

Abstract
We previously determined that augmented EGFR tyrosine kinase (EGFRtk) impairs vascular function in type 2 diabetic mouse (TD2). Here we determined that EGFRtk causes vascular dysfunction through NADPH oxidase activity in TD2.
Mesenteric resistance arteries (MRA) from C57/BL6 and db−/db− mice were mounted in a wired myograph and pre-incubated for one hour with either EGFRtk inhibitor (AG1478) or exogenous EGF. The inhibition of EGFRtk did not affect the contractile response to phenylephrine-(PE) and thromboxane-(U46619) or endothelium-dependent relaxation (EDR) to acetylcholine in MRA from control group. However, in TD2 mice, AG1478 reduced the contractile response to U46619, improved vasodilatation and reduced p22phox-NADPH expression, but had no effect on the contractile response to PE. The incubation of MRA with exogenous EGF potentiated the contractile response to PE in MRA from control and diabetic mice. However, EGF impaired the EDR and potentiated the vasoconstriction to U46619 only in the control group. Interestingly, NADPH oxidase inhibition in the presence of EGF restored the normal contraction to PE and improved the EDR but had no effect on the potentiated contraction to U46619. Vascular function improvement was associated with the rescue of eNOS and Akt and reduction in phosphorylated-RhoKinase, NOX4 mRNA levels and NADPH oxidase activity. MRA from p47phox−/− mice incubated with EGF potentiated the contraction to U46619 but no effect to PE or ACh responses.
The present study provides evidence that augmented EGFRtk impairs vascular function by NADPH oxidase-dependent mechanism. Therefore, EGFRtk and oxidative stress should be potential targets to treat vascular dysfunction in TD2.
INTRODUCTION
Several clinical studies showed that endothelial and SMC dysfunction plays an important role in the development of cardiovascular diseases. (8,29) Additionally, a growing body of evidence reveals the presence of an impaired vascular function in both animal models of type 2 diabetes (T2D) and in T2D patients. (10,11) Interestingly, upregulation of NADPH oxidase activity and EGFRtk has been frequently observed in diabetic patients, and the combination between these two features (NADPH oxidase and EGF) potentiates the severity of vascular dysfunction. (25,24)
Different studies stated the beneficial effect of EGFRtk inhibition in mature adult diabetic animals. (2) Others and we have demonstrated that increased EGFRtk phosphorylation contributes to resistance artery dysfunction in type 1 and type 2 diabetes mellitus. (2,13,7,21) Additionally it is well known that oxidative stress is increased in diabetes and it also plays an important role in damaging the vascular function. (12,22) Several studies provide evidence for a relationship between diabetes, EGFR, and oxidative stress. (1,14,20) The significance and role of exacerbated EGFRtk and oxidative stress in microvascular dysfunction in diabetes mellitus remains unclear. Thus, the aim of this study was to determine the link between EGFRtk and NADPH oxidase activity in microvascular dysfunction in type 2 diabetic mouse.
MATERIALS AND METHODS
General protocol in mice
All experiments were performed according to the American Guidelines for the Ethical Care of Animals and were approved by EVMS Animal Care and Use Committee. Type 2 diabetic, male mice (db−/db−, 8 to 10 week-old) and their homologous controls, as well as knockout mice of the p47phox expression (p47phox−/−, 8 to 10 weeks-old males) were purchased from Jackson Laboratories (Bar Harbor, ME). All mice were housed in groups of five mice, maintained at a temperature of 23 °C with 12 h light/dark cycles and fed a solid standard diet (Na+ content 0.4%) and water. Blood glucose, body weight and SBP were measured and then mice were sacrificed by isoflurane and were not flushed before harvesting the arteries, and then mesenteric resistance arteries (MRA) were harvested immediately and placed in PSS solution (composition in mM: NaCl 118; KCl 4.7; CaCl2 2.5; KH2PO4 1.2; MgSO4×7H2O 1.2; NaHCO3 25 and glucose 11, pH=7.4) and processed appropriately for further studies.
Vascular Reactivity
Mesenteric resistance arteries (MRAs) from control, type 2 diabetic and p47phox−/− mice were carefully cleaned of fat and connective tissue and then cut into rings (2 mm in length). MRAs were mounted in a small vessel dual chamber myograph for measurement of isometric tension. After a 30 minute equilibration period in PSS solution bubbled with carbogen, at 37°C and pH=7.4, arteries were stretched to their optimal lumen diameter for active tension development.
EGFR kinase inhibition in MRA from control and type 2 diabetic mice
MRAs rings from control and type 2 diabetic mice were incubated with and without AG1478, a specific inhibitor of EGFRtk. After one-hour incubation, cumulative concentration responses to phenylephrine (PE, 10−8–10−4 M), thromboxane (TXA) analogue (U46619, 10−8–10−4 M) and acetylcholine (ACh, 10−8–3.10−5 M) were performed. To further assess the role of EGFRtk, MRAs from control and type 2 diabetic mice were incubated or not with exogenous epidermal growth factor (EGF, Sigma Aldrich). After one hour of incubation, cumulative concentration responses to PE (10−8–10−4 M), U46619 (10−8–10−4 M) and ACh (10−8–3.10−5 M) were obtained. Additionally, vascular constriction to KCl in diabetic mice and control mice was performed.
Another set of experiments was performed to evaluate whether NADPH oxidase could be involved in the contractile response to PE and U46619 and the relaxant response to ACh. MRAs rings from control and type 2 diabetic mice were incubated or not with exogenous EGF in the presence and absence of gp91 ds-tat (a specific inhibitor of NADPH oxidase). After 30 minutes of incubation, cumulative concentration responses to PE (10−8–10−4 M), U46619 (10−8–10−4 M) and ACh (10−8–3.10−5 M) were performed.
To strengthen our results, MRAs rings from p47phox−/− were incubated with exogenous EGF. After one hour of incubation, cumulative concentration responses to PE (10−8–10−4 M), U46619 (10−8–10−4 M) and ACh (10−8–3.10−5 M) were obtained.
Western Blot Analysis
Western blot analysis for endothelial nitric oxide synthase (eNOS), Akt, P-ERK1/2, phospho-RhoKinase, p22phox and p47phox (1:1000 dilution; Cell Signaling Technology, Inc) was performed in lysates of MRA as previously described. (16,17)
NADPH oxidase activity
NADPH oxidase activity in MRA was measured as previously described. (16,17,13)
Reverse Transcription Polymerase Chain Reaction Real-Time Assay
Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 2 (Nox2) and Nox4 mRNA levels were determined in MRA from as previously described. (16,17,13)
In vitro studies with primary cultured cells and smooth muscle cells
Primary cultured vascular endothelial cells and SMC were used. After confluency, cells were stimulated with exogenous EGF for 1 hour with and without endoplasmic reticulum stress and NADPH oxidase activity inhibitors (Tudca and gp91ds-tat). Then cells were harvested and Western blot analysis and NADPH oxidase activity were performed.
Drugs
Phenylephrine hydrochloride and acetylcholine was obtained from Sigma-Aldrich (St. Louis, MO). The gp91 ds-tat was obtained from Eurogentec (USA). Thromboxane analogue (U46619) was purchased from Calbiochem (USA).
Statistical analysis
Results are expressed as mean ± SEM. Concentration-response curves were analyzed using the GraphPad Prism 4.0 software (GraphPad, USA). One-way or 2-way ANOVA was used to compare parameters, when appropriate. Comparisons between groups were performed with t- tests when the ANOVA test was statistically significant. Values of P < 0.05 were considered significant. Differences between specified groups were analyzed using the Student’s t test (two-tailed) for comparing two groups with P < 0.05 considered statistically significant.
RESULTS
EGFR kinase inhibition in MRA from control and type 2 diabetic mice
Body weight and glucose level were highly increase in diabetic mice compared to control mice (23.4 ± 0.51 vs 42.6 ± 0.92; 129.1 ± 3.27 vs 481.4 ± 24.6) however SBP was similar between both groups (103.8 ± 1.1 vs 100.2 ± 2.33) (Fig Supp 1A, B, C). The incubation of MRAs from control mice with AG1478 did not affect the contraction response to PE and U46619 neither the endothelium-dependent relaxation (EDR) response to ACh (Figure 1A, B, C; Fig Supp 2A). However, the contractile response of MRAs rings from type 2 diabetic mice in response to U46619 was significantly reduced after incubation with AG1478 (Figure 1D; Fig Supp 2A) but the contractile response to PE was not affected (Figure 1E; Fig Supp 2A). The impaired EDR in MRA from diabetic mice was significantly improved after inhibition of EGFRtk (Figure 1F, Fig Supp 2A).
Figure 1. Effect of AG1478 on Mesenteric resistance arteries reactivity (n=6).
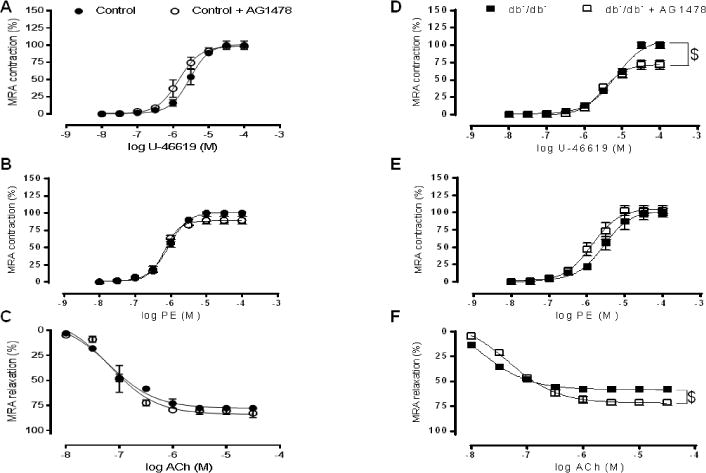
Percentage of contraction to thromboxane analogue (U46619) and phenylephrine (PE) and endothelium dependent relaxation to acetylcholine (ACh) in MRA from control (A, B, C) and type 2 diabetic mice (D, E, F) incubated with or without AG1478. $P< 0.05 for db−/db− vs. db−/db− + AG1478.
In other set of experiments and in order to study the direct effect of EGFRtk on the contractile and relaxation responses, MRAs from control and diabetic mice were incubated with exogenous EGF. In the control group, incubation with exogenous EGF potentiates the contractile response to PE and U46619 (Figure 2A, B; Fig Supp 2B) and impairs the EDR (Figure 2C; Fig Supp 2B). In the diabetic group, MRA incubated with exogenous EGF did not affect the EDR, in response to ACh, neither in the contractile responses to U46619 (Figure 2D, F; Fig Supp 2B). However, incubation with exogenous EGF significantly potentiates the contractile response to PE in MRA from type 2 diabetic mice (Figure 2E; Fig Supp 2B).
Figure 2. Effect of exogenous EGF and gp91 ds-tat on Mesenteric resistance arteries reactivity (n=6).
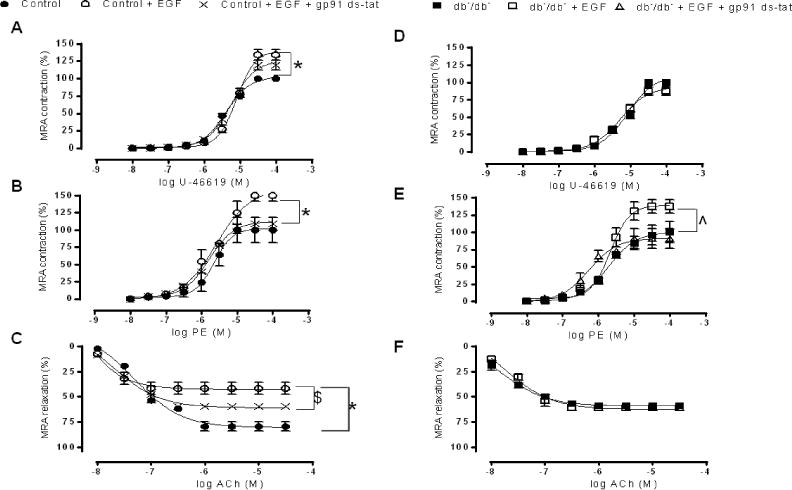
Percentage of contraction to thromboxane analogue (U46619) and phenylephrine (PE) and endothelium dependent relaxation to acetylcholine (ACh) in MRA from control (A, B, C) and type 2 diabetic mice (D, E, F) incubated with or without exogenous EGF and gp91 ds-tat. *P< 0.05 for Control vs. Control EGF. $P< 0.05 for Control vs. Control EGF+ gp91 ds-tat. ˆP< 0.05 for db−/db− vs. db−/db− + EGF and db−/db− + EGF + gp91 ds-tat.
To study the role of NADPH oxidase in the contractile and relaxation response, MRAs from control and type 2 diabetic mice were pre-incubated with gp91 das-tat (specific inhibitor of NADPH oxidase). Then, MRAs from both groups of mice were incubated with exogenous EGF. Our results indicate that the potentiation of the contractily to PE was reduced in the diabetic group and in the control group as well (Figure 2B, E; Fig Supp 2B). Additionally, the impaired EDR was significantly improved in the control group (Figure 2C; Fig Supp 2B). In contrast, gp91 ds-tat had no effect on the potentiated contraction to U46619 in the control group (Figure A, Fig supp 2B). The contraction to KCl was similar between control and diabetic group (Fig Supp 2D).
To strengthen our results, we used MRAs from p47phox knockout mice (p47phox−/−). Our data illustrate that the contractile response to U44619 was increased in MRA of p47phox−/− mice after incubation with exogenous EGF (Figure 3A; Fig Supp 2C) indicating that metabolic stimulus induced-contraction independently of the NADPH oxidase subunit p47phox. However, the contractility responses to PE and the relaxation responses to acetylcholine were not affected after incubation with exogenous EGF (Figure 3B, C; Fig supp 2C) suggesting that p47phox plays an important role in the contractile response, to sympathetic stimulation, and relaxation in response to ACh.
Figure 3. Effect of exogenous EGF on Mesenteric resistance arteries reactivity (n=6).
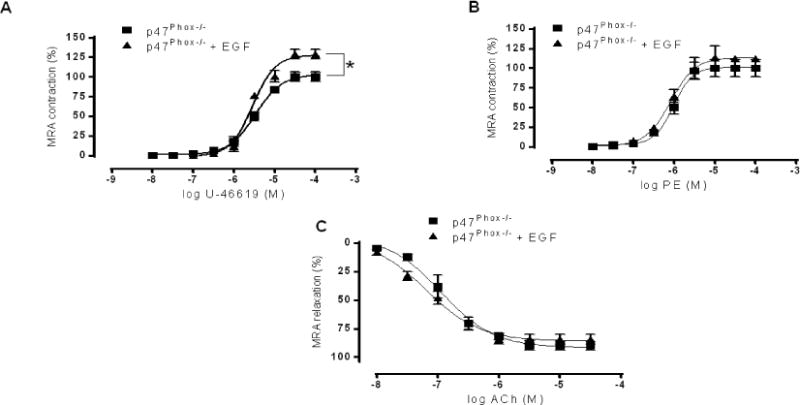
Percentage of contraction to thromboxane analogue (U46619, A) and phenylephrine (PE, B) and endothelium dependent relaxation to acetylcholine (ACh, C) in MRA from p47Phox−/− mice incubated with or without exogenous EGF. *P< 0.05 for p47Phox−/− vs. p47Phox−/− EGF.
Western blot
Western blot analysis showed that expression of p22phox, p47phox and phosphorylated ERK1/2 and Rho-kinase were increased in MRA from control mice after incubation with exogenous EGF and were significantly reduced in the presence of gp91 ds-tat (Figure 4A). Furthermore, the phosphorylated eNOS and Akt levels were reduced in MRA from control mice after incubation with EGF and were significantly increased in the presence of gp91 (Figure 4A).
Figure 4. Western blot and NADPH oxidase activity in mesenteric resistance arteries (n=4).
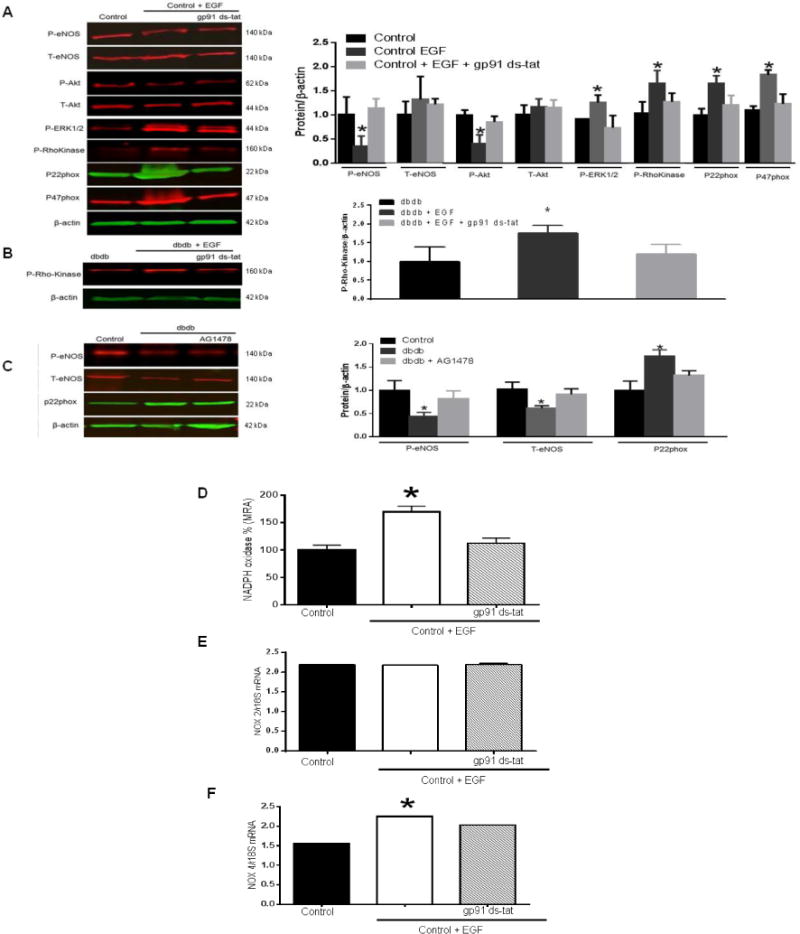
(A) Western blot analysis showing the phosphorylated (P) and total (T)-eNOS, Akt, phosphorylated (P)-ERK1/2, Rho Kinase, p22phox and p47phox expression in MRA from control mice incubated with exogenous EGF in the presence or absence of gp91 ds-tat; (B) Western blot analysis showing phosphorylated-Rho Kinase in MRA from db−/db− mice incubated with exogenous EGF in the presence or absence of gp91 ds-tat; (C) Western blot analysis showing the total and phosphorylated-eNOS, p22phox expression in MRA from control and db−/db− mice incubated with AG1478.
NADPH oxidase activity (D), RT PCR for NOX2 and NOX4 (E, F) in lysates of mesenteric resistance arteries from control mice incubated with exogenous EGF in the presence or absence of gp91 ds-tat; *p< 0.05 for control EGF vs. control and control EGF + gp91 ds-tat.
In MRA from type 2 diabetic mice group, we observed an increase in the expression of phosphorylated Rho-kinase after incubation with exogenous EGF, which was significantly reduced in the presence of gp91 ds-tat (Figure 4B).
Moreover, the phosphorylated eNOS, which was reduced in MRA from diabetic mice compared to control group, was not improved after incubation with AG1478 (Figure 4C). However, p22phox, which was increased in MRA from diabetic group compared to control group, was significantly reduced in the presence of AG1478 (Figure 4C).
NADPH oxidase activity
Our results showed an increase in NADPH oxidase activity in MRA from control mice incubated with exogenous EGF (Figure 4D). Importantly, NADPH oxidase activity was significantly reduced after incubation with gp91 ds-tat (Figure 4D). In MRA from control mice incubated with EGF, mRNA levels of NOX2 were intact however the mRNA levels of NOX4 were enhanced (Figure 4E, F). Interestingly, NOX4 mRNA levels were significantly reduced in the presence of gp91 ds-tat (Figure 4F).
In vitro studies with primary cultured cells
Primary cultured endothelial cells and VSMC were stimulated with EGF for 1 hour in the presence or absence of ER stress inhibitor (TUDCA) or NADPH oxidase activity inhibitor (gp91 ds-tat). Western blot analysis for ERK1/2 MAP kinase and endoplasmic reticulum stress marker (CHOP) as well as NADPH oxidase activity were determined in these cells. Our results revealed that EGF enhances ERK1/2 phosphorylation and CHOP expression (Figure 5A, C) and NAPDH oxidase activation (Figure 5B, D) in VSMC and endothelial cells. All these data were reversed after incubation with either TUDCA or gp91 ds-tat (Figure 5A, B, C, D).
Figure 5.
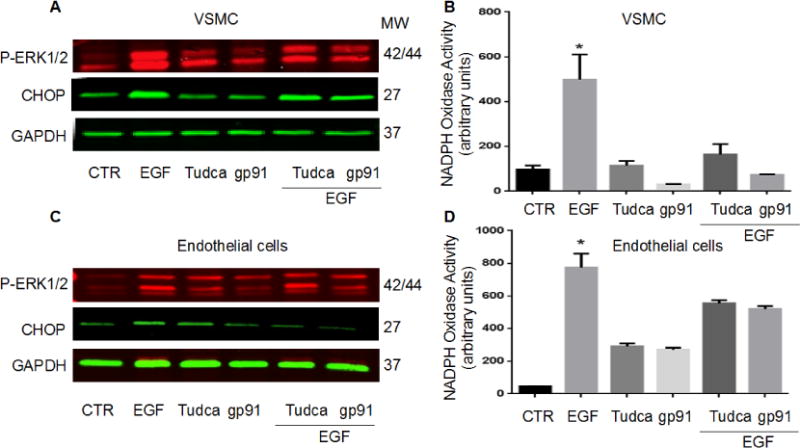
Western blot analysis and NADPH oxidase activity in EC and SMC (n=3): NADPH oxidase activity and Western blot analysis for ERK1/2 phosphorylation, ER stress CHOP and GAPDH in primary cultured endothelial cells (A, B) and VSMC (C, D), *p<0.05 for EGF vs. control and EGF + Tudca, EGF + gp91 ds-tat, n=3.
DISCUSSION
In the present study we determined that incubation with exogenous EGF impairs VSMC contractility of mesenteric resistance arteries (MRAs) in response to sympathetic (PE) and metabolic (thromboxane) stimulation and reduces vascular MRAs endothelium-dependent relaxation. This was associated with a reduction in eNOS and Akt signaling and an up-regulation of NADPH oxidase expression and activity. Interestingly, in the presence of NADPH oxidase inhibitor, we restored the contractile response to PE, improved the EDR but there was no effect on the impaired contraction to thromboxane analogue. These experiments provide evidence on the link between EGFRtk and NADPH oxidase activity in the regulation of vascular function.
It has been reported that multiple cardiovascular complications are associated with elevated EGFRtk activity. Experimental and clinical studies indicate an important role for EGFRtk activation in vascular dysfunction in type 1 and type 2 diabetes. (19,27,2,3) Additionally, it has been reported that the inhibition of EGFRtk activity promotes vasodilatation and reduces elevated arterial blood pressure in hypertensive animal models with or without insulin resistance. (15,23) Although, all these studies showed the involvement of EGFRtk in vascular disease, however the mechanism of EGFRtk in vascular dysfunction in diabetes is not fully understood.
Several mechanisms have been proposed to explain the relationship between exacerbate EGFRtk and vascular dysfunction. (2,13,7,19) Others and we have reported a potential mechanism that link EGFRtk to NADPH oxidase activity and endothelial function in type 1 and 2 diabetes. (2,7,13) In these in vivo studies, the inhibition of EGFRtk was accompanied by a reduction in blood glucose and NADPH oxidase activity and an improvement in vascular function. However it is difficult to speculate whether NADPH oxidase activity reduction and vascular improvement is a direct consequence of EGFRtk inhibition or it is indirect due to the reduction of blood glucose. In order to determine the link between EGFR, NADPH oxidase activity and endothelial function independently of blood glucose levels, we performed in vitro studies by incubating MRA from control and diabetic mice with or without exogenous EGF in the presence or absence of NADPH oxidase inhibitor.
It has been reported that smooth muscle cells and endothelial cells express all four membranes of EGFRtk family (18,9,26) indicating that EGFRtk over-expression, during diseases, may affect the vascular reactivity. Additionally, it has been demonstrated that EGF causes VSMC contraction. (4) Our in vitro data are in accordance with these studies since we showed a potentiated contraction to phenylephrine and TXA analogue and an impaired EDR after incubation of MRA from control mice with exogenous EGF. It is well known that sympathetic stimulation increases Rho-Kinase activity. (5) Our data indicate that the active Rho-kinase activity was increased after incubation with exogenous EGF. In the other hand, it has been demonstrated that TXA, once it binds to its receptor it produce the contractile effect through the ERK1/2 activation. (31) Our data indicates that the incubation with exogenous EGF was associated with an increased in PERK1/2 expression. Our data are in accordance with several studies showing that activation of EGFR leads to ERK1/2 activation. (7,28,6,30) Additionally, the EDR impairment was accompanied by a decrease in the phosphorylated eNOS. Interestingly, in our previous studies, we reported that in vivo chronic inhibition of EGFRtk by the AG1478 rescued eNOS phosphorylation and expression. (2,13) However, in the present in vitro acute study, the inhibition of EGFRtk using AG1478 did not rescue eNOS indicating the differential effect of AG1478 chronic in vivo vs. acute in vitro. Part of the reduction of NO bioavailability is due to ROS production by NADPH oxidase, which is the main producer of ROS in vessels. Additionally, data from our group have identified activation of EGFRtk in the mechanism of increased NOX expression in vessels. (13) Consistent with these observations, NADPH oxidase expression and activity were determined. Our data indicate an increase in the p22phox and p47phox-NADPH oxidase subunit as well as NADPH oxidase activity after incubation with exogenous EGF. These data in one hand explain part of the EDR impairment and on the other hand determine a direct link between EGF/EGFRtk and NADPH oxidase activity. To further confirm the direct link between EGF/EGFRtk and NADPH oxidase activity, pharmacological approach using gp91 ds-tat, a specific inhibitor of NADPH oxidase, and genetics approach using p47phox knockout mice were used. Data from these approaches indicate that NADPH oxidase inhibition decreases the potentiated contraction to PE and this was accompanied by a decrease in the active Rho-Kinase. These results provide evidence on the role of NADPH oxidase activity linked to EGF/EGFRtk signaling in the potentiation of the contractile response to the alpha-adrenergic receptor. Also, NADPH oxidase inhibition improved the EDR indicating that EGF/EGFRtk pathway impairs vascular relaxation by a mechanism that involved NADPH oxidase along with other mechanisms independently of ROS production. Interestingly, the potentiated contraction to thromboxane TXA analogue was not reduced after NADPH oxidase inhibition, indicating that TXA is not linked to EGF/EGFRtk in the regulation of vascular function and independently of NADPH oxidase.
To determine the mechanism at the cellular levels, we used primary endothelial cells and VSMC that were stimulated with exogenous EGF with and without ER stress and NADPH oxidase inhibitors. Data revealed that EGF enhances NADPH oxidase activity partially through ER stress-mechanism and more likely through ERK1/2 MAP-Kinase.
In summary, these studies indicate a direct link between EGF/EGFRtk and NADPH oxidase in the regulation of vascular function in response to sympathetic stimulation and acetylcholine-induced relaxation independently of the in vivo glucose levels. We also determined that EGF/EGFRtk and metabolic stimulation causes vascular dysfunction independently of NADPH oxidase activity. Therefore, EGFRtk inhibition is a potential target for novel therapeutic strategies to improve vascular function in cardiovascular diseases.
Supplementary Material
Acknowledgments
N/A
Sources of Funding
This work was supported by the National Institutes of Health (HL095566; PI: Dr. Matrougui) and (HL097111; PI: Dr. Trebak)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
None
Conflict of interest
None
References
- 1.Akhtar S, Benter IF. The role of epidermal growth factor receptor in diabetes-induced cardiac dysfunction. Bioimpacts. 2013;3:5–9. doi: 10.5681/bi.2013.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Belmadani S, Palen DI, Gonzalez-Villalobos RA, Boulares HA, Matrougui K. Elevated epidermal growth factor receptor phosphorylation induces resistance artery dysfunction in diabetic db/db mice. Diabetes. 2008;57:1629–37. doi: 10.2337/db07-0739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Benter IF, Yousif MH, Griffiths SM, Benboubetra M, Akhtar S. Epidermal growth factor receptor tyrosine kinase-mediated signalling contributes to diabetes-induced vascular dysfunction in the mesenteric bed. Br J Pharmacol. 2005;145:829–836. doi: 10.1038/sj.bjp.0706238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berk BC, Brock TA, Webb RC, Taubman MB, Atkinson WJ, Gimbrone MA, Jr, Alexander RW. Epidermal growth factor, a vascular smooth muscle mitogen, induces rat aortic contraction. J Clin Invest. 1985;75:1083–1086. doi: 10.1172/JCI111772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Budzyn K, Marley PD, Sobey CG. Opposing roles of endothelial and smooth muscle phosphatidylinositol 3-kinase in vasoconstriction: effects of rho-kinase and hypertension. J Pharmacol Exp Ther. 2005;313:1248–53. doi: 10.1124/jpet.104.082784. [DOI] [PubMed] [Google Scholar]
- 6.Chen CH, Cheng TH, Lin H, Shih NL, Chen YL, Chen YS, Cheng CF, Lian WS, Meng TC, Chiu WT. Reactive oxygen species generation is involved in epidermal growth factor receptor transactivation through the transient oxidization of src homology2-containing tyrosine phosphatase in endothelin-1 signaling pathway in rat cardiac fibroblasts. Mol Pharmacol. 2006;69:1347–1355. doi: 10.1124/mol.105.017558. [DOI] [PubMed] [Google Scholar]
- 7.Choi SK, Galán M, Partyka M, Trebak M, Belmadani S, Matrougui K. Chronic inhibition of epidermal growth factor receptor tyrosine kinase and extracellular signal-regulated kinases 1 and 2 (ERK1/2) augments vascular response to limb ischemia in type 2 diabetic mice. Am J Pathol. 2012;180:410–8. doi: 10.1016/j.ajpath.2011.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deanfield JE1, Halcox JP, Rabelink TJ. Endothelial function and dysfunction: testing and clinical relevance. Circulation. 2007;115:1285–95. doi: 10.1161/CIRCULATIONAHA.106.652859. [DOI] [PubMed] [Google Scholar]
- 9.Dreux AC, Lamb DJ, Modjtahedi H, Ferns GA. The epidermal growth factor receptors and their family of ligands: Their putative role in atherogenesis. Atherosclerosis. 2006;186:38–53. doi: 10.1016/j.atherosclerosis.2005.06.038. [DOI] [PubMed] [Google Scholar]
- 10.Earney MT, Duncan ER, Kahn M, Wheatcroft SB. Insulin resistance and endothelial cell dysfunction: studies in mammalian models. Exp Physiol. 2008;93:158–163. doi: 10.1113/expphysiol.2007.039172. [DOI] [PubMed] [Google Scholar]
- 11.El Assar M, Ruiz de Adana JC, Angulo J, Pindado Martínez ML, Hernández Matías A, Rodríguez-Mañas L. Preserved endothelial function in human obesity in the absence of insulin resistance. J Transl Med. 2013;11:263. doi: 10.1186/1479-5876-11-263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferdinando Giacco, Michael Brownlee. Oxidative Stress and Diabetic Complications. Circulation Research. 2010;107:1058–1070. doi: 10.1161/CIRCRESAHA.110.223545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Galán M, Kassan M, Choi SK, Partyka M, Trebak M, Henrion D, Matrougui K. A novel role for epidermal growth factor receptor tyrosine kinase and its downstream endoplasmic reticulum stress in cardiac damage and microvascular dysfunction in type 1 diabetes mellitus. Hypertension. 2012;60:71–80. doi: 10.1161/HYPERTENSIONAHA.112.192500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gao P, Wang XM, Qian DH, Qin ZX, Jin J, Xu Q. Induction of Oxidative stress by Oxidized LDL via Meprinalpha-Activated Epidermal Growth Factor Receptor in Macrophages. Cardiovasc Res. 2012 doi: 10.1093/cvr/cvs369. [DOI] [PubMed] [Google Scholar]
- 15.Hao L, Du M, Lopez-Campistrous A, Fernandez-Patron C. Agonistinduced activation of matrix metalloproteinase-7 promotes vasoconstriction through the epidermal growth factor-receptor pathway. Circ Res. 2004;94:68–76. doi: 10.1161/01.RES.0000109413.57726.91. [DOI] [PubMed] [Google Scholar]
- 16.Kassan M, Galán M, Partyka M, Saifudeen Z, Henrion D, Trebak M, Matrougui K. Endoplasmic reticulum stress is involved in cardiac damage and vascular endothelial dysfunction in hypertensive mice. Arterioscler Thromb Vasc Biol. 2012;32:1652–1661. doi: 10.1161/ATVBAHA.112.249318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kassan M, Galan M, Partyka M, Trebak M, Matrougui K. Interleukin-10 released by CD4(+)CD25(+) natural regulatory T cells improves microvascular endothelial function through inhibition of NADPH oxidase activity in hypertensive mice. Arterioscler Thromb Vasc Biol. 2011;31:2534–2542. doi: 10.1161/ATVBAHA.111.233262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kato M, Inazu T, Kawai Y, Masamura K, Yoshida M, Tanaka N, Miyamoto K, Miyamori I. Amphiregulin is a potent mitogen for the vascular smooth muscle cell line, A7r5. Biochem Biophys Res Commun. 2003;301:1109–1115. doi: 10.1016/s0006-291x(03)00093-7. [DOI] [PubMed] [Google Scholar]
- 19.Kim JA, Montagnani M, Koh KK, Quon MJ. Reciprocal relationships between insulin resistance and endothelial dysfunction: molecular and pathophysiological mechanisms. Circulation. 2006;113:1888–1904. doi: 10.1161/CIRCULATIONAHA.105.563213. [DOI] [PubMed] [Google Scholar]
- 20.Kobayashi T, Eguchi S. The epidermal growth factor receptor: a missing link between endoplasmic reticulum stress and diabetic complications? Hypertension. 2012;60:20–1. doi: 10.1161/HYPERTENSIONAHA.112.197038. [DOI] [PubMed] [Google Scholar]
- 21.Lucchesi PA, Sabri A, Belmadani S, Matrougui K. Involvement of metalloproteinases 2/9 in epidermal growth factor receptor transactivation in pressure-induced myogenic tone in mouse mesenteric resistance arteries. Circulation. 2004;110:3587–3593. doi: 10.1161/01.CIR.0000148780.36121.47. [DOI] [PubMed] [Google Scholar]
- 22.Matough FA, Budin SB, Hamid ZA, Alwahaibi N, Mohamed J. The role of oxidative stress and antioxidants in diabetic complications. Sultan Qaboos Univ Med J. 2012;12:5–18. doi: 10.12816/0003082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nagareddy PR, MacLeod KM, McNeill JH. GPCR agonist-induced transactivation of the EGFR upregulates MLC II expression and promotes hypertension in insulin-resistant rats. Cardiovasc Res. 2010;87:177–186. doi: 10.1093/cvr/cvq030. [DOI] [PubMed] [Google Scholar]
- 24.Rajendran P, Rengarajan T, Thangavel J, Nishigaki Y, Sakthisekaran D, Sethi G, Nishigaki I. The vascular endothelium and human diseases. Int J Biol Sci. 2013;9:1057–69. doi: 10.7150/ijbs.7502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Savoia C, Sada L, Zezza L, Pucci L, Lauri FM, Befani A, Alonzo A, Volpe M. Vascular inflammation and endothelial dysfunction in experimental hypertension. Int J Hypertens. 2011;2011:281240. doi: 10.4061/2011/281240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stanic B, Pandey D, Fulton DJ, Miller FJ., Jr Increased epidermal growth factor-like ligands are associated with elevated vascular nicotinamide adenine dinucleotide phosphate oxidase in a primate model of atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32:2452–2460. doi: 10.1161/ATVBAHA.112.256107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Triggle CR, Ding H. A review of endothelial dysfunction in diabetes: a focus on the contribution of a dysfunctional eNOS. J Am Soc Hypertens. 2010;4:102–115. doi: 10.1016/j.jash.2010.02.004. [DOI] [PubMed] [Google Scholar]
- 28.Ushio-Fukai M, Griendling KK, Becker PL, Hilenski L, Halleran S, Alexander RW. Epidermal growth factor receptor transactivation by angiotensin II requires reactive oxygen species in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2012;21:489–495. doi: 10.1161/01.atv.21.4.489. [DOI] [PubMed] [Google Scholar]
- 29.Versari D, Daghini E, Virdis A, Ghiadoni L, Taddei S. Endothelial Dysfunction as a Target for Prevention of Cardiovascular Disease. Diabetes Care November. 2009;32:S314–S321. doi: 10.2337/dc09-S330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang CM, Lin MI, Hsieh HL, Sun CC, Ma YH, Hsiao LD. Bradykinin-induced p42/p44 MAPK phosphorylation and cell proliferation via Src, EGF receptors, and PI3-K/Akt in vascular smooth muscle cells. J Cell Physiol. 2005;203:538–546. doi: 10.1002/jcp.20250. [DOI] [PubMed] [Google Scholar]
- 31.Zhang W, Zhang Y, Edvinsson L, Xu CB. Transcriptional down-regulation of thromboxane A(2) receptor expression via activation of MAPK ERK1/2, p38/NF-kappaB pathways. J Vasc Res. 2009;46:162–74. doi: 10.1159/000153247. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.