

Abstract
To enhance the accumulation and penetration of nanomedicines in tumor tissue, we developed and evaluated the biological properties of matrix metallo-proteinase 2 (MMP-2)-responsive N-(2-hydroxypropyl)-methacrylamide (HPMA) copolymer drugs and tumor-penetrating peptide conjugates (P-DOX-PLGLAG-iRGD). Two different spacers were used in the design: a lysosomally (cathepsin B) cleavable tetrapeptide GFLG spacer conjugated doxorubicin (DOX) to HPMA copolymer, and an MMP-2-degradable linker (PLGLAG) connected tumor-homing and -penetrating cyclic peptide iRGD to HPMA copolymer. The accumulation of DOX in P-DOX-PLGLAG-iRGD-treated monolayer (2D) and multilayer (3D) DU-145 prostate cancer cells was higher than that of control groups (P-DOX and P-DOX + iRGD). The cell cycle arrest analysis and cytotoxicity data demonstrated that P-DOX-PLGLAG-iRGD produced a higher G2/M arrest and possessed stronger cytotoxicity against DU-145 cells than P-DOX + iRGD or P-DOX, which was consistent with the drug uptake results. Similarly, P-DOX-PLGLAG-iRGD demonstrated the highest penetration ability in 3D multicellular DU-145 tumor cell spheroids. The results indicate that covalent conjugation of iRGD via MMP-2-sensitive bonds enhances accumulation and penetration of nanomedicines into tumor cell monolayers and spheroids.
The potential for nanomedicines to translate into the clinic for cancer treatment is limited by their lower accumulation in desired organs and poor penetration into solid tumors.1 Most nanomedicines rely on enhanced permeability and retention (EPR) effect to enter into tumor areas, but the EPR efficacy is often compromised by the tumor microenvironment.2 The dense stroma, increased interstitial fluid pressure (IFP), and extracellular matrix hamper the convection of nanomedicines into the tumor.3 The high IFP and the tight structure of tumors also hinder nanomedicines’ transport across more than one or two cell layers.4 Therefore, there are several ways to enhance the efficacy of nanomedicines and the speed of their translation into the clinic. One strategy is to increase the accumulation of nanomedicines in tumors by attaching targeting moieties to drug carriers.5 Another method is to enhance the tumor-penetrating ability of nanomedicines by conjugation with cell-penetrating peptides (CPPs).6 The third approach is to decrease the IFP by co-administration of poly(ethylene glycol)-modified hyaluronidase3a or hedgehog pathway inhibitors.3c
The newly developed cyclic peptide iRGD (CRGDKGPDC), which is cleaved by cell-surface-associated protease(s) to expose the neuropilin-1-binding RGDK sequence, can function as both a tumor-homing and -penetrating peptide.7 However, the short half-life (a few minutes due to proteolysis and rapid renal clearance) of free iRGD limits its clinical application.8 Conjugation of peptides to larger polymers is a general strategy to prolong their half-life.8a However, another group in addition to our own has shown that the conjugated form of iRGD may lose its cell-penetrating/targeting ability as compared to its free form.9 Therefore, we aimed to develop a smart drug delivery system that can prolong the half-life of iRGD as well as preserve its penetrating/targeting ability.
To this end, we designed a matrix metalloproteinase 2 (MMP-2)-responsive drug delivery system for treatment of prostate cancer (Figure 1). N-(2-Hydroxypropyl)methacrylamide (HPMA) copolymer was used as the drug carrier because it is water-soluble and biocompatible.1c Doxorubicin (DOX) was selected as the model drug for two major reasons: first, the fluorescence of DOX makes it easy to track its fate; second, DOX is one of the most commonly used anti-cancer drugs, but it has only limited penetration in prostate cancer.10 DOX was conjugated to HPMA copolymer via a well-defined lysosomally cleavable linker (glycylphenylalanylleucylglycine, GFLG) to limit nonspecific cytotoxicity because the drug is only released from the conjugate after it is taken up into cells.1c iRGD was conjugated to HPMA copolymer via a PLGLAG peptide spacer, which is cleaved by MMP-2 in the tumor microenvironment.11 MMP-2 was selected as the cleavage enzyme because the expression of MMP-2 is associated with the growth and progression of prostate cancer.12
Figure 1.
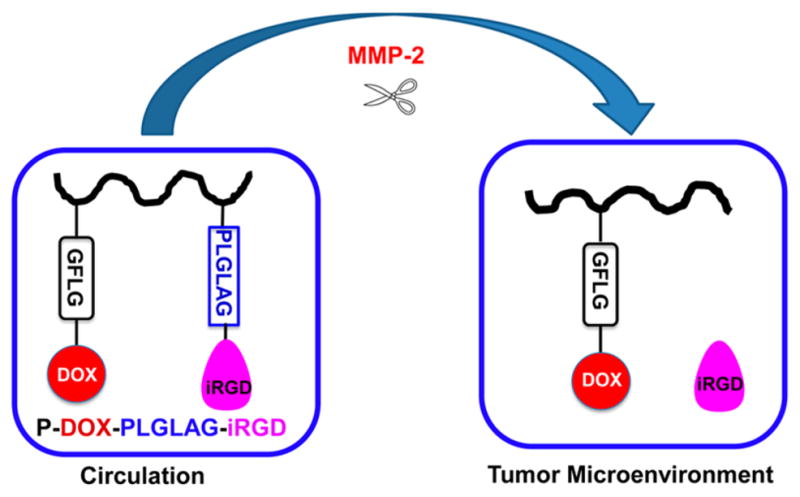
Schematic illustration and proposed fate of P-DOX-PLGLAG-iRGD.
This strategy has at least three potential advantages. First, the conjugation of iRGD to the HPMA copolymer–drug conjugate enhances accumulation of drug conjugates at the tumor site due to an active targeting effect. Second, the large molecular weight of the HPMA copolymer–iRGD conjugate increases the half-life of iRGD, resulting in increased opportunities to interact with integrins on tumor vessel endothelial cells and tumor cells. This also augments the accumulation of drug conjugates in the tumor. Third, the penetration ability of free iRGD will be regained after the iRGD is released from the HPMA copolymer conjugate in the tumor microenvironment. The accumulation and penetration of DOX conjugates were tested in DU-145 prostate cancer monolayer cells and multicellular spheroids. The activities of DOX conjugates were evaluated and compared by measuring cell cycle arrest and viability.
Because iRGD requires the exposure of C-terminus to preserve its activity, we prepared the monomer MA-GG-PLGLAG-iRGD by conjugation starting from the N-terminus of iRGD using solid-phase synthesis (Figure 2).7 The synthesis of MA-GG-PLGLAG-iRGD started from manual attachment of the Fmoc-Cys(Acm)-OH to 2-chlorotrityl chloride resin. After the active group on the resin was capped with methanol, protected amino acids (Fmoc-Asp(OBut)-OH, Fmoc-Pro-OH, Fmoc-Gly-OH, Fmoc-Lys(Boc)-OH, Fmoc-Asp(OBut)-OH, Fmoc-Gly-OH, Fmoc-Arg(Pbf)-OH, Fmoc-Cys(Acm)-OH, Fmoc-Gly-OH, Fmoc-Ala-OH, Fmoc-Leu-OH, Fmoc-Gly-OH, Fmoc-Leu-OH, and Fmoc-Pro-OH) or MA-GG-OH were loaded to resin sequentially via classic peptide coupling reactions. The resin-bound linear peptide was then cyclized with thallium(III) trifluoroacetate (Tl(OOCF3)3). The cyclization reaction on resin is able to avoid the formation of intermolecular disulfide bonds between two linear iRGDs. Finally, the crude product MA-GG-PLGLAG-iRGD was cleaved from resin by mixing the dried resin-bound peptide with a mixture solution (TFA/TIPS/H2O= 95/2.5/2.5) and purified with a semipreparative HPLC. The structure of monomer MA-GG-PLGLAG-iRGD was confirmed by matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectroscopy. As shown in Figure S1, the peak at m/z 1638.79 corresponds to [M+H]+ of the monomer MA-GG-PLGLAG-iRGD. The procedure for the synthesis of iRGD was similar to that of MA-GG-PLGLAG-iRGD with minor modification (Figure S2). The peak at m/z 948.37 in Figure S3 corresponds to [M+H]+ of the free iRGD.
Figure 2.

Scheme showing the synthesis of monomer MA-GG-PLGLAG-iRGD.
The conjugates P-DOX-PLGLAG-iRGD (Figure 3) and PDOX (Figure S4) were prepared by copolymerization of HPMA13 with MA-GFLG-DOX14 and MA-GG-PLGLAG-iRGD, or HPMA and MA-GFLG-DOX, respectively. The number- and weight-average molecular weights and molecular weight distribution (Mw/Mn) of conjugates P-DOX and P-DOX-PLGLAG-iRGD were measured with size exclusion chromatography. The DOX content in conjugates P-DOX and P-DOX-PLGLAG-iRGD was calculated by measuring the DOX UV absorbance in methanol. The iRGD content in conjugate P-DOX-PLGLAG-iRGD was determined by amino acid analysis. Aspartic acid and alanine were used for calibration. The molecular weights, Mw/Mn, DOX, and iRGD content in DOX conjugates are summarized in Table 1. The average number of iRGD units in one polymer chain was 4.59. For detailed descriptions, see the SI.
Figure 3.

Scheme showing the synthesis of polymer conjugate P-DOX-PLGLAG-iRGD.
Table 1.
Physicochemical Properties of DOX Conjugates
| DOX conjugate | Mn (kDa) | Mw (kDa) | Mw/Mn | DOX (wt%) | iRGD (wt%) |
|---|---|---|---|---|---|
| P-DOX | 69.8 | 120.0 | 1.72 | 8.35 | n/a |
| P-DOX-PLGLAG-iRGD | 71.9 | 134.3 | 1.87 | 3.87 | 6.05 |
To test the cleavability of PLGLAG spacer in a tumor microenvironment, polymer P-DOX-PLGLAG-iRGD was incubated with human recombinant MMP-2 enzyme, and the cleavage product was analyzed with LC-MS. As shown in Figure S5B, a new peak appeared at a retention time (tR) of 6.80 min; it was attributed to the cleaved product LAG-iRGD (Figure S6). The detected active MMP-2 concentration in DU-145 cell culture media was 463 ± 144 pg/mL, as measured by ELISA assay (Life Technologies), according to the manufacturer’s protocol. To test the cleavability of GFLG spacer, polymer P-DOX-PLGLAG-iRGD was incubated with lysosomal enzyme cathepsin B. Monomer MA-GG-DOX (Figure S7) with noncleavable spacer was used as the control. DOX was released from P-DOX-PLGLAG-iRGD but not from MA-GG-DOX (Figure S8).
To assay the accumulation of DOX in monolayer prostate cancer cells, we used confocal microscopy (Figure 4A) and flow cytometry (Figure 4B) to quantitatively measure DOX fluorescence intensity in DU-145 cells after incubation with P-DOX-PLGLAG-iRGD and controls. Higher fluorescence intensity was detected in P-DOX and P-DOX plus iRGD-treated groups compared to nontreated and iRGD-treated group. The P-DOX-PLGLAG-iRGD-treated group showed the highest cellular uptake and accumulation of DOX. In contrast, the mixture of iRGD with P-DOX only resulted in a slight increase of uptake and accumulation of P-DOX in DU-145 cells at the tested concentration and incubation time.
Figure 4.

Accumulation of DOX conjugates and controls in DU-145 monolayer cells as determined by DOX fluorescence: (A) confocal microscopy and (B) flow cytometry.
It has been reported that the treatment of prostate cancer DU-145 cells with DOX resulted in G2/M arrest.15 We performed a cell cycle arrest assay to check whether the enhanced accumulation of DOX in the P-DOX-PLGLAG-iRGD-treated group leads to increased G2/M cell cycle arrest. Consistent with DOX accumulation results, the highest percentage (41.6 ± 7.2%) of cells in the G2/M phase was found in the P-DOX-PLGLAG-iRGD- treated group, whereas a mixture of iRGD and P-DOX induced a milder increase in G2/M arrest (32.9 ± 6.4%) (Figure 5). These results further demonstrated that the conjugated form of iRGD enhanced the accumulation of HPMA copolymer–DOX conjugates in DU-145 cells.
Figure 5.

(A) Average percentages of DU-145 cell cycle distribution after treatment with DOX conjugates and related controls. Percentage of cells in (B) G1 phase and (C) G2/M phase. Averaged data are presented as mean ± SD. Statistics: one-way ANOVA plus Dunnett’s post-hoc test (P < 0.05 = *; not significant = n.s).
The cytotoxicity of DOX conjugates and related controls against DU145 cells was determined using the CCK-8 assay after the cells were incubated with P-DOX-PLGLAG-iRGD and related controls for 12 or 24 h. As expected, the conjugate P-DOX-PLGLAG-iRGD was more toxic to DU-145 cells than P-DOX or the mixture of P-DOX and iRGD (Figure 6). The high cytotoxicity of P-DOX-PLGLAG-iRGD could be attributed, at least in part, to its fast intracellular uptake. The fact that the conjugate P-DOX-PLGLAG-iRGD is more toxic than the mixture of P-DOX and iRGD further supports the conclusion that the biorecognition of RGD by integrins on the DU-145 cell surface possibly facilitates internalization of the drug conjugate.
Figure 6.

In vitro cytotoxicity of P-DOX-PLGLAG-iRGD and related controls against DU-145 prostate cancer cells: (A) 12 h and (B) 24 h. Statistics: one-way ANOVA plus Tukey’s post-hoc test (P < 0.001 = ***, 0.001 < P < 0.01 = **, 0.01 < P < 0.05 = *, not significant = n.s).
Multicellular tumor spheroids (MTSs) are considered one of the primary tools for drug development.16 Because the VEGF production, Ki-67, and gene expression profiles in multicellular prostate cancer spheroids are similar to those of the solid tumors in immune-deprived mice, we next tested the penetration and accumulation of DOX conjugates in DU-145 MTS.17 The penetration of DOX conjugates was monitored by measuring DOX’s intrinsic fluorescence in formalin-fixed DU-145 MTS via confocal microscopy. The images were converted to surface plots using Image-J software. As shown in Figure 7, the highest fluorescence intensity was observed in P-DOX-PLGLAG-iRGD-treated spheroids over other control-treated spheroids. The accumulation of DOX conjugates in DU-145 MTS was measured by analyzing the DOX fluorescence in disassociated cells with flow cytometry after the spheroids were incubated with P-DOX-PLGLAG-iRGD and controls for 24 h (Figure 8). Consistent with monolayer cell accumulation results, the cells in the P-DOX-PLGLAG-iRGD-treated group possessed the strongest fluorescence intensity. Spheroids exposed to a mixture of P-DOX and iRGD, or to P-DOX alone, also had enhanced fluorescence intensity compared to nontreated or iRGD-only-treated groups.
Figure 7.

Penetration of DOX conjugates and controls in DU-145 MTS (surface plots of images): (A) control, (B) iRGD, (C) P-DOX, (D) PDOX + iRGD, and (E) P-DOX-PLGLAG-iRGD.
Figure 8.

Accumulation of DOX conjugates and controls in DU-145 cells from MTS.
In this study, a stimuli-responsive drug delivery system with iRGD conjugated to a HPMA copolymer–DOX conjugate via an MMP-2 cleavable spacer was developed. The chemistry for preparing MA-GG-PLGLAG-iRGD can be applied for iRGD derivatization. Conjugation of iRGD to a drug carrier via a PLGLAG spacer enhanced the accumulation and penetration of our DOX conjugate in both monolayer and multicellular spheroid models of prostate cancer, as evidenced by enhanced cell cycle arrest and cell death. Consistent with the results from another research group,18 we further demonstrated that tumor spheroids are an excellent model for testing drug penetration before moving to in vivo evaluation. The results presented here open up avenues for modifying nanoconjugates with iRGD to enhance their tumor-targeting and -penetrating ability.
Supplementary Material
Acknowledgments
We acknowledge support by NIH grant RO1 CA132831 (to J.K.) and P30 CA042014 (to Huntsman Cancer Institute, University of Utah).
Footnotes
Notes
The authors declare no competing financial interest.
Materials, experimental procedures, and supplemental figures. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.5b00922.
References
- 1.(a) Petros RA, DeSimone JM. Nat Rev Drug Discovery. 2010;9:615. doi: 10.1038/nrd2591. [DOI] [PubMed] [Google Scholar]; (b) Banga RJ, Chernyak N, Narayan SP, Nguyen ST, Mirkin CA. J Am Chem Soc. 2014;136:9866. doi: 10.1021/ja504845f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Yang J, Kopeček J. J Controlled Release. 2014;190:288. doi: 10.1016/j.jconrel.2014.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Park K. ACS Nano. 2013;7:7442. doi: 10.1021/nn404501g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Venditto VJ, Szóka FC., Jr Adv Drug Delivery Rev. 2013;65:80. doi: 10.1016/j.addr.2012.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Shrestha R, Elsabahy M, Luehmann H, Samarajeewa S, Florez-Malaver S, Lee NS, Welch MJ, Liu Y, Wooley KL. J Am Chem Soc. 2012;134:17362. doi: 10.1021/ja306616n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Markman JL, Rekechenetskiy A, Holler E, Ljubimova JY. Adv Drug Delivery Rev. 2013;65:1866. doi: 10.1016/j.addr.2013.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tong R, Chiang HH, Kohane DS. Proc Natl Acad Sci USA. 2013;110:19048. doi: 10.1073/pnas.1315336110. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Schroeder A, Heller D, Winslow M, Dahlman J, Pratt G, Langer R, Jacks T, Anderson DG. Nat Rev Cancer. 2012;12:39. doi: 10.1038/nrc3180. [DOI] [PubMed] [Google Scholar]
- 3.(a) Provenzano P, Cuevas C, Chang A, Goel V, Von Hoff D, Hingorani S. Cancer Cell. 2012;21:418. doi: 10.1016/j.ccr.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Prabhakar U, Maeda H, Jain RK, Sevick-Muraca EM, Zamboni W, Farokhzad OC, Barry ST, Gabizon A, Grodzinski P, Blakey DC. Cancer Res. 2013;73:2412. doi: 10.1158/0008-5472.CAN-12-4561. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA. Clin Cancer Res. 2012;18:4266. doi: 10.1158/1078-0432.CCR-11-3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Dreher MR, Liu W, Michelich CR, Dewhirst MW, Yuan F, Chilkoti A. J Natl Cancer Inst. 2006;98:335. doi: 10.1093/jnci/djj070. [DOI] [PubMed] [Google Scholar]; (b) Wong C, Stylianopoulos T, Cui J, Martin J, Chauhan VP, Jiang W, Popović Z, Jain RK, Bawendi MG, Fukumura D. Proc Natl Acad Sci USA. 2011;108:2426. doi: 10.1073/pnas.1018382108. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Desoize B, Gimonet D, Jardiller JC. Anticancer Res. 1998;18:4147. [PubMed] [Google Scholar]
- 5.Corti A, Curnis F. Curr Pharm Biotechnol. 2011;12:1128. doi: 10.2174/138920111796117373. [DOI] [PubMed] [Google Scholar]
- 6.(a) Dubikovskaya EA, Thorne SH, Pillow TH, Contag CH, Wender PA. Proc Natl Acad Sci USA. 2008;105:12128. doi: 10.1073/pnas.0805374105. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lu H, Wang D, Kazane S, Javahishvili T, Tian F, Song F, Sellers A, Barnett B, Schultz PG. J Am Chem Soc. 2013;135:13885. doi: 10.1021/ja4059525. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Blum A, Kammeyer J, Yin J, Crystal D, Rush A, Gilson M, Gianneschi NC. J Am Chem Soc. 2014;136:15422. doi: 10.1021/ja5088216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sugahara K, Teesalu T, Karmali P, Kotamraju V, Agemy L, Girard O, Hanahan D, Mattrey R, Ruoslahti E. Cancer Cell. 2009;16:510. doi: 10.1016/j.ccr.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Pollaro L, Heinis C. MedChemComm. 2010;1:319. [Google Scholar]; (b) Pang HB, Braun G, She ZG, Kotamraju V, Sugahara K, Teesalu T, Ruoslahti E. J Controlled Release. 2014;175:48. doi: 10.1016/j.jconrel.2013.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Peng ZH, Kopeček J. Bioorg Med Chem Lett. 2014;24:1928. doi: 10.1016/j.bmcl.2014.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sugahara K, Teesalu T, Karmali P, Kotamraju V, Agemy L, Greenwald D, Ruoslahti E. Science. 2010;328:1031. doi: 10.1126/science.1183057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Primeau AJ, Rendon A, Hedley D, Lilge L, Tannock IF. Clin Cancer Res. 2005;11:8782. doi: 10.1158/1078-0432.CCR-05-1664. [DOI] [PubMed] [Google Scholar]
- 11.(a) Jiang T, Olson ES, Nguyen QT, Roy M, Jennings PA, Tsien RY. Proc Natl Acad Sci USA. 2004;101:17867. doi: 10.1073/pnas.0408191101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhu L, Wang T, Perche F, Taigind A, Torchilin VP. Proc Natl Acad Sci USA. 2013;110:17047. doi: 10.1073/pnas.1304987110. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wang H, Yang X, Sun C, Mao C, Zhu Y, Wang J. Biomaterials. 2014;35:7622. doi: 10.1016/j.biomaterials.2014.05.050. [DOI] [PubMed] [Google Scholar]
- 12.(a) Dos Reis S, Villanova F, Andrade P, Pontes J, de Sousa-Canavez J, Sañudo A, Antunes A, Dall’oglio M, Srougi M, Moreira Leite K. Urol Oncol. 2010;28:624. doi: 10.1016/j.urolonc.2008.10.012. [DOI] [PubMed] [Google Scholar]; (b) Singh S, Singh U, Grizzle W, Lillard JW., Jr Lab Investig. 2004;84:1666. doi: 10.1038/labinvest.3700181. [DOI] [PubMed] [Google Scholar]
- 13.Kopeček J, Bažilová H. Eur Polym J. 1973;9:7. [Google Scholar]
- 14.Ulbrich K, Šubr V, Strohalm J, Plocová D, Jelínková M, Říhová B. J Controlled Release. 2000;64:63. doi: 10.1016/s0168-3659(99)00141-8. [DOI] [PubMed] [Google Scholar]
- 15.Tyagi AK, Singh RP, Agarwal C, Chan DCF, Agarwal R. Clin Cancer Res. 2002;8:3512. [PubMed] [Google Scholar]
- 16.(a) Hirschhaeuser F, Menne H, Dittfeld C, West J, Mueller-Klieser W, Kunz-Schughart L. J Biotechnol. 2010;148:3. doi: 10.1016/j.jbiotec.2010.01.012. [DOI] [PubMed] [Google Scholar]; (b) Yip D, Cho CH. Biochem Biophys Res Commun. 2013;433:327. doi: 10.1016/j.bbrc.2013.03.008. [DOI] [PubMed] [Google Scholar]; (c) Mehta G, Hsiao AY, Ingram M, Luker GD, Takayama S. J Controlled Release. 2012;164:192. doi: 10.1016/j.jconrel.2012.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takagi A, Watanabe M, Ishii Y, Morita J, Hirokawa Y, Matsuzaki T, Shiraishi T. Anticancer Res. 2007;27:45. [PubMed] [Google Scholar]
- 18.Ai-Jamal KT, Ai-Jamal WT, Wang J, Rubio N, Buddle J, Gathercole D, Zloh M, Kostarelos K. ACS Nano. 2013;7:1905. doi: 10.1021/nn305860k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.