

Abstract
Background
Fabry disease (FD) is an X-linked multisystemic disorder with a heterogeneous phenotype. Especially atypical or late-onset type 2 phenotypes present a therapeutical dilemma.
Methods
To determine the clinical impact of the alpha-Galactosidase A (GLA) p.A143T/ c.427G > A variation, we retrospectively analyzed 25 p.A143T patients in comparison to 58 FD patients with other missense mutations.
Results
p.A143T patients suffering from stroke/ transient ischemic attacks had slightly decreased residual GLA activities, and/or increased lyso-Gb3 levels, suspecting FD. However, most male p.A143T patients presented with significant residual GLA activity (~50 % of reference), which was associated with normal lyso-Gb3 levels. Additionally, p.A143T patients showed less severe FD-typical symptoms and absent FD-typical renal and cardiac involvement in comparison to FD patients with other missense mutations. Two tested female p.A143T patients with stroke/TIA did not show skewed X chromosome inactivation. No accumulation of neurologic events in family members of p.A143T patients with stroke/transient ischemic attacks was observed.
Conclusions
We conclude that GLA p.A143T seems to be most likely a neutral variant or a possible modifier instead of a disease-causing mutation. Therefore, we suggest that p.A143T patients with stroke/transient ischemic attacks of unknown etiology should be further evaluated, since the diagnosis of FD is not probable and subsequent ERT or chaperone treatment should not be an unreflected option.
Electronic supplementary material
The online version of this article (doi:10.1186/s13023-016-0441-z) contains supplementary material, which is available to authorized users.
Keywords: Fabry disease, Lyso-Gb3, Variant of unknown significance, Late-onset, GLA mutation, Stroke, Genotype
Background
Fabry disease (FD; OMIM #301500) is an X-linked (Xq22.1) inborn error of glycosphingolipid catabolism due to deficient α-galactosidase A activity (GLA; 300644; [EC 3.2.1.22]). GLA mutations may lead to a classical or non-classical FD phenotype [1]. A clear link between genotype and phenotype in FD has not yet been established. Classical FD manifestations lead to clinical manifestations such as early stroke, malignant cardiac arrhythmia, myocardial infarction, cardiac failure, left ventricular hypertrophy (LVH) as well as progressive renal impairment, associated with differential systemic cellular accumulation of globotriaoslyceramide (Gb3). Several missense and nonsense mutations are accompanied by nearly absent GLA activities and increased lyso-Gb3 levels [2, 3]. Most non-classical FD phenotypes result either from missense mutations or from mutations within intronic or regulatory GLA regions, lacking classical FD symptoms. Affected patients suffer from late-onset organ manifestations such as cardiomyopathy, kidney failure, stroke, or neuropathic pain accompanied by high residual GLA activities and lyso-Gb3 values within the reference values, as recently shown for several non-classical FD mutations [4–12]. Due to the high frequencies of private mutations in FD patients, most genotype-phenotype relationships of controversially discussed mutations have been based on single case studies, necessitating multicenter approaches with larger patient groups carrying identical mutations. This approach will also allow for the assessment of whether affected patients may benefit from enzyme replacement therapy (ERT).
Recent studies considered the c.427G > A (p.A143T) GLA mutation to be more likely a neutral variant or a possible modifier instead of a disease causing mutation [10, 13]. In the current work, we retrospectively analyzed the first visit of 25 ERT naïve adult female and male patients, carrying the controversially discussed p.A143T variation in a multicenter study of four German FD centers and compared these data gender-specific to 58 ERT naïve patients (39 females) with other missense GLA mutations to determine a potential clinical impact of p.A143T.
Methods
Study design and patients
Between July 2006 and March 2014 twenty-five genetically confirmed adult heterozygous (females) and hemizygous (males) FD patients with the p.A143T mutation were consecutively recruited at Fabry centers of the University hospitals in Muenster, Wuerzburg, Cologne, and the Charité Berlin. Patients were retrospectively analyzed in an open cohort study and compared gender-specifically to 58 FD patients with other missense mutations (n = 24). All patients including patients with p.A143T within this study were either heterozygous (females) or hemizygous (males) carriers for the corresponding GLA mutations. Missense mutations included single nucleotide substitutions within the GLA coding region, resulting in a single substitution of amino acids, generally associated with a milder FD phenotype, compared to patients with nonsense mutations. A detailed overview of all analyzed mutations is provided in the supplement (Additional file 1: Table S1). All investigations were performed after approval of the Ethical Committees of the participating centers (Medical Association of Westfalian-Lippe and the Ethical Committee of the Medical Faculty of the University of Muenster; Ethical Committee of the Medical Faculty of the University of Cologne; Ethical Committee of the Medical Faculty of the University of Wuerzburg; Ethical Committee of the Charite Berlin, project-no: 2011–347-f; 14–328) and written informed consent of the patients for molecular analysis and publication was obtained.
A comprehensive diagnostic work-up had been performed in all centers, including medical history and cardiac, renal, and neurologic evaluation. The documentation of assessments follows the clinical practice of the German Fabry Expert Centers for a rare multisystemic disorder [14]. Cardiac assessment included echocardiography and electrocardiography. LVH was defined as an interventricular septum thickness in diastole (IVSd) >12 mm. Renal function was quantified by the estimated glomerular filtration rate (eGFR) using the Chronic Kidney Disease-Epidemiology Collaboration equation (CKD-EPI [15]) and the albumin-to-creatinine ratio (ACR) from spot urine. Renal impairment was defined as eGFR <90 ml/min/1.73 m2 according to KDIGO guidelines [16] and albuminuria as ACR >30 mg albumin per g creatinine. All patients underwent neurologic examination and a clinical interview focusing on a history of stroke or transient ischemic attack (TIA), and neuropathic pain. Strokes/TIAs were classified according to Trial of ORG 10172 in Acute Stroke Treatment (TOAST) criteria [17]. Disease severity scores were assessed using the Mainz Severity Score Index (MSSI [18]) and the total Disease Severity Scoring System (DS3 [19]).
GLA sequencing, measurement of GLA activity and plasma lyso-Gb3
Genotyping for GLA gene mutations was performed by direct sequencing of all 7 coding exons including adjacent intron-exons boundaries as previously reported [20]. GLA activity was determined using 4-methylumbelliferyl-α-D-galactopyranoside (Santa Cruz Biotechnology, Heidelberg, Germany) as previously described [21]. N-acetylgalactosamine (Santa Cruz Biotechnology) was used as specific inhibitor of endogenous α-Galactosidase B activity [22]. GLA enzyme activity was determined as nanomoles (nmol) of substrate hydrolyzed per hour (h) per mg protein. For plasma lyso-Gb3, lysoCeramide was used as reference (Matreya LLC, Pleasant Gap, USA) and D5Fluticasone Propionate (EJY Tech, Inc., Rockville, USA) served as internal standard as described elsewhere. Plasma lyso-Gb3 levels were measured in the lab of Arndt Rolfs (University of Rostock, Germany). Reference values for lyso-Gb3 were <0.9 ng/ml in plasma and for GLA activity >32 nmol/h/mg protein in leukocytes. ERT-naïve lyso-Gb3 values were available for 10 male and 14 female p.A143T patients and 14 male and 26 female patients with missense mutations. X chromosome inactivation analysis was performed with genomic DNA from leukocytes using a modified human androgen receptor (HUMARA) assay. In short, 150 ng DNA were either incubated for 12 h at 37 °C with 4 units RsaI, or RsaI with HpaII (all New England Biolabs, Frankfurt am Main, Germany). Thirty ng DNA were used as template for subsequent amplification with KAPAHifi (Peqlab, Erlangen, Germany) using the primer combination HUM_fw: 5’-GCGCGAAGTGATCCAG-3’ and HUM_rev: 5’GCCTCTACGATGGGCTTG-3’. Amplicons were separated on a 3.5 % agarosegel and stained with ethidium bromide. Subsequent analysis according to Allen and colleagues [23] was performed with ImageJ (NIH, Bethesda, USA; http://imagej.nih.gov/ij/).
Statistical analysis
Eighty-three patients from the participating FD centers were included in the analysis. If not stated otherwise, continuous variables were expressed as mean with standard deviation, or in case of unequal distribution as median [range]. Categorical data were expressed as numbers and relative frequencies in percent. Differences between groups were analyzed with the unpaired Student’s t or Mann–Whitney U test for continuous data, and the Fisher’s exact test for categorical data. Statistical significance was considered at a 2-sided p < 0.05.
All results are reported with their respective 95 % confidence intervals (CI). SAS version 9.3 (SAS Institute Inc, Cary, USA) and GraphPad PRISM V5.0 software (GraphPad Software Inc., La Jolla, USA) were used for all statistical analyses.
Results
An index patient for the current study was a female, 30 years of age, presenting at the IFAZ Muenster in 2012 after a stroke (media infarction) of unknown etiology (Fig. 1a). The patient revealed an unrevealing cardiovascular risk profile and no evidence of extracerebral arteriopathy, vasculitis or other inflammatory disease. Direct GLA sequencing identified the p.A143T (c.427G > A, SNP# rs10489845) mutation, but plasma lyso-Gb3 level was normal. A family screening identified her father (age: 64 years), her uncle (father’s brother; age: 63 years) with his daughter (age: 32 years) also with the p.A143T variant. Although both males showed a slightly reduced GLA activity of ~50 % of the reference level none of the three relatives presented with increased plasma lyso-Gb3 levels, nor other FD-typical organ manifestations. The family history revealed strokes of her paternal grandparents at a higher age, but not in any further relatives of the index patient (neither in p.A143T affected, nor unaffected). Since at this time-point no other stroke-causing diseases could be detected within the index patient, FD was diagnosed and ERT with agalsidase-alfa was initiated, although the patient showed no further FD-typical renal or cardiac manifestations.
Fig. 1.
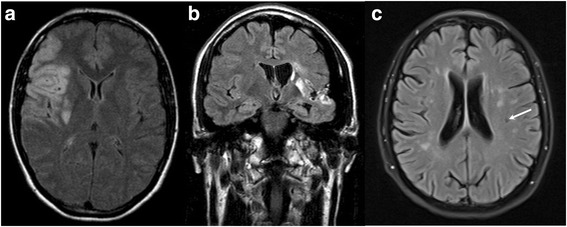
Magnetic resonance (MR) images of p.A143T patients with cerebrovascular events. a Axial MRI of the index female patient (30 years of age) suffering from an embolic stroke within the vascular territory of the right middle cerebral artery. b Coronal FLAIR-MRI of a male patient (38 years of age) suffering from an embolic stroke within the vascular territory of the left middle cerebral artery. c FLAIR-MR of a female patient (46 years of age) with TIAs and periventricular and subcortical microangiopathic and lacunar ischemic lesions (white arrow). FLAIR: fluid attenuated inversion recovery. TIA: transient ischemic attack
Since the effect of the GLA p.A143T variation is discussed controversially and it has been recently described as a non-pathogenic GLA variant instead of a FD-causing mutation [10, 13], we retrospectively analyzed each first visit between July 2006 and March 2014 of p.A143T patients, to determine its potential clinical impact. Twenty-one additional patients of another 11 unrelated families with the p.A143T mutation were consecutively recruited at Fabry centers in Muenster, Wuerzburg, Cologne and Berlin. A summary of clinical symptoms and manifestations in patients with p.A143T and other missense mutations is provided in Table 1. Even if females with p.A143T suffered more often from unspecific GI symptoms such as abdominal pain (p = 0.0133) and diarrhea (p = 0.0048), all other analyzed parameters did not differ (Table 1). For subsequent analyses, naïve p.A143T patients were gender-specifically compared to naïve FD patients with other GLA missense mutations concerning organ manifestations and clinical events (Table 2). A detailed overview of the analyzed GLA mutations is provided in the supplement (Additional file 1: Table S1). GLA activity in all tested hemizygous males with p.A143T was below the reference, but significantly higher compared to males with other missense mutations (Table 2; Fig. 2a). GLA activity values in heterozygous females with the p.A143T variant were lower compared to heterozygous females with other missense mutations (p < 0.05), but still above reference (Table 2; Fig. 2a). In contrast to previous studies [13] no tissue biopsies have been performed.
Table 1.
Symptoms and manifestations in p.A143T patients and patients with missense GLA mutations
| p.A143T Fabry patients | Missense mutation patients | |||||
|---|---|---|---|---|---|---|
| Total (n = 25) | Female (n = 15) | Male (n = 10) | Total (n = 58) | Female (n = 39) | Male (n = 19) | |
| gastrointestinal pain, n | 12 (48.0) | 8 (53.3)* | 4 (40.0) | 11 (20.0) | 6 (16.2) | 5 (27.8) |
| diarrhea, n | 8 (32.0) | 6 (40.0)** | 2 (20.0) | 5 (9.1) | 2 (5.4) | 3 (16.7) |
| tinnitus, n | 7 (29.2) | 4 (28.6) | 3 (30.0) | 6 (10.7) | 6 (16.2) | 0 (0.0) |
| neuropathic pain, n | 7 (28.0) | 4 (26.7) | 3 (30.0) | 26 (45.6) | 18 (47.4) | 8 (42.1) |
| hypohidrosis, n | 6 (24.0) | 4 (26.7) | 2 (20.0) | 18 (32.1) | 11 (29.0) | 7 (38.9) |
| fatigue, n | 5 (20.0) | 4 (26.7) | 1 (10.0) | 10 (18.9) | 8 (23.5) | 2 (10.5) |
| hypacusis, n | 3 (12.5) | 1 (7.1) | 2 (20.0) | 5 (8.8) | 5 (13.2) | 0 (0.0) |
| dyspnea, n | 3 (12.0) | 3 (20.0) | 0 (0.0) | 12 (21.1) | 8 (21.1) | 4 (21.1) |
| edema, n | 1 (4.2) | 1 (6.7) | 0 (0.0) | 2 (3.6) | 0 (0.0) | 2 (10.5) |
| cornea verticillata, n | 0 (0.0) | 0 (0.0) | 0 (0.0) | 9 (19.6) | 8 (29.6)* | 1 (5.3) |
| LVEF, % | 63.1 ± 6.4 | 60.9 ± 6.2 | 66.2 ± 5.6 | 63.2 ± 9.6 | 64.5 ± 9.8 | 60.6 ± 8.8 |
Values are given as mean ± SD for continuous data or n (%) for categorical data. Continuous values were compared using unpaired Student’s t test and categorical values have been tested with Fisher’s exact test (both two-sided). LVEF: left ventricular ejection fraction. Patients have been gender-specific compared. *p < 0.05, **p < 0.01
Table 2.
Clinical data and parameters
| p.A143T patients | Missense mutation patients | |||||
|---|---|---|---|---|---|---|
| Total (n = 25) | Female (n = 15) | Male (n = 10) | Total (n = 58) | Female (n = 39) | Male (n = 19) | |
| age, years | 46.3 ± 13.7 | 48.7 ± 14.3 | 42.7 ± 12.6 | 44.1 ± 16.6 | 47.7 ± 17.3 | 36.8 ± 12.6 |
| body weight, kg | 76.6 ± 20.4 | 65.8 ± 10.7 | 92.7 ± 21.4 | 75.8 ± 19.6 | 71.6 ± 19.1 | 83.8 ± 18.3 |
| body height, cm | 172.0 ± 10.9 | 166.1 ± 5.1 | 181.0 ± 11.3 | 171.9 ± 9.4 | 167.0 ± 5.9 | 181.6 ± 7.2 |
| BMI, kg/m2 | 25.6 ± 4.8 | 24.0 ± 4.3 | 28.0 ± 4.5 | 25.5 ± 5.8 | 25.6 ± 6.3 | 25.3 ± 4.8 |
| GLA activity (normal range 100–250 %) , % of reference | 65 [25–181] | 100 [54–181] | 48 [25–72] | 80 [0–272] | 166 [34–272]* | 13 [0–69]** |
| lyso-Gb3 within reference range (<0.9 ng/ml), n | 19 (82.6) | 12 (85.7) | 7 (77.8) | 9 (22.5) | 8 (30.8)** | 1 (7.1)*** |
| lyso-Gb3, ng/ml | 0.7 ± 0.3 | 0.6 ± 0.3 | 0.8 ± 0.3 | 13.0 ± 16.0 | 4.5 ± 4.8** | 30.2 ± 16.9**** |
| ERT, na | 10 (40.0) | 4 (26.7) | 6 (60.0) | 25 (43.1) | 12 (30.8) | 13 (68.4) |
| Fabry crisis, n | 2 (8.0) | 2 (13.3) | 0 (0.0) | 8 (15.1) | 5 (14.7) | 3 (15.8) |
| stroke/TIA, n | 7 (28.0) | 3 (20.0) | 4 (40.0) | 5 (8.6) | 2 (5.1) | 3 (15.8) |
| IVSd, mm | 9.0 ± 1.8 | 8.3 ± 1.8 | 9.8 ± 1.4 | 11.6 ± 5.0 | 10.6 ± 3.2** | 14.0 ± 7.3* |
| LVH (IVSd > 12 mm), n | 3 (12.0) | 1 (6.7) | 2 (20.0) | 19 (40.4) | 11 (33.3) | 8 (57.1) |
| pacemaker, n | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (1.7) | 1 (2.6) | 0 (0.0) |
| NYHA class, n | ||||||
| 0 | 8 (33.3) | 3 (21.4) | 5 (50.0) | 9 (15.5) | 7 (18.0) | 2 (10.5)* |
| I | 15 (62.5) | 10 (71.4) | 5 (50.0) | 36 (62.1) | 23 (59.0) | 13 (68.4) |
| II | 1 (4.2) | 1 (7.1) | 0 (0.0) | 10 (17.2) | 8 (20.5) | 2 (10.5) |
| III | 0 (0.0) | 0 (0.0) | 0 (0.0) | 3 (5.2) | 1 (2.6) | 2 (10.5) |
| IV | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| ACR, mg albumin/ g creatinine | 7 [0–14323] | 12 [0–14323] | 1 [0–12] | 82 [0–4080] | 59 [0–4080]*** | 195 [0–2668]**** |
| albuminuria (>30 mg/g), n | 4 (16.7) | 4 (28.6) | 0 (0.0) | 37 (82.2) | 22 (75.9)** | 15 (93.8)**** |
| creatinine, mg/dl | 0.86 ± 0.31 | 0.80 ± 0.35 | 0.96 ± 0.20 | 0.86 ± 0.38 | 0.73 ± 0.18 | 1.12 ± 0.53 |
| eGFR, ml/min/1.73 m2 | 95.6 ± 17.6 | 95.0 ± 16.5 | 96.4 ± 20.1 | 97.3 ± 27.5 | 98.3 ± 21.4 | 95.2 ± 37.6 |
| dialysis, n | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (1.7) | 0 (0.0) | 1 (5.3) |
| NTX, n | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (1.7) | 0 (0.0) | 1 (5.3) |
| CKD stage, n | ||||||
| G1 (≥90 ml/min/1.73 m2) | 18 (72.0) | 11 (73.3) | 7 (70.0) | 37 (67.3) | 26 (70.3) | 11 (61.1) |
| G2 (60–89 ml/min/1.73 m2) | 6 (24.0) | 4 (26.7) | 2 (20.0) | 12 (21.8) | 9 (24.3) | 3 (16.7) |
| G3 (30–59 ml/min/1.73 m2) | 1 (4.0) | 0 (0.0) | 1 (10.0) | 6 (10.9) | 2 (5.4) | 4 (22.2) |
| G4 (15–29 ml/min/1.73 m2) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| G5 (<15 ml/min/1.73 m2) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| RAAS blockers, n | 8 (32.0) | 5 (33.3) | 3 (30.0) | 18 (54.6) | 10 (43.5) | 8 (80.0)* |
| diuretics, n | 2 (8.0) | 0 (0.0) | 2 (20.0) | 5 (16.1) | 5 (23.8) | 0 (0.0) |
| analgesics, n | 2 (8.0) | 1 (6.7) | 1 (10.0) | 6 (21.4) | 6 (30.0) | 0 (0.0) |
| antidepressants, n | 3 (12.0) | 2 (13.3) | 1 (10.0) | 4 (14.8) | 4 (21.1) | 0 (0.0) |
| MSSI score (max. 76) | 8.5 ± 7.8 | 8.1 ± 6.7 | 9.0 ± 9.7 | 8.9 ± 6.7 | 8.1 ± 7.5 | 10.4 ± 4.7 |
| MSSI general (max. 18) | 2.4 ± 1.9 | 2.8 ± 2.3 | 1.8 ± 1.2 | 1.8 ± 1.6 | 1.4 ± 1.6* | 2.4 ± 1.3 |
| MSSI cardiac (max. 20) | 0.9 ± 2.5 | 0.6 ± 2.4 | 1.2 ± 2.6 | 2.7 ± 4.2 | 2.9 ± 4.1* | 2.3 ± 4.4 |
| MSSI renal (max. 18) | 2.1 ± 3.6 | 2.3 ± 3.8 | 1.8 ± 3.5 | 2.4 ± 2.8 | 1.8 ± 2.8 | 3.6 ± 2.6* |
| MSSI neurologic (max. 20) | 3.1 ± 4.1 | 2.4 ± 3.7 | 4.1 ± 4.6 | 2.1 ± 2.2 | 2.1 ± 2.3 | 2.2 ± 2.2 |
| total DS3 score (max. 80) | 6.8 ± 6.5 | 6.3 ± 6.3 | 7.6 ± 7.1 | 9.0 ± 6.2 | 7.4 ± 6.0 | 12.1 ± 5.6 |
| total DS3 cardiac (max. 24) | 0.6 ± 1.8 | 0.4 ± 1.3 | 1.0 ± 2.3 | 2.8 ± 3.3 | 2.6 ± 3.2* | 3.3 ± 3.5 |
| total DS3 renal (max. 24) | 1.3 ± 2.1 | 1.6 ± 2.3 | 1.0 ± 1.7 | 2.4 ± 3.4 | 1.9 ± 3.0 | 3.6 ± 3.9* |
| total DS3 neurologic (max. 16) | 4.4 ± 4.7 | 3.9 ± 4.4 | 5.2 ± 5.3 | 3.4 ± 3.5 | 2.8 ± 3.3 | 4.8 ± 3.7 |
Missense mutations were restricted to amino acid substitutions due to single nucleotide mutations within the coding region absent of the catalytic active protein sites. Values are given as mean ± SD, median [range] for continuous data or n (%) for categorical data. Continuous values were compared using unpaired Student’s t test or Mann Whitney U test if unequal distribution was observed (both two-sided). Categorical values have been tested with Fisher’s exact test (two-sided). Reference values for plasma lyso-Gb3 < 0.9 ng/ml and GLA activity >32 nmol/h/mg protein in leukocytes. To compare GLA activities between laboratories, values are presented as the percentage of the laboratory reference value. ACR: albumin-to-creatinine ratio; BMI: body mass index; CKD: chronic kidney disease; DS3: total Disease Severity Scoring System; eGFR: estimated glomerular filtration rate (calculated via CKD-EPI formula); ERT: enzyme replacement therapy; IVSd: interventricular septum thickness in diastole; LVH: left ventricular hypertrophy; MSSI: Mainz Severity Score Index; NTX: renal transplantation; NYHA: New York Heart Association; RAAS: renin-angiotensin-aldosterone-system. TIA: transient ischemic attack. aERT initialization within 12 months after 1.st visit. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
Fig. 2.
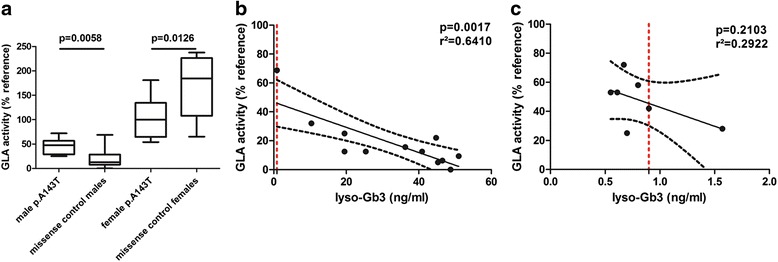
a Distribution of residual GLA activities in patients with p.A143T and Fabry disease patients with other missense GLA mutations. b Correlation of residual GLA activities with lyso-Gb3 levels in male Fabry disease patients with other missense GLA mutations and c in male patients with p.A143T. The dotted line represents the upper reference value of measured lyso-Gb3 (0.9 ng/ml). Values are given as medians with 5–95 % confidence intervals
Mean plasma lyso-Gb3 levels in p.A143T patients were significantly lower than in patients with classical missense mutations, so that most p.A143T patients showed normal lyso-Gb3 values (lyso-Gb3 < 0.9 ng/ml: females: 85.7 %; males: 77.8 %, Table 2). Linear regression analysis revealed that male FD patients with missense mutations showed decreased GLA activities, which were significantly associated with increased lyso-Gb3 values (p = 0.0017; r2 = 0.6410; Fig. 2b). In this analysis a residual GLA activity of <40–50 % was always associated with increased lyso-Gb3 levels (Fig. 2b). In contrast, in male p.A143T patients such a correlation was statistically not significant (p = 0.2103), since most males showed only a slightly decreased GLA activity with a normal lyso-Gb3 level (Fig. 2c).
In addition, p.A143T patients (females as well as males) had lower values for IVSd (p < 0.01 and p < 0.05, respectively; Table 2), resulting in lower frequencies of LVH. This mild cardiac involvement is also reflected by the distribution of the New York Heart Association (NYHA) classes, as half of the male p.A143T patients were classified in NYHA class 0, whereas ~90 % of the typical FD patients suffered from a more severe heart failure (NYHA class II-IV, Table 2). The PR intervals seen on ECG in p.A143T patients were also in a normal range (mean: 157 ± 20 ms) and not shortened as typical for FD [24]. All p.A143T patients had a lower ACR (p < 0.001) compared to other FD missense mutation patients, resulting in a lower risk for albuminuria (females: p = 0.0031, males: p < 0.0001; Table 2). The eGFR values and raw serum creatinine levels were comparable in all analyzed patients (Table 2). Male patients with missense mutation were more often treated with RAAS blockers (Table 2). The mild organ involvement was also demonstrated by the evaluation of disease scores in that patients with p.A143T (females as well as males) presented with lower values of MSSI and DS3 subscores as patients with other missense mutations (Table 2). In detail, female p.A143T patients had lower cardiac scores (both, MSSI and DS3) and males lower renal scores (both, MSSI and DS3). Furthermore, in contrast to patients with other missense mutations (MSSI: p = 0.0076), increasing MSSI scores were not associated with increasing age (MSSI: p = 0.4593).
According to the TOAST criteria [17] six additional (2 heterozygous females and 4 hemizygous males) unrelated p.A143T patients with a mean age of 41.3 ± 5.6 years (age at event) suffered from stroke (n = 5) or TIA (n = 1) of either unknown (n = 4), macroangiopathic (n = 1), or other origin (A. carotis interna dissection, n = 1) before their first assessment. Of note, similar to the index patient, four patients with cryptogenic stroke/TIA were the first of their families presenting at the participating FD centers. Although subsequent family screening revealed more p.A143T relatives, none of these suffered from stroke/TIA. Additionally, family members without p.A143T were free of neurological events.
Figure 1 shows representative magnet resonance images (MRI) of three p.A143T patients (1 male/ 2 females) suffering from stroke/TIA. Both females showed random X chromosome inactivation (allele ratios for index patient 50:50 and TIA patient: 60:40) in blood samples (leukocytes) (Additional file 2: Figure S1).
Of note, p.A143T patients suffering from stroke/TIA had slightly decreased residual GLA activities, and/or increased lyso-Gb3 levels.
Discussion
To determine the clinical impact of the GLA p.A143T variation, we retrospectively analyzed 25 heterozygous (females) and hemizygous (males) adult Fabry patients with this variation and compared gender-specific their clinical symptoms and manifestations with a well-characterized group of 58 FD patients (39 heterozygous females) with other classical missense mutations.
Our main findings are: 1) Most male p.A143T patients had only slightly decreased residual GLA activities and normal lyso-Gb3 levels, while GLA activity in heterozygous females was normal; 2) Female and male p.A143T patients showed a less organ involvement in comparison to FD patients with other missense mutations; 3) Female and male p.A143T patients suffering from stroke/TIA showed no further FD-typical organ manifestations; 4) No accumulation of neurologic events in family members of p.A143T patients with stroke/TIA was observed.
Recent studies suggested the p.A143T variation to be more likely a neutral variant or a possible modifier than a disease causing mutation [10, 13]. In our study, the risk for FD-typical manifestations such as albuminuria, increased IVSd, and increased disease severity scores was significantly lower in male as well as female p.A143T patients when compared to groups with classical FD missense mutations. The higher disease burden of FD patients with other missense mutations is also demonstrated by severely decreased GLA activities and/or increased lyso-Gb3 levels, while most p.A143T patients (females and males) showed normal values. However, the residual enzymatic activities in male p.A143T patients revealed a bright variance between 25 to 72 % of normal GLA activity, which has also been observed by other investigators especially in cultured T cells [24]. The only moderately decreased GLA activity in leukocytes of p.A143T males seems to be sufficient to keep plasma lyso-Gb3 levels below the reference value. The ultimate presence or absence of cellular Gb3 deposits can be diagnosed by appropriate tissue biopsies (“gold standard”), which have not been performed in our study. However, Terryn and colleagues performed cardiac biopsy in one female patient with p.A143T with LVH and kidney biopsies in two p.A143T males with renal failure and microalbuminuria, demonstrating no Gb3 deposits [13]. Hence, the authors concluded that the presence of this mutation is not directly associated with FD-pathology (i.e. Gb3 deposits) and should not be labeled as “pathogenic” [13]. The authors also stated that the lack of Gb3 deposits does not preclude elevated plasma lyso-Gb3 levels that could be pathogenic and cause endothelial cell dysfunction, and suggested that this should be analyzed in further studies [13]. Similar to recent studies [9, 10, 13] the risk for patients with p.A143T in our study with only slightly reduced GLA activities (males) and/or normal lyso-Gb3 levels (males and females) for renal and cardiac end organ damage was reduced in comparison to other FD patients with classical missense mutations. In addition, instead of a shortened PR interval on ECG, which would be typical for a storage disease [25, 26], the PR interval in our p.A143T patients was normal, demonstrating also no FD-typical cardiac involvement. The lack of FD-typical manifestations might be also reflected by the absence of an age effect on the MSSI score within the p.A143T patients.
Including the index patient, we identified at least 5 neurologic events (stroke or TIA) in heterozygous (females) as well as hemizygous (males) patients with p.A143T of unknown etiology, which might be assigned to FD. This would be in line with recent observations suggesting that p.A143T may be associated with a "stroke-only" phenotype [8, 27]. Due to the fact that the neurologic system seems to be highly sensitive to reduced GLA activity with neuropathic pain as one of the first symptoms and early neurologic manifestations such as stroke and TIA [28, 29], the slightly reduced GLA activities observed in p.A143T patients might predominantly manifest in the central nervous system. However, strokes in FD look like strokes from other causes and the high frequency of neurological events in our p.A143T patients (20.0 %) in comparison to missense mutation patients (8.6 %) seems to be based on a screening bias. According to the Exome Aggregation Consortium (ExAC, http://exac.broadinstitute.org/) the population frequency of c.427G > A is 0.078 % in the European population, demonstrating a not infrequent finding of this variation. If the p.A143T variation alone would be pathogenic, significant more patients with even this variation with FD-typical signs and symptoms should have been identified by the participating FD centers. Our GLA expression analysis revealed no decreased mRNA expression compared to healthy controls, suggesting that p.A143T seems to be more likely a neutral variant. However, it might be possible, that the variant acts as a modifier of a yet unknown causal factor.
Conclusions
With respect to our current findings and in accordance with previous reports, which demonstrate the lack of Gb3 deposits within affected organs [13], we conclude that p.A143T seems not to be causal for FD but rather a genetic variant of unknown significance or a genetic modifier. Our data confirm previous findings that lyso-Gb3 levels seem to be mostly normal in atypical FD patients [30] or patients with genetics variants of unknown significance [10].
We suggest that p.A143T patients with stroke/ TIA of unknown etiology should be further evaluated, since the diagnosis of FD is not probable and ERT or chaperone treatment should not be an unreflected option.
Limitations
Possible restrictions of this study are the retrospective design and the limited number of patients. Due to the multicenter approach, some parameters (such as lyso-Gb3 values) and DNA samples for X-chromosome inactivation analysis were not available for the entire study cohort. Since DNA for the two analyzed females was only available from leukocytes, according to Echevarria and colleagues a skewed X chromosome inactivation within other tissues cannot be excluded [31]. The lack of tissue biopsies demonstrating the ultimately presence or absence of cellular Gb3 deposits is a limitation. A clear link between genotype and phenotype in FD is difficult to be established especially in young patients presented with stroke since it is usually not possible to document GB3 deposit in the central nervous system.
Acknowledgments
We thank the patients whose participation made this work possible. We thank Arndt Rolfs (University of Rostock, Germany) for GLA activity and lyso-Gb3 measurements. We acknowledge support by the Deutsche Forschungsgemeinschaft and Open Access Publication Fund of the University of Muenster. Parts of this manuscript were supported by Genzyme GmbH, Germany. The funder had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Abbreviations
- ACR
albumin/creatinine ratio
- BMI
body mass index
- CI
confidence interval
- CKD
chronic kidney disease
- DS3
Disease severity scoring system
- ECG
electrocardiogram
- eGFR
estimated glomerular filtration rate
- ERT
enzyme replacement therapy
- FD
Fabry disease
- Gb3
globotriaosylceramide
- GLA
alpha-galactosidase A
- IVSd
inter ventricular systolic diameter
- LVH
left ventricular hypertrophy
- MSSI
Mainz Severity Score Index
- NTX
renal transplantation
- NYHA
New York Heart Association
- OR
odds ratio
- PCR
polymerase chain reaction
- RAAS
renin-angiotensin-aldosterone-system
- TIA
transient ischemic attack
- TOAST
Trial of ORG 10172 in Acute Stroke Treatment
Additional files
Overview of different missense GLA mutations (n = 24) within the study cohort served as controls. (DOC 57 kb)
X chromosome inactivation analysis in two female p.A143T patients suffering from stroke and TIA. (DOC 3993 kb)
Footnotes
Competing interests
Authors have received honoraria, and/or research grants from Genzyme Corporation and/or Shire Corporation. BS, DB and SR have nothing to declare.
Authors’ contributions
All authors have contributed to the article by participating in the conception and design (ML, SMB, EB), acquisition of data (ML, BS, JS, SR, FW, JK, CK, SC, EB) or analysis and interpretation of data (ML, BS, SMB, EB), drafting the article (ML, SMB, EB) or revising it critically for important intellectual content (FW, CW, CK, BS, SCK, JS, TD). All authors read and approved the final version of the manuscript.
References
- 1.Zarate YA, Hopkin RJ. Fabry’s disease. Lancet. 2008;372:1427–1435. doi: 10.1016/S0140-6736(08)61589-5. [DOI] [PubMed] [Google Scholar]
- 2.Mehta A, Ricci R, Widmer U, Dehout F, Garcia de Lorenzo A, Kampmann C, Linhart A, Sunder-Plassmann G, Ries M, Beck M. Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry Outcome Survey. Eur J Clin Invest. 2004;34:236–242. doi: 10.1111/j.1365-2362.2004.01309.x. [DOI] [PubMed] [Google Scholar]
- 3.Niemann M, Rolfs A, Störk S, Bijnens B, Breunig F, Beer M, Ertl G, Wanner C, Weidemann F. Gene mutations versus clinically relevant phenotypes: lyso-Gb3 defines Fabry disease. Circ Cardiovasc Genet. 2014;7:8–16. doi: 10.1161/CIRCGENETICS.113.000249. [DOI] [PubMed] [Google Scholar]
- 4.Oliveira JP, Ferreira S, Reguenga C, Carvalho F, Månsson JE. The g.1170C > T polymorphism of the 5’ untranslated region of the human alpha-galactosidase gene is associated with decreased enzyme expression--evidence from a family study. J Inherit Metab Dis. 2008;31:S405–S413. doi: 10.1007/s10545-008-0972-0. [DOI] [PubMed] [Google Scholar]
- 5.Rombach SM, Dekker N, Bouwman MG, Linthorst GE, Zwinderman AH, Wijburg FA, Kuiper S, Vd Bergh Weerman MA, Groener JE, Poorthuis BJ, Hollak CE, Aerts JM. Plasma globotriaosylsphingosine: diagnostic value and relation to clinical manifestations of Fabry disease. Biochim Biophys Acta. 2010;1802:741–748. doi: 10.1016/j.bbadis.2010.05.003. [DOI] [PubMed] [Google Scholar]
- 6.Mitobe S, Togawa T, Tsukimura T, Kodama T, Tanaka T, Doi K, Noiri E, Akai Y, Saito Y, Yoshino M, Takenaka T, Saito S, Ohno K, Sakuraba H. Mutant α-galactosidase A with M296I does not cause elevation of the plasma globotriaosylsphingosine level. Mol Genet Metab. 2012;107:623–626. doi: 10.1016/j.ymgme.2012.07.003. [DOI] [PubMed] [Google Scholar]
- 7.Lenders M, Duning T, Schelleckes M, Schmitz B, Stander S, Rolfs A, Brand SM, Brand E. Multifocal white matter lesions associated with the D313Y mutation of the α-galactosidase A gene. PLoS One. 2013;8:e55565. doi: 10.1371/journal.pone.0055565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rolfs A, Fazekas F, Grittner U, Dichgans M, Martus P, Holzhausen M, Böttcher T, Heuschmann PU, Tatlisumak T, Tanislav C, Jungehulsing GJ, Giese AK, Putaala J, Huber R, Bodechtel U, Lichy C, Enzinger C, Schmidt R, Hennerici MG, Kaps M, Kessler C, Lackner K, Paschke E, Meyer W, Mascher H, Riess O, Kolodny E, Norrving B, Stroke in Young Fabry Patients (sifap) Investigators Acute cerebrovascular disease in the young: The stroke in Fabry patients study. Stroke. 2013;44:340–349. doi: 10.1161/STROKEAHA.112.663708. [DOI] [PubMed] [Google Scholar]
- 9.De Brabander I, Yperzeele L, Groote CC, Brouns R, Baker R, Belachew S, Delbecq J, De Keulenaer G, Dethy S, Eyskens F, Fumal A, Hemelsoet D, Hughes D, Jeangette S, Nuytten D, Redondo P, Sadzot B, Sindic C, Sheorajpanday R, Thijs V, Van Broeckhoven C, De Deyn PP. Phenotypical characterization of α-galactosidase A gene mutations identified in a large Fabry disease screening program in stroke in the young. Clin Neurol Neurosurg. 2013;115:1088–1093. doi: 10.1016/j.clineuro.2012.11.003. [DOI] [PubMed] [Google Scholar]
- 10.Smid BE, Hollak CE, Poorthuis BJ, van den Bergh Weerman MA, Florquin S, Kok WE, Lekanne Deprez RH, Timmermans J, Linthorst GE. Diagnostic dilemmas in Fabry disease: a case series study on GLA mutations of unknown clinical significance. Clin Genet. 2015;88:161–166. doi: 10.1111/cge.12449. [DOI] [PubMed] [Google Scholar]
- 11.Schelleckes M, Lenders M, Guske K, Schmitz B, Tanislav C, Ständer S, Metze D, Katona I, Weis J, Brand SM, Duning T, Brand E. Cryptogenic stroke and small fiber neuropathy of unknown etiology in patients with alpha-galactosidase A -10 T genotype. Orphanet J Rare Dis. 2014;9:178. doi: 10.1186/s13023-014-0178-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu HC, Lin HY, Yang CF, Liao HC, Hsu TR, Lo CW, Chang FP, Huang CK, Lu YH, Lin SP, Yu WC, Niu DM. Globotriaosylsphingosine (lyso-Gb3) might not be a reliable marker for monitoring the long-term therapeutic outcomes of enzyme replacement therapy for late-onset Fabry patients with the Chinese hotspot mutation (IVS4 + 919G > A) Orphanet J Rare Dis. 2014;9:111. doi: 10.1186/s13023-014-0111-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Terryn W, Vanholder R, Hemelsoet D, Leroy BP, Van Biesen W, De Schoenmakere G, Wuyts B, Claes K, De Backer J, De Paepe G, Fogo A, Praet M, Poppe B. Questioning the Pathogenic Role of the GLA p.Ala143Thr “Mutation” in Fabry Disease: Implications for Screening Studies and ERT. JIMD Rep. 2013;8:101–108. doi: 10.1007/8904_2012_167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weidemann F, Krämer J, Duning T, Lenders M, Canaan-Kühl S, Krebs A, Guerrero González H, Sommer C, Üçeyler N, Niemann M, Störk S, Schelleckes M, Reiermann S, Stypmann J, Brand SM, Wanner C, Brand E. Patients with Fabry disease after enzyme replacement therapy dose reduction versus treatment switch. J Am Soc Nephrol. 2014;25:837–849. doi: 10.1681/ASN.2013060585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Levey AS, Stevens LA, Schmid CH, Zhang YL, Castro AF, 3rd, Feldman HI, Kusek JW, Eggers P, Van Lente F, Greene T, Coresh J, CKD-EPI (Chronic Kidney Disease Epidemiology Collaboration) A new equation to estimate glomerular filtration rate. Ann Intern Med. 2009;150:604–612. doi: 10.7326/0003-4819-150-9-200905050-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group KDIGO 2012 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease. Kidney Int Suppl. 2013;3:1–150. doi: 10.1038/kisup.2012.73. [DOI] [Google Scholar]
- 17.Amarenco P, Bogousslavsky J, Caplan LR, Donnan GA, Hennerici MG. Classification of stroke subtypes. Cerebrovasc Dis. 2009;27:493–501. doi: 10.1159/000210432. [DOI] [PubMed] [Google Scholar]
- 18.Whybra C, Kampmann C, Krummenauer F, Ries M, Mengel E, Miebach E, Baehner F, Kim K, Bajbouj M, Schwarting A, Gal A, Beck M. The Mainz Severity Score Index: a new instrument for quantifying the Anderson-Fabry disease phenotype, and the response of patients to enzyme replacement therapy. Clin Genet. 2004;65:299–307. doi: 10.1111/j.1399-0004.2004.00219.x. [DOI] [PubMed] [Google Scholar]
- 19.Giannini EH, Mehta AB, Hilz MJ, Beck M, Bichet DG, Brady RO, West M, Germain DP, Wanner C, Waldek S, Clarke JT, Mengel E, Strotmann JM, Warnock DG, Linhart A. A validated disease severity scoring system for Fabry disease. Mol Genet Metab. 2010;99:283–290. doi: 10.1016/j.ymgme.2009.10.178. [DOI] [PubMed] [Google Scholar]
- 20.Rolfs A, Martus B, Heuschmann PU, Grittner U, Holzhausen M, Tatlisumak T, Böttcher T, Fazekas F, Enzinger C, Ropele S, Schmidt R, Riess O, Norrving B, sifap1 Investigators Protocol and methodology of the stroke in young Fabry patients (sifap1) study: A prospective multicenter European study of 5,024 young stroke patients aged 18–55 years. Cerebrovasc Dis. 2011;31:253–262. doi: 10.1159/000322153. [DOI] [PubMed] [Google Scholar]
- 21.Desnick RJ, Allen KY, Desnick SJ, Raman MK, Bernlohr RW, Krivit W. Fabry’s disease: enzymatic diagnosis of hemizygotes and heterozygotes. J Lab Clin Med. 1973;81:157–171. [PubMed] [Google Scholar]
- 22.Mayes JS, Scheerer JB, Sifers RN, Donaldson ML. Differential assay for lysosomal alpha-galactosidases in human tissues and its application to Fabry’s disease. Clin Chim Acta. 1981;112:247–251. doi: 10.1016/0009-8981(81)90384-3. [DOI] [PubMed] [Google Scholar]
- 23.Allen RC, Zoghbi HY, Moseley AB, Rosenblatt HM, Belmont JW. Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am J Hum Genet. 1992;51:1229–1239. [PMC free article] [PubMed] [Google Scholar]
- 24.Shin S, Murray GJ, Kluepfer-Stahl S, Cooney AM, Quirk JA, Schiffmann R, Brady RO, Kaneski CR. Screening for pharmacological chaperones in Fabry disease. Biochem Biophys Res Commun. 2007;359:168–173. doi: 10.1016/j.bbrc.2007.05.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mehta J, Tuna N, Moller JH, Desnick RJ. Electrocardiographic and vectorcardiographic abnormalities in Fabry’s disease. Am Heart J. 1977;93:699–705. doi: 10.1016/S0002-8703(77)80064-1. [DOI] [PubMed] [Google Scholar]
- 26.Gambarin FI, Disabella E, Narula J, Diegoli M, Grasso M, Serio A, Favalli BM, Agozzino M, Tavazzi L, Fraser AG, Arbustini E. When should cardiologists suspect Anderson-Fabry disease? Am J Cardiol. 2010;106:1492–1499. doi: 10.1016/j.amjcard.2010.07.016. [DOI] [PubMed] [Google Scholar]
- 27.Brouns R, Thijs V, Eyskens F, Van den Broeck M, Belachew S, Van Broeckhoven C, Redondo P, Hemelsoet D, Fumal A, Jeangette S, Verslegers W, Baker R, Hughes D, De Deyn PP, BeFaS Investigators Belgian Fabry study: prevalence of Fabry disease in a cohort of 1000 young patients with cerebrovascular disease. Stroke. 2010;41:863–868. doi: 10.1161/STROKEAHA.110.579409. [DOI] [PubMed] [Google Scholar]
- 28.Desnick RJ, Brady RO. Fabry disease in childhood. J Pediatr. 2004;144:S20–S26. doi: 10.1016/j.jpeds.2004.01.051. [DOI] [PubMed] [Google Scholar]
- 29.Dubuc V, Moore DF, Gioia LC, Saposnik G, Selchen D, Lanthier S. Prevalence of Fabry disease in young patients with cryptogenic ischemic stroke. J Stroke Cerebrovasc Dis. 2013;22:1288–1292. doi: 10.1016/j.jstrokecerebrovasdis.2012.10.005. [DOI] [PubMed] [Google Scholar]
- 30.Ferraz MJ, Marques AR, Appelman MD, Verhoek M, Strijland A, Mirzaian M, Scheij S, Ouairy CM, Lahav D, Wisse P, Overkleeft HS, Boot RG, Aerts JM. Lysosomal glycosphingolipid catabolism by acid ceramidase: formation of glycosphingoid bases during deficiency of glycosidases. FEBS Lett. 2016 doi: 10.1002/1873-3468.12104. [DOI] [PubMed] [Google Scholar]
- 31.Echevarria L, Benistan K, Toussaint A, Dubourg O, Hagege AA, Eladari D, Jabbour F, Beldjord C, De Mazancourt P, Germain D. X-chromosome inactivation in female patients with Fabry disease. Clin Genet. 2016;89:44–45. doi: 10.1111/cge.12613. [DOI] [PubMed] [Google Scholar]