradiation or reactive oxygen species activate Nrf2 in keratinocytes in vitro and in vivo. J Invest Dermatol. 2007; 127: 646–653. [DOI] [PubMed] [Google Scholar]
- 27. Zhan Q,, Carrier F,, Fornace AJ,, Jr. Induction of cellular p53 activity by DNA-damaging agents and growth arrest. Mol Cell Biol. 1993; 13: 4242–4250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Seite S,, Moyal D,, Verdier MP,, Hourseau C,, Fourtanier A. Accumulated p53 protein and UVA protection level of sunscreens. Photodermatol Photoimmunol Photomed. 2000; 16: 3–9. [DOI] [PubMed] [Google Scholar]
- 29. Santamaria AB,, Davis DW,, Nghiem DX,, et al. p53 and Fas ligand are required for psoralen and UVA-induced apoptosis in mouse epidermal cells. Cell Death Differ. 2002; 9: 549–560. [DOI] [PubMed] [Google Scholar]
- 30. McFeat GD,, Allinson SL,, McMillan TJ. Characterisation of the p53-mediated cellular responses evoked in primary mouse cells following exposure to ultraviolet radiation. PLoS One. 2013; 8: e75800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kappes UP,, Luo D,, Potter M,, Schulmeister K,, Runger TM. Short- and long-wave UV light (UVB and UVA) induce similar mutations in human skin cells. J Invest Dermatol. 2006; 126: 667–675. [DOI] [PubMed] [Google Scholar]
- 32. Bitar MS,, Liu C,, Ziaei A,, Chen Y,, Schmedt T,, Jurkunas UV. Decline in DJ-1 and decreased nuclear translocation of Nrf2 in Fuchs endothelial corneal dystrophy. Invest Ophthalmol Vis Sci. 2012; 53: 5806–5813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liu C,, Chen Y,, Kochevar IE,, Jurkunas UV. Decreased DJ-1 leads to impaired Nrf2-regulated antioxidant defense and increased UV-A-induced apoptosis in corneal endothelial cells. Invest Ophthalmol Vis Sci. 2014; 55: 5551–5560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Griffith M,, Osborne R,, Munger R,, et al. Functional human corneal equivalents constructed from cell lines. Science. 1999; 286: 2169–2172. [DOI] [PubMed] [Google Scholar]
- 35. Chen CH,, Rama P,, Chen AH,, et al. Efficacy of media enriched with nonlactate-generating substrate for organ preservation: in vitro and clinical studies using the cornea model. Transplantation. 1999; 67: 800–808. [DOI] [PubMed] [Google Scholar]
- 36. Wang Z,, Handa JT,, Green WR,, Stark WJ,, Weinberg RS,, Jun AS. Advanced glycation end products and receptors in Fuchs' dystrophy corneas undergoing Descemet's stripping with endothelial keratoplasty. Ophthalmology. 2007; 114: 1453–1460. [DOI] [PubMed] [Google Scholar]
- 37. Ziaei A,, Schmedt T,, Chen Y,, Jurkunas UV. Sulforaphane decreases endothelial cell apoptosis in fuchs endothelial corneal dystrophy: a novel treatment. Invest Ophthalmol Vis Sci. 2013; 54: 6724–6734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hseu YC,, Lo HW,, Korivi M,, Tsai YC,, Tang MJ,, Yang HL. Dermato-protective properties of ergothioneine through induction of Nrf2/ARE-mediated antioxidant genes in UVA-irradiated Human keratinocytes. Free Radic Biol Med. 2015; 86: 102–117. [DOI] [PubMed] [Google Scholar]
- 39. Gao X,, Talalay P. Induction of phase 2 genes by sulforaphane protects retinal pigment epithelial cells against photooxidative damage. Proc Natl Acad Sci U S A. 2004; 101: 10446–10451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ito T,, Kimura S,, Seto K,, et al. Peroxiredoxin I plays a protective role against UVA irradiation through reduction of oxidative stress. J Dermatol Sci. 2014; 74: 9–17. [DOI] [PubMed] [Google Scholar]
- 41. Waster PK,, Ollinger KM. Redox-dependent translocation of p53 to mitochondria or nucleus in human melanocytes after UVA- and UVB-induced apoptosis. J Invest Dermatol. 2009; 129: 1769–1781. [DOI] [PubMed] [Google Scholar]
- 42. Iida K,, Itoh K,, Maher JM,, et al. Nrf2 and p53 cooperatively protect against BBN-induced urinary bladder carcinogenesis. Carcinogenesis. 2007; 28: 2398–2403. [DOI] [PubMed] [Google Scholar]
- 43. Chen W,, Jiang T,, Wang H,, et al. Does Nrf2 contribute to p53-mediated control of cell survival and death? Antioxid Redox Signal. 2012; 17: 1670–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Prekeges JL. Radiation hormesis or, could all that radiation be good for us? J Nucl Med Technol. 2003; 31: 11–17. [PubMed] [Google Scholar]
- 45. Halilovic A,, Schmedt T,, Benischke A,, et al. Menadione-induced DNA damage leads to mitochondrial dysfunction and fragmentation during rosette formation in Fuchs Endothelial Corneal Dystrophy [published online ahead of print March 2, 2016]. Antioxid Redox Signal . doi:http://dx.doi.org/10.1089/ars.2015.6532. [DOI] [PMC free article] [PubMed]
- 46. Sun SY. N-acetylcysteine reactive oxygen species and beyond. Cancer Biol Ther. 2010; 9: 109–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zafarullah M,, Li WQ,, Sylvester J,, Ahmad M. Molecular mechanisms of N-acetylcysteine actions. Cell Mol Life Sci. 2003; 60: 6–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ji L,, Liu R,, Zhang XD,, et al. N-acetylcysteine attenuates phosgene-induced acute lung injury via up-regulation of Nrf2 expression. Inhal Toxicol. 2010; 22: 535–542. [DOI] [PubMed] [Google Scholar]
- 49. Cai Z,, Lou Q,, Wang F,, et al. N-acetylcysteine protects against liver injure induced by carbon tetrachloride via activation of the Nrf2/HO-1 pathway. Int J Clin Exp Pathol. 2015; 8: 8655–8662. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.