

Abstract
Xenografting primary tumor cells allows modeling of the heterogeneous natures of malignant diseases and the influences of the tissue microenvironment. Here, we demonstrate that xenografting primary chronic lymphocytic leukemia (CLL) B lymphocytes with activated autologous T cells into alymphoid mice results in considerable CLL B cell division and sizable T cell expansion. Nevertheless, most/all CD5+CD19+ cells are eventually lost, due in part to differentiation into antibody-secreting plasmablasts/plasma cells. CLL B cell differentiation is associated with isotype class switching and development of new IGHV-D-J mutations and occurs via an activation-induced deaminase-dependent pathway that upregulates IRF4 and Blimp-1 without appreciable levels of the expected Bcl-6. These processes were induced in IGHV-unmutated and IGHV-mutated clones by Th1-polarized T-bet+ T cells, not classical T follicular helper (Tfh) cells. Thus, the block in B cell maturation, defects in T cell action, and absence of antigen-receptor diversification, which are often cardinal characteristics of CLL, are not inherent but imposed by external signals and the microenvironment. Although these activities are not dominant features in human CLL, each occurs in tissue proliferation centers where the mechanisms responsible for clonal evolution operate. Thus, in this setting, CLL B cell diversification and differentiation develop by a nonclassical germinal center–like reaction that might reflect the cell of origin of this leukemia.
Xenografting chronic lymphocytic leukemia cells with activated T cells into alymphoid mice leads to diversification and terminal differentiation via an apparent non-classical germinal center-like reaction.
Introduction
Chronic lymphocytic leukemia (CLL) CD5+ B lymphocytes are frozen at the surface membrane Ig (smIg) stage of B cell maturation (1). Most CLL clones exhibit gene expression (2, 3) and sm (4) phenotypes resembling antigen-experienced B lymphocytes. The importance of antigen selection in CLL is highlighted by findings relating to the smIg expressed by the leukemic cells. These include use of specific Ig heavy chain variable (IGHV) genes that differs from that of B cells from healthy subjects (5), dichotomy among patients in the presence of IGHV somatic hypermutation (SHMs) (5), and the presence of remarkably similar VH and VL CDR3s, often due to association of specific IGHVs with IGHD and IGHJ segments (6), referred to as stereotyped B cell receptors (BCRs) (7). Each of these parameters can identify patients with more severe clinical courses and outcomes (1), as can expression of CD38 (4), CD49d (8), and ZAP-70 (9), and the presence of cytogenetic (10) and molecular (11) abnormalities.
Although recent studies suggest that CLL originates from the human equivalent of murine B-1a cells (12) or from subsets of human CD5+ B lymphocytes (13), it is still controversial whether different disease subgroups originate from a distinct or common B cell subtype and at what B cell developmental stage transformation begins and completes (14). Adding to this complexity is the interplay of CLL cells with nonleukemic cells within the microenvironments in the BM, lymph nodes (LNs), and spleen (15), where the main tumor burden exists. Only a small fraction of CLL cells divide (16), occurring principally in “proliferation centers” of primary and secondary lymphoid tissues (17), where contact with antigen (18) and other elements, including T cells (19, 20), occurs.
Due to this underlying heterogeneity and complexity, there is no genetically modified animal model that recapitulates all features of CLL. This has created interest in xenogeneic transfers utilizing primary patient material. We have shown that transferring patient-derived peripheral blood (PB) cells into NOD/Shi-scid,γcnull (NSG) mice leads to reproducible engraftment and proliferation of CLL cells only if concomitant T cell activation occurs (21). Although this model faithfully recapitulated many aspects of the disease, CLL B cell engraftment did not persist long-term due, in part, to the development of graft versus host disease (GvHD) promoted by the presence of human antigen-presenting cells allogeneic to patient T and B cells; this led to the loss of B lymphocytes and premature death of recipient animals (21). Recently, we improved this model by using only CLL cells (thereby eliminating human vs. human GvHD) and by activating autologous T cells in vitro prior to transfer with CLL cells (22). This leads to CLL B cell engraftment and expansion at levels at least equivalent to our initial report.
Despite these improvements, however, CLL B cell engraftment still does not persist long-term. Here, we show that this is the consequence, at least in part, of leukemic B cell maturation to plasmablasts/plasma cells (PCs). Differentiation is associated with IGH-class switch recombination (CSR) and the development of new IGHV mutations, even in IGHV-unmutated (U-CLL) clones. Notably, this occurs via a maturation pathway that involves activation-induced cytosine deaminase (AID), as well as IRF4 and Blimp-1, but not appreciable levels of Bcl-6. Finally, these processes are induced by Th1-polarized, T-bet+ T cells either acting on a subset of initially T-bet+ CLL B cells or inducing T-bet in the responding leukemia cells.
Results
Transfer of CLL B cells into NSG mice can lead to terminal differentiation.
Supplemental Figure 1 (supplemental material available online with this article; doi:10.1172/jci.insight.86288DS1) illustrates the details of the xenograft system. In brief, spleens from 70 mice, each having received approximately 20 × 106 peripheral blood mononuclear cells (PBMCs) from 1 of 13 CLL patients (6 U-CLL, and 7 IGHV-mutated [M-CLL]), along with 0.5 × 106 in vitro–activated autologous T cells, were analyzed at least 28 days after cell transfer (median mice/sample, 4; range, 3–10; median assessment time after transfer, 38 days; range, 29–105 days; Table 1). Analysis of PB at euthanasia revealed circulating human T cells in 75.7% (51/70) of mice (Table 1).
Table 1. Characteristics of CLL samples before and after xenograftingA.

Flow cytometry (FC) analysis of dispersed spleen cells from these same mice identified, in addition to CD3+ cells, 2 sets of human (h) CD45+CD19+ cells that were present in varying proportions depending on the sample and the time of analysis. One population resembled the originally transferred CLL cells in size and phenotype (CD5+CD19+), while the second was larger and exhibited intense smCD38 expression (CD38++), along with lower mean densities of smCD19 and smCD5 (Figure 1A). A subset of these cells also expressed CD138.
Figure 1. Transfer of CLL B cells into NSG mice can lead to terminal differentiation of cells into plasmablasts/plasma cells.

(A) Analysis of mCD45–hCD4–hCD8– cells from spleens reveals a hCD45+ subpopulation strongly expressing CD38 (CD38++) that are larger but with less CD19 (MFI: CD38++ = 1,178 vs. CD38– = 7,157) and CD5 (MFI: CD38++ = 8,508 vs. CD38– = 9,763) intensities. A CD38++ subset expresses CD138 (19.7%). When FACS-sorted, this population (*) used the clonal, patient-specific IGHV-D-J rearrangement. (B) Representative immunohistology (IH) of a CD20+PAX5+ perivascular aggregate (PVA). Arrow identifies vessel. Scale bar: 250 μm. (C) Representative IH of human IgM, IgG, Igκ, and Igλ in a CD20+PAX5+PVA. Scale bar: 250 μm. (D) Igκ staining of area indicated by arrow in C showing denser Ig at the CD20+PAX5+PVAs rims. H&E staining reveals a plasmablast/plasma cell (PC) morphology. Scale bar: 10 μm. (E) Representative H&E and IH of area with cells having PC morphology shows expression of CD38, PC-marker VS38c, and CD138 in a subset of cells. Scale bar: 50 μm. (F) Representative immunofluorescence staining of a CD20+PAX5+PVA rim, as indicated by arrows in C. Blue, nuclear stain; red, CD20; and green, Igκ. Scale bar: 10 μm. Preceding data derived from 13 chronic lymphocytic leukemia (CLL) cases in 13 independent experiments involving 51 mice with T cell expansion (Table 1). m, murine; h, human; MFI, mean fluorescence intensity; NSG, NOD/Shi-scid,γcnull; PVA, perivascular aggregate.
Immunohistology (IH) showed aggregates of CD20+ cells that also displayed the pan–B cell marker PAX5. Since these aggregates were always localized around blood vessels (Figure 1B), as reported (21), we hereafter refer to these perivascular aggregates as CD20+PAX5+ perivascular aggregates (PVAs).
By IH, CD20+PAX5+PVAs contained cells with the same L chain isotype as the original CLL clone (Igκ in 9 and Igλ in 4 of 13 cases; Table 1 and Figure 1, C and D). More intensely stained Ig+ cells were also identified, often at the rims of CD20+PAX5+PVAs or near other vascular structures (Figure 1C, arrows); both IgM+ and IgG+ cells were seen (Figure 1C). High-power views indicated that these cells had PC morphology (Figure 1, D and E), with intense cytoplasmic Ig (cIg) staining (Figure 1D); were strongly positive for CD38 and the PC marker VS38c; and were variably stained with anti-CD138 (Figure 1E).
Immunofluorescent imaging of cells at the margins of PVAs confirmed large amounts of cIg (Figure 1F) and demonstrated reduced smCD20 compared with CD20+ cells located within CD20+PAX5+PVAs (not shown). In a subset of experiments (8/13), the presence of cells with a differentiated phenotype (determined by FC and/or IH) correlated with plasma IgM and IgG in recipient mice with T cell expansion (Table 1). No obvious associations existed between the generation of PCs and clinically relevant prognostic factors (Table 1).
Derivation of PCs from the CLL B cell clone.
The CLL B cell clone, and not other B cells in the transferred PBMCs, was the PC precursor. In all instances, only one L chain was expressed in the spleen-residing PCs, and this matched the L chain of the injected CLL cell (Figure 1C). Second, IGHV-D-J cDNA sequence analyses of FACS-sorted CD5+CD38++CD138+ cells from NSG spleens revealed the patient-specific rearrangement (Figure 1A, marked with *). Finally, CD5+CD38++CD138+ cells were found after adoptive transfer of highly purified, FACS-sorted CD5+CD19+ cells, and these exhibited the appropriate Ig L chain and the clonal, patient-specific rearrangement when studied (not shown).
CLL-derived PCs and plasma Ig become apparent after leukemic B cells have undergone many divisions.
The temporal relationship between CLL-cell xenografting and PC appearance was assessed in splenic tissue at days 3 and 7 after transfer and then at weekly intervals thereafter (n = 2 cases, U-CLL1122 and M-CLL1164, in independent experiments; for each, 30 recipients, with euthanasia of 5 at each time point; Table 1 and Supplemental Table 2). Appreciable numbers of CD5+CD19+CD38++ cells were found by FC from week 3 onward (Figure 2A; 1-way ANOVA test; % CD38++ week 2 = 1.9% vs. week 3 = 32.4%, P < 0.05; vs. week 4 = 51.4%, P < 0.0001; vs. week 5 = 38.0%, P < 0.01).
Figure 2. CLL-derived plasma cells and plasma Ig only become apparent after leukemic cells have undergone many divisions.

(A) Human (h) CD45+CD5+CD19+CD38++ cells are only identified in spleen-residing cells 21 days after chronic lymphocytic leukemia (CLL) cell transfer. Cells increase in number afterward. Mean and SEM shown; 1-way ANOVA; n = 60 mice. (B) Representative FC plots of CFSE-labeled hCD45+CD5+CD19+ cells in spleen following transfer of U-CLL1122 (upper) and M-CLL1164 (lower). Note development of CD38++ phenotype with progressive division. (C) hCD45+CD5+CD19+ cells with a CD38++ phenotype are only apparent after >60% of CLL cells have undergone ≥6 divisions. Curve shows exponential relationship (r2 = 0.76; n = 60). (D) Plasma Igs are not detectable until CD38++ cells appear. Mean μg/ml and SEM shown for IgM and IgG; n = 60 mice. Above results obtained from 5 mice euthanized at each time point in 2 independent experiments involving U-CLL1122 and M-CLL1164 (total mice = 60; Table 1 and Supplemental Table 2). U-CLL, CLL clone with IGHV sequence differing ≤2% from most similar germline gene; M-CLL, CLL clone with IGHV sequence differing >2% from most similar germline gene; FC, flow cytometry.
Next, we assessed the relationship between development of CD38++ cells and CLL B cell division determined by CFSE dilution. The development of CD38++ cells required multiple divisions. In Figure 2B, each plot represents a different mouse, showing that, as cells divide, higher levels of CD38 are achieved. High percentages of CD5+CD19+CD38++ cells were only found after the majority of CD5+CD19+ cells had divided at least 6 times (Figure 2C).
Moreover, appreciable plasma Ig levels were detected after finding splenic CD5+CD19+CD38++ cells (Figure 2D). From week 3 onward, plasma IgM and IgG levels correlated with the percent of CD5+CD19+CD38++ cells (Spearman r = 0.61 and 0.78, respectively; P < 0.0001). Notably, the levels of IgG were higher than IgM at all 3 time points.
Expression of AID correlates with CSR and new IGHV-D-J mutations.
Because CLL cells maturing to antibody-secreting cells produced switched Igs, despite 12 of 13 clones being IgM+ (Table 1), we searched for AID protein, the enzyme necessary for CSR and SHM (23). FC analysis of spleen-residing CLL B cells demonstrated increasing levels of AID as division progressed, with a significant difference between undivided and highly divided (>5) cells (Figure 3A). IH further confirmed the presence of AID protein in CD20+PAX5+PVAs from both U-CLL and M-CLL clones (Figure 3B). AID+ cells ranged from 5%–50% (Figure 3C illustrates examples of AID protein extremes).
Figure 3. Dividing CLL B cells express activation-induced cytosine deaminase (AID) protein that correlates with class switch recombination (CSR).

(A) AID expression in spleen-residing human (h) CD45+CD5+CD19+ cells increases as cells divide. Representative FC of single cell splenic suspensions. Significantly higher AID protein is found in the most divided chronic lymphocytic leukemia (CLL) B cells. Mean and SEM shown. Data from 3 independent experiments involving 12 mice; mice euthanized between days 14 and 28; Mann-Whitney test result. (B) AID+ cells are localized in CD20+PAX5+PVAs. ×20 images of CD20, PAX5, and AID; scale bars: 125 μm. (C) ×60 images of AID in areas indicated by arrows in B for U-CLL1122 and M-CLL1164 showing approximately 50% and 5% AID+ cells, respectively. Scale bar: 50 μm. For B and C, representative data of mice sampled from 11 independent experiments showing approximately highest and lowest extremes of AID+ cells identified in vivo. (D) Switching to IgG becomes evident after CLL B cells have divided multiple times. FC and matching IH of CFSE-labeled CD5+CD19+ cells for IgM and IgG. Undivided cells produce only IgM and not IgG (upper), whereas multiply divided CLL B cells make both isotypes (lower). Representative data of mice sampled from 5 independent experiments. Scale bar: 50 μm (E). FC confirms splenic-residing hCD45+CD5+CD19+ cells undergo CSR. hCD5+CD19+ cells with only minimal division (day 14) do not express smIgG, whereas CD5+CD19+ cells from mice receiving the same CLL clone express smIgG after multiple divisions (day 28). Representative data of mice sampled from 3 independent experiments. U-CLL, CLL clone with IGHV sequence differing ≤2% from most similar germline gene; M-CLL, CLL clone with IGHV sequence differing >2% from most similar germline gene; FC, flow cytometry; IH, immunohistology; sm, surface membrane; MFI, mean fluorescence intensity.
Splenic tissue assessment early after transfer indicated that switched Ig+ cells were not apparent by IH in those transfers where no/minimal CLL division occurred (Figure 3D, upper panel); this was in marked contrast to transfers in which cells divided multiple times (Figure 3D, lower panel). Parallel FC studies confirmed that smIgG was present only on CD5+CD19+ cells that had divided several times (Figure 3E). Reverse-transcription PCR (RT-PCR) and cDNA sequencing indicated that the switched transcripts were of original patient origin (not shown).
Using a standard 40-cycle PCR (24), we detected switched transcripts in only 37.5% of the original patients’ samples prior to transfer. Therefore, since the majority of samples did not exhibit evidence for CSR prior to cell transfer, isotype switching most often occurred after xenografting.
Next, we looked for another hallmark of AID’s action, SHM. Ultra-deep sequencing of human IGHV-D-J cDNAs from mice that received cells from 7 patients (4 U-CLL, 3 M-CLL) was carried out. Sequence reads that occurred only once were discarded and, of the remaining sequences, any that were found in the pretransfer sample were also discarded. This left only IGHV-D-J rearrangements with new mutations occurring in vivo. Using this approach, new IGHV-D-J mutations were found in every instance (Table 2). These mutations occurred in IgM at frequencies ranging from 24.01 × 10–4 to 1.07 × 10–4. Remarkably, mutation frequencies did not differ between U-CLL and M-CLL samples (P = 0.2844). For 3 samples (U-CLL1122, U-CLL1279, M-CLL1164), IgM, IgG, and IgA cDNAs were sequenced (Table 2), allowing a comparison of mutation frequencies in IgM, IgG, and IgA subclones. Pair-wise comparison (9 IgM vs. 5 IgG + 1 IgA) indicated that the frequency of newly acquired mutations was higher in isotype-switched than companion IgM subclones (P = 0.0361).
Table 2. New IGHV-D-J mutations in xenografted CLL B cells exhibit hallmarks of AID action.
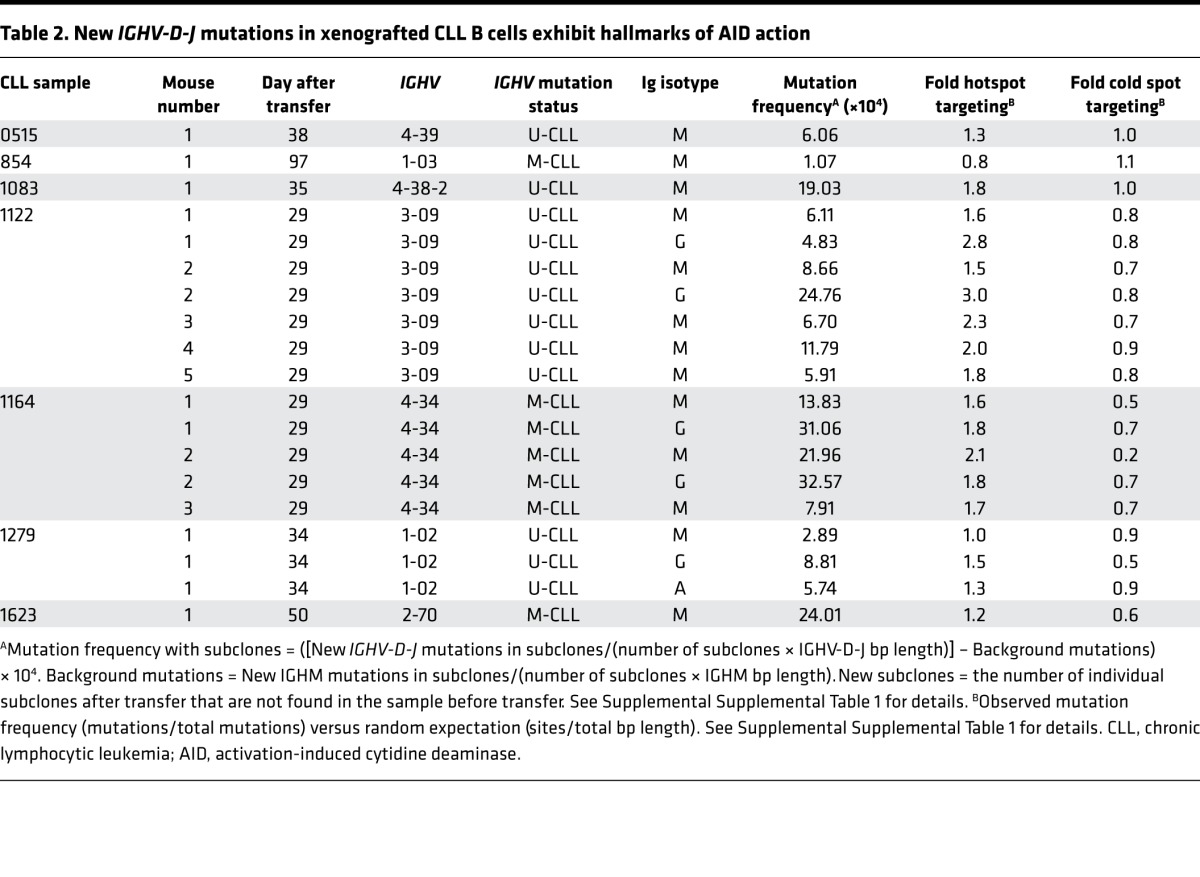
AID preferentially targets cytidines (C) within specific nucleotide motifs (WRC), referred to as “AID hotspots”, and avoids other motifs (SYC), “AID coldspots” (25). New mutations were focused preferentially at AID hotspots in 18 of 19 NSG recipients of 6 of 7 CLL clones (Table 2). This hotspot versus coldspot targeting comparison was significant (P = 0.0002; Figure 4A, left), as was the case for IgM+ (P = 0.0007; Figure 4A, center) and for switched (IgG+ + IgA+) transcripts (P = 0.0355; Figure 4A, right).
Figure 4. Xenografted chronic lymphocytic leukemia (CLL) B cells demonstrate clonally related IgM and IgG IGHV-D-J mutations that exhibit hallmarks of AID action.

(A) AID hotspot and coldspot mutation frequencies were calculated from new IGHV-D-J mutations in 19 xenografted samples and log2 transformed for statistical analysis by Wilcoxon test (P = 0.0002, left). This indicated that AID hotspot mutation frequencies are significantly higher. The same was observed among IgM subclones (n = 13; P = 0.0007, center) and among Ig isotype-switched subclones (n = 6; P = 0.0355, right). (B) Representative phylogenetic relationships of new subclones are illustrated using a polar tree layout, with each branch tip representing a distinct subclone. The length that each branch extends from the circle is roughly proportional to the number of mutations (K80 phylogenetic distance, scale bar: 0.03). Subclones with single mutations are indicated by branch tips extending a short distance from the circle, while multiple-mutated subclones are represented by tips extending farther out. The branches connecting each tip illustrate the sequence-relatedness between subclones, with closely related sequences clustered together. Total number of subclones, defined by changes in DNA sequence from that of the initial clone and from those of subclones present in vivo at the time of sampling can be determined by counting the number of terminal branch tips at various lengths from the circle’s center. U-CLL515-1 IgM subclone IGHV-D-J sequences describe a tree with large multibranched relationships (386 subclones with 1–25 mutations). M-CLL1623-1 IgM subclone IGHV-D-J sequences define a tree with few branched relationships (91 subclones with 1–7 mutations).(C) Comparison of IgM and IgG phylogenetic relationships from same sample are shown as in B. IgM and IgG relationships are shown for U-CLL1122-1 (173 subclones with 1–26 mutations and 225 subclones with 1–28 mutations, respectively) and M-CLL1164-2 (135 subclones with 1–35 mutations and 144 subclones with 1–32 mutations, respectively) with shared sequences indicated as red branches. U-CLL, CLL clone with IGHV sequence differing ≤2% from most similar germline gene; M-CLL, CLL clone with IGHV sequence differing >2% from most similar germline gene.
Finally, the occurrence of new shared IGHV-D-J mutations permitted definition of phylogenies among subclones. Multiple clonally related sequences, some with considerable and others with minimal branching, were found (Figure 4B). In some instances, these phylogenies exhibited extensive progression, suggesting selection by antigen(s) in vivo (Figure 4B, U-CLL515-1). This was the case for IgM- and IgG/IgA-expressing subclones from both U-CLL and M-CLL (Table 2 and Figure 4C).
Hence, AID protein and its functional consequences (CSR and SHM) were found in all CLL xenografts, regardless of the original IGHV mutation status of the leukemic clone.
CLL B cell differentiation and diversification occur in the absence of appreciable levels of Bcl-6 protein.
AID is most intensively expressed during germinal center (GC) reactions that classically occur in anatomic structures within secondary lymphoid tissues (26–28). At these sites, centroblasts coexpress AID and Bcl-6 in the absence of IRF4 (29), and this combination promotes SHM (30). Without Bcl-6, classical GC reactions do not occur (31).
To assess the relationships between AID, Bcl-6, and IRF4, we first determined Bcl-6 protein expression by IH in the original 13 xenografted samples (Table 1). Compared with tonsillar GC B cells that strongly coexpress AID and Bcl-6 in the majority of cells (Figure 5A), Bcl-6 protein was never identified in ≥1% of cells within CD20+PAX5+PVAs (Figure 5, A and B), despite the concomitant synthesis of AID. Indeed, Bcl-6 levels were less than levels seen in tonsil T cell areas (Figure 5B).
Figure 5. CLL B cell diversification and differentiation occurs without appreciable levels of Bcl-6 protein but in the presence of increasing levels of T-bet protein.

(A) Bcl-6+ cells are rarely found in CD20+PAX5+PVAs and are found less than in tonsillar T cell areas. Human tonsil shows Bcl-6+ cells within germinal centers (GCs) (arrow), whereas chronic lymphocytic leukemia (CLL) cells within CD20+PAX5+PVAs (*) are Bcl-6–. Conversely, AID+ cells are found on serial sections in both tissues. Scale bar: 250 μm. (B) High-power views of Bcl-6 staining in tonsillar GC, tonsillar T cell area, and xenografted CLL cells within CD20+PAX5+PVA. Scale bar: 50 μm. For A and B, representative data of mice sampled from 13 independent experiments. (C) Representative FC indicates that ≤1% Bcl-6+ cells are found among CD5+CD19+ cells before and after xenografting. This is compared with Ramos cells and from human tonsil cells (CD19+ only). Data are representative of 7 CLL samples analyzed pretransfer and 5 independent xenograft experiments sampling 41 mice. (D) Bcl-6 production does not appear/change upon commencement of B cell division. In contrast, T-bet expression within CD20+PAX5+PVAs becomes more intense. Analysis of CLL transfers at 3 days, 2 weeks, and 3 weeks. Each shows FC of CFSE-labeled CD5+CD19+ cells with companion IH images for PAX5, Bcl-6, CD3, and T-bet. Representative FC and IH images from 5 mice euthanized at each time point in 2 independent experiments involving U-CLL1122 and M-CLL1164. Scale bar: 50 μm. U-CLL, CLL clone with IGHV sequence differing ≤2% from most similar germline gene; M-CLL, CLL clone with IGHV sequence differing >2% from most similar germline gene; PVA, perivascular aggregate; FC, flow cytometry; IH, immunohistology; NSG, NOD/Shi-scid,γcnull; AID, activation-induced cytidine deaminase.
Because this result was unexpected, we analyzed by FC expression of Bcl-6 protein in CLL cells prior to and following transfer to assure that synthesis of Bcl-6 was not missed temporally. Analyses of 7 samples prior to cell transfer indicated the presence of Bcl-6 protein in 0.5% (median; range, 0.1%–3.7%) of CD5+CD19+ cells (Figure 5C).
Using cells from U-CLL1122 and M-CLL1164, we assessed Bcl-6 protein levels by IH after xenografting at days 3 and 7 and at weekly intervals thereafter (30 recipients each, with euthanasia of 5 at each time point; Table 1 and Supplemental Table 2). Subsequently, we focused on the early periods after transfer (days 3, 7, and 14, M-CLL0827 and M-CLL1024; for all 15 recipients, with euthanasia of 5 at each time point, Supplemental Table 2), this time examining cells by both FC and IH. Only 0.5% of cells (median; range, 0%–4%) were found to express Bcl-6 by FC, regardless of the time of testing (Figure 5, C and D). This finding was confirmed using 2 additional anti–Bcl-6 mAbs in other IH studies (Supplemental Figure 2). Thus, appreciable levels of Bcl-6 protein were never documented for 15 CLL patient samples in a total of 117 mice (Table 1 and Supplemental Table 2), regardless of the time of euthanasia (i.e., days 3–105), the extent of division of CLL B cells (0 to ≥6), or the presence or absence of differentiated cells. In addition, Bcl-6 was not seen in other cells in or around the PVAs by IH or FC, including T lymphocytes.
In contrast, IRF4 expression was detectable in cells from NSG spleens at all stages of CLL B cell division and maturation, although the numbers of IRF4+ cells and the intensity of protein expression varied for and within samples. Dim expression was observed in a limited number of cells within CD20+PAX5+PVAs prior to appreciable CLL division (Figure 6A, upper panels) but became intense upon CD5+CD19+ cell replication (Figure 6A, lower panels). In those cases that demonstrated extensive CLL B cell maturation, IRF4 was visualized both within and outside of CD20+PAX5+PVAs (Figure 6B, arrow and asterisk); less intense expression was seen in CD20+ cells, and stronger expression was seen in cIg+ cells. Finally, terminal differentiation of cIg+ and CD138+ cells was confirmed by coexpression of both IRF4 and the PC master regulator Blimp-1 (Figure 6C).
Figure 6. IRF4 and Blimp-1 protein are expressed during CLL B cell differentiation and diversification.

(A) Upper panels show FC of undivided CFSE-labeled human (h) CD45+CD5+CD19+ cells with companion IH images of IRF4 and PAX5 (day 3). Lower panels show FC of dividing CFSE-labeled hCD45+CD5+CD19+ cells with companion IH images of IRF4 and PAX5 (day 14). Scale bar: 50 μm. Representative data of mice sampled from 5 independent experiments. (B) IRF4 staining is seen within (arrow, upper left) and outside of (*, upper left) CD20+PAX5+PVAs, being expressed dimly by CD20+ cells and more intensely by cIg+ cells. Upper panels: dual IH images of CD20 (black) and IRF4 (brown); left hand panel (scale bar: 250 μm), with high-power view of area marked with arrow on right (scale bar: 50 μm). Lower panels: dual IH images of Igκ (black) and IRF4 (brown); left hand panel (scale bar: 250 μm), with high-power view of area marked with arrow on right (scale bar: 50 μm). Representative data of dual IH performed on mice sampled from 5 independent experiments. (C) Dual IH of spleen-residing cells 6 weeks following transfer indicates chronic lymphocytic leukemia (CLL) plasmablasts/plasma cells express IRF4 and Blimp-1. Scale bar: 10 μm. Representative data of mice sampled from 6 independent experiments. FC, flow cytometry; PVA, perivascular aggregate; IH, immunohistology; cIg, cytoplasmic immunoglobulin.
Th1 cells are involved in CLL B cell division and maturation.
The phenotype of CD4+ T cells at the time of xenografting and at euthanasia was studied, focusing on the possibility that these were Tfh cells (32). FC analysis of anti-CD3/28 bead-activated CD4+ cells at the time of transfer showed high numbers of ICOS+ (~90%) and CXCR5+ (~45%) cells. However, at the time of euthanasia, negligible numbers of CD4+CD5+CD19–ICOS+ (<1%) and CD4+CD5+CD19–CXCR5+ (~2%) cells were detected in recipient spleens. However, CD57+ cells (number and density) and PD1+ cells (density) were increased (Figure 7A). In line with this, cells within CD20+PAX5+PVAs exhibited weak to absent ICOS expression and more appreciable levels of PD1 and CD57 by IH than those in tonsillar GCs (Figure 7B). The paucity of ICOS and CXCR5 expression, along with the previous inability to identify Bcl-6 in T (and B) cells (Figure 5), indicated that these CD4+ T cells were not Tfh cells (32).
Figure 7. Th1 cells are involved in CLL B cell division and maturation.

(A) FC plots of pretransfer in vitro–activated chronic lymphocytic leukemia (CLL) CD4+ cells (upper) compared with spleen-residing CD4+ cells (lower) stained for ICOS, CXCR5, CD57 and PD1. Red, isotype control; blue, CD5+CD4+ cells. Numbers represent percentage of cells expressing each surface protein over isotype control. (B) IH of human tonsil and CLL cells from NSG spleen stained for ICOS, CD57, and PD1. Scale bar: 250 μm. Representative IH and FC from 5 (PD1), 6 (CD57), and 5 (ICOS) independent experiments. (C) IFNγ is the most frequent and abundant T cell–derived cytokine, and its levels correlate with T cell numbers and degree of expansion in mice. Better correlation exists for IFNγ than for IL-5. Data from 7 independent experiments, n = 77 mice. IFNγ becomes detectable (>10 pg/ml) 2 weeks following cell transfer and increases thereafter. Data from 30 mice for U-CLL1122, with 5 mice euthanized at each time point. Representative of 2 independent experiments. Dotted line indicates plasma level of 10 pg/ml. (D) Spleen-residing CD4+ cells express IFNγ from 2 weeks after transfer. Data from 2 independent experiments with 2 CLL cases (M-CLL0827 and M-CLL1024), 5 mice euthanized per case at 1 and 2 weeks after transfer, unpaired t test result, mean and SEM shown. Red, isotype control; blue, CD5+CD4+IFNγ+ cells. (E) T-bet expression increases in CD4+ cells and B cells and is maintained in CD8+ cells following xenografting. Pretransfer data from 8 CLL cases subsequently xenografted in 8 independent experiments. Wilcoxon test result. At euthanasia, all cases showed evidence for plasma cell maturation. U-CLL, CLL clone with IGHV sequence differing ≤2% from most similar germline gene; M-CLL, CLL clone with IGHV sequence differing >2% from most similar germline gene; FC, flow cytometry; IH, immunohistology; NSG, NOD/Shi-scid,γcnull.
Tfh cells produce large amounts of IL-4 and IL-21 and lesser amounts of IL-2, TNFα, and IFNγ (32). We tested plasmas of 77 mice from 7 experiments with different CLL clones for IL-2, IL-4, IL-5, IL-10, TNFα, and IFNγ. IL-2 and IL-4 levels were undetectable, and those of IL-10 and TNFα were very low to negligible (not shown). This contrasted with IFNγ, which was detected in 59% of plasmas and was the most abundant cytokine (mean 434 pg/ml; Figure 7C, left graph). IL-5 was also found, albeit in <30% of plasmas analyzed and at concentrations ~5× less than IFNγ (mean 78.2 pg/ml; Figure 7C, center graph). IFNγ levels closely correlated with the numbers of spleen-residing T cells (Spearman r = 0.7835, P < 0.0001; Figure 7C); a weaker correlation was present for IL-5 (Spearman r = 0.5699, P < 0.0001; Figure 7C). Furthermore, IFNγ levels were found elevated 3 weeks after cell transfer (Figure 7C, right graph), associating with the time when T cells had undergone ≥6 divisions. Importantly, a significant increase in the percentage of spleen CD4+IFNγ+ cells was identified by FC between weeks 1 and 2 (1.6% vs. 26.2%, P = 0.0002; Figure 7D). Finally, analyzing a separate set of plasmas from 30 mice injected with 2 different CLL clones, IL-21 was not detected (not shown).
Because T cells expanding in recipient mice exhibited a ICOS–CXCR4–Bcl-6– phenotype and plasma did not contain appreciable levels of IL-4 and/or IL-21 but large amounts of IFNγ, we examined CLL T lymphocytes for synthesis of the Th1 master regulator, T-bet. Prior to cell transfer, a small subset of CD4+ and a larger subset of CD8+ cells expressed T-bet (median = 9% and 64.4%, respectively; n = 8). However, at euthanasia, the percent of T-bet+ cells was significantly higher in CD4+ cells and was maintained in CD8+ cells (median = 54.2% [P = 0.0156] and 74.5% [NS], respectively; n = 8; Figure 7E). Thus, T cell phenotype, including T-bet expression, and plasma cytokine profiles indicated that the predominant population of T cells was Th1-polarized (33).
T-bet is also expressed by CLL B cells within CD20+PAX5+PVAs.
In the process of analyzing T-bet in T lymphocytes, we found that maturing CD5+CD19+ cells displayed T-bet by FC (Figure 8A), and T-bet intensity in these cells was at least equal to that in CD4+ and CD8+ cells. Of note, only a few CLL B cells expressed T-bet before xenografting (median = 1.8%), which increased significantly at euthanasia (median = 89.6%, P = 0.0078, n = 8; Figure 7E). Correspondingly, CD20+PAX5+PVAs were universally T-bet+ (mice = 33; median number of mice/patient = 3; Figure 8B) by IH. In contrast, T-bet was weak to absent in tonsillar GCs (Figure 8B) with a T-bet protein intensity in PVAs comparable with cells in the tonsillar T cell zone (Figure 8, B and C). Dual-color IH confirmed that T-bet was found principally in CD20+ cells of CD20+PAX5+PVAs, although some CD20– cells were also T-bet+ (Figure 8D).
Figure 8. Both B and T cells within CD20+PAX5+ perivascular aggregates (PVAs) express T-bet protein.

(A) T-bet is expressed by spleen-residing chronic lymphocytic leukemia (CLL) T cell and B cell populations at euthanasia. Data from 9 independent experiments with 9 CLL clones. Mean and SEM shown. (B and C) T-bet expression localizes to CD20+PAX5+PVAs and is of similar intensity to that in tonsillar T cell areas. (B) Human (h) CD20+, hCD3+, and hT-bet+ cells from NSG spleen. T-bet is also shown in tonsil. Scale bar: 250 μm. (C) High-power images of T-bet+ cells in a CD20+PAX5+PVA (area marked with * in B) and in the T cell area of tonsil (area marked with § in B). Scale bar: 250 μm. Spleen data representative of 13 independent experiments (n = 33 mice). (D) Dual IH shows that CD20+ and CD20– cells within CD20+PAX5+PVAs are T-bet+. Left panel: CD20 (black) and T-bet (brown) protein in CD20+PAX5+PVA. Scale bar: 250 μm. Right panel: High-power view of area marked by arrow. Scale bar: 50 μm. NSG, NOD/Shi-scid,γcnull; PVA, perivascular aggregate; IH, immunohistology; MFIR, mean fluorescence intensity ratio.
To determine if T-bet expression in CLL cells changed over time, we compared protein levels within CD20+PAX5+PVAs and related these to the extent of CD5+CD19+ cell division (Figure 5D). T-bet expression changed from initially dim to marked, as leukemia cells began to divide (Figure 5D); as noted above, no such change in expression was seen for Bcl-6 in the same cell populations (Figure 5D).
Discussion
Here, we document that CLL B cells have the potential to undergo CSR, SHM, and differentiation to antibody-secreting cells in vivo when given “T cell help” and a supportive microenvironment. Although our xenografting system exaggerates the extent to which these processes occur in the disease setting, each process occurs in patients, usually within proliferation centers where the actions responsible for clonal expansion and disease evolution happen. Thus, our results provide insights into the latent activities of CLL B and T cells, imply mechanisms that execute (or inhibit) them, and suggest effects for patients that may come about from inducing them.
CLL B cell differentiation to antibody-secreting PCs was manifested by phenotypic and functional changes that occur with terminal B cell maturation: appearance of large cells expressing surface markers of plasmablasts/PCs (Figure 1) and synthesis and secretion of Igs (Figure 1 and Figure 2). These findings are consistent with certain CLL clones differentiating and secreting Ig naturally in vivo (34) and being inducible to do the same in vitro (35–37). However, whereas the frequency and degree of terminal B cell maturation occurring in vivo and in vitro varies among patient clones, in these transfer studies, all CLL clones undergo this process if T cell help is provided (Table 1 and Figure 7). Notably, we did not observe a difference among CLL clones differing in IGHV mutation status, CD38 expression, or genomic abnormalities (Table 1).
CLL B cell maturation to PCs involves the synthesis of AID protein in a sizable fraction of leukemic B cells from all samples studied (~5%–50%, Figure 3, A–C). AID protein appears when CSR (Figure 3, D and E) and acquisition of new IGHV-D-J mutations that localize to AID hot spots (ref. 25, Table 2, and Figure 4) develop. Notably, these IGHV-D-J mutations occur in both U-CLL and M-CLL B cells, indicating that SHM can occur in all leukemia cells if they receive sufficient costimulation in a supportive microenvironment. Finally, the presence of AID protein associates with increasing synthesis of IRF4 (Figure 6) and eventually Blimp-1 (Figure 6), as expected (38, 39),
Surprisingly, the maturation process that upregulates AID, IRF4, and Blimp-1 does not induce appreciable levels of Bcl-6 protein (Figure 5), which is required for standard follicular maturation (30). Although primary CLL cells express Bcl-6 mRNA and can be the target of SHM (13, 40), Bcl-6 protein production is negligible in xenografts despite the occurrence of leukemia-cell diversification and differentiation (Figure 5).
In several ways, the maturation process we observed here resembles extrafollicular reactions described in autoimmune mice (41) and humans (42), where SHM occurs without typical GC-type structures. Murine B lymphocytes responding to Salmonella typhimurium infection in vivo undergo a similar process (43). Like our findings with CLL B cells, this latter response involves extensive and rapid CSR and SHM that suggest antigen selection and affinity maturation, based on genealogic trees and improved binding of mAbs to Salmonella target antigens. Although we have not tested the CLL Abs diversifying in our system for antigen reactivity, the presence of phylogenies of varying sizes (Figure 4) suggests antigen-selection and affinity maturation, possibly for (auto)antigens expressed in recipient mice. At this point, a need for Bcl-6 in extrafollicular diversification and differentiation responses has not been documented, and our findings would suggest the opposite. Of relevance, T cells can have a facultative role in extrafollicular responses (44, 45).
Maturation of CLL B cells to PCs requires activated T cells with a Th1 phenotype (Figure 7). In the human disease, requirement for T cell activation and help is inferred by leukemia cell division occurring in proliferation centers of secondary lymphoid tissues and BM (17) among other elements such as activated T lymphocytes (19, 20). Furthermore, the absence of T cell help (46, 47) may explain the blocked maturation in patients and its lack of occurrence in most cells of a CLL clone and in all patients. Our observation that, without in vivo expansion of T cells, CLL B cells neither proliferate nor differentiate in vivo (Table 1, Figure 7, and ref. 21) and the finding that only with T cells can normal B cells developing from xenografts of healthy human CD34+ cells acquire a mature phenotype and become Ig-secreting cells (48) support this possibility. Our studies, however, do not indicate T cell cognate recognition and interactions as mechanisms for B cell maturation, as suggested by others (49), and a cytokine-mediated (e.g., IFNγ) process is possible. In this regard, passage of normal naive T cells into alymphoid mice can lead to 2 T cell subsets: a slowly proliferating population and a rapidly dividing one that quickly develops a memory phenotype and secretes IFNγ (50), as in our findings (Figure 7, C and D). Indeed, T lymphocytes located in CD20+PAX5+PVAs express T-bet, the master regulator of Th1-cells that suppresses Th2 and Th17 programs (33), but do not express Bcl-6, which promotes Tfh maturation (51). The lack of ICOS, CXCR5, and Bcl-6 expression assigns these T cells to the Th1 and not the Tfh lineage (Figure 7 and ref. 32). However, because the T cells expanding in NSG recipients do express more PD-1 and CD57 (Figure 7), the possibility that this is an unappreciated Th1-Tfh hybrid cell cannot be excluded. Alternatively, PD-1 and CD57 expression levels before transfer that increase very significantly after extensive proliferation in vivo could reflect the “pseudo-exhausted” nature of CLL T lymphocytes induced by chronic stimulation (52).
It is possible that the NSG microenvironment is the primary determinant of the Th1-dependent, apparently Bcl-6–deficient pathway for B cell maturation and differentiation. However, we propose that this is the fulfillment of the leukemic cells’ physiological maturational programs that take place in patients, but usually to a lesser extent. First, as mentioned, CLL cells can differentiate and secrete Ig spontaneously in vivo (34) and in vitro (35–37). Second, in vivo and in vitro stimulation of CLL cells can lead to expression of AID mRNA (53–56) and protein (57), promoting CSR and SHM (56–59). Indeed, all samples tested here underwent B cell maturation, AID upregulation, and development of IGHV-D-J mutations, even if the CLL clones were classified as U-CLL prior to transfer. Third, proliferation of CLL B cells in patients occurs principally in proliferation centers (17), where AID+ (57, 60), IRF4+ (61–63), and T-bet+ (64) cells can be found, although the levels of all 3 vary and are not always concomitantly present. Moreover, previous work indicates that this NSG transfer system recapitulates many features of human CLL, including the numbers of CD38+ cells within the clone (21) and a gene expression profile resembling cells in the human LN microenvironment (65). Finally, passage of normal human CD34+ hematopoietic stem cells into alymphoid mice leads to the development of B cells that follow a follicular maturation pathway resulting in GC-like structures that contain Bcl-6+ B cells (66).
What then prevents this Th1-mediated maturation from happening to a greater degree in patients? Circulating T cells of CLL patients are often dysfunctional (67), unable to form effective immune synapses (68) due to leukemic B cell expression of ligands that trigger T cell inhibitory receptors (69). In contrast, after transfer into NSG mice, the expanding T cell compartment is clearly functional, at least with regard to facilitating B cell diversification and differentiation. The outgrowth of Th1 cells could reflect in vivo selection for these cells, release from an inhibitory milieu present in patients but not in the NSG environment, or other cellular interactions modifying production of inhibitory molecules by the leukemic cells. Loss of the inhibitory effect of CD200, known to downregulate Th1 responses (70), would be an example of this, and infusion of soluble CD200 can modify CLL cell growth in vivo (71). Additionally, other human-derived cellular and soluble elements that are found in proliferation centers, such as nurse-like cells and certain human nonhematopoietic stromal elements (15), are lacking in our system, and their modulating effects on Th1-influenced B cell proliferation — either directly or through other T cell subsets (e.g., Tregs) — may be relevant. Finally, our data suggest that CLL clones differ in the ability to accept and/or respond to diversification and differentiation signals. Only a minor subset of CLL B cells in patients express T-bet prior to xenografting (Figures 5 and 8), whereas after expansion, diversification, and differentiation in NSG mice, the CLL clone is mostly T-bet+. Thus, either the initial minor subset is selected for maturation or in vivo activation induces T-bet production in CLL B cells. Supporting the former, a subset of CLL B cells expresses T-bet (64, 72), and our study confirms and places this finding into a framework suggesting involvement in B cell diversification and differentiation. T-bet expression is involved in T cell subset directionality since T-bet interacts with and downregulates Bcl-6, affecting the balance between Th1 and Tfh pathways (73). Similarly, T-bet might be responsible for the absence of Bcl-6 protein in CLL B cells maturing in our transfer model.
Our findings also have implications for the cell(s) of origin of CLL, as the maturation pathway they follow is similar to that of several B cell subsets including immature/transitional B cells (74), marginal zone B cells (75), and those involved in early memory B cell responses (76, 77) and in physiologic responses to Salmonella (43). The 2 latter B cell subsets generate switched and unswitched antibody-secreting cells in the absence of GC B cells. It is interesting that transitional B cells can express both T-bet and AID (74), as our CLL cells can. Expression of T-bet and AID by the other subsets — and the effects of Th1 cells on hCD5+ cells and hB-1–like cells, potential precursors of CLL (12, 13) — are not known at this point.
Finally, our studies indicate that the block in B cell maturation, the defect in T cell action, and the absence of antigen-receptor diversification (each a cardinal feature of CLL) are not inherent but context-dependent, leaving open therapeutic options. Might differentiation therapy be of value in CLL as in acute promyelocytic leukemia (78)? This possibility is both provocative and precarious. For example, the enhanced B cell proliferation and AID action induced by Th1 cell help could lead to acquisition of new genetic aberrations outside the IG locus, as implied by ultra-deep sequencing signatures (79). However, on the positive side, terminally differentiated CLL B cells may be less dangerous, since the genetic abnormalities in CLL B lymphocytes differ from those in myeloma PCs. Moreover, CLL B cells induced to terminally differentiate lose expression of the survival factor Lef-1 (37), which could lead to death. Finally, some of the newer therapeutic agents might be altering T cell programs in CLL patients in a way analogous to what we report here. For example, ibrutinib, a Bruton’s tyrosine kinase inhibitor, promotes Th1 responses in CLL patients by inhibiting IL-2–inducible kinase, ITK (80), and lenalidomide, an immunomodulatory drug, increases IFNγ production by CLL T cells (81). Hence, polarization to Th1 cells with IFNγ production, which are involved in CLL B cell maturation in our xenograft system (Figure 7), might be a component the therapeutic actions of these drugs. Thus, altering T cell subset and/or cytokine balance in a manner that promotes PC maturation but avoids new genetic lesions could be a therapeutic option in CLL.
Methods
CLL samples.
PB was collected from 15 patients with available clinical information, laboratory data, and IGHV-D-J DNA sequences (5). PBMCs were separated by density gradient centrifugation and cryopreserved prior to use.
Xenogeneic transplantation (see Supplemental Figure 1).
CD3-enriched cells were obtained from frozen CLL PBMCs using anti-CD3 microbeads (Milteyni Biotec) and were activated for 3–14 days with 25 μl Human T-Activator CD3/CD28 Dynabeads (Invitrogen) and 30 units IL-2 (R&D Systems) per ml of cells. In those experiments where cells were transferred after 7 days, beads were removed using a magnet and cells recultured with fresh CD3/28 beads. Activated CD3+ cells (0.5 × 106) were transferred with 20 × 106 freshly thawed CLL PBMCs into 4- to 8-week-old male NSG mice (the Jackson Laboratory) via the retro-orbital vein. To determine division, cells (2 × 107/ml) were incubated 10 minutes at 37°C with CFSE (5 μM; Invitrogen) before transfer. Following injection, mice were bled biweekly, and PBMCs were separated by density gradient centrifugation. Human cells were identified and murine cells excluded using a panel of antibodies (Supplemental Table 3).
Assessment of splenic tissue at euthanasia.
Spleens were bisected equally for FC and IH studies (Supplemental Figure 1). For FC of single cell suspensions, antigens were identified using antibodies listed in Supplemental Table 3, with fixation and permeabilization performed using a Cytofix/Cytoperm kit (BD Biosciences) prior to intracellular staining. FC data were acquired with a BD LSRII flow cytometer (BD Immunocytometry Systems) and analyzed by FlowJo V7.2.4 software (TreeStar Inc.). The gating strategy for hCD45 cells is shown in Figure 1A and Supplemental Figure 1.
For IH, spleens were formalin fixed and 5 micron sections prepared with subsequent antigen retrieval using a high-pressure decloaking chamber (Biocare Medical). Primary antibodies are listed in Supplemental Table 4. Antibodies for light microscopy were visualized using the Vectastain ABC system with ImmPACT DAB substrate and haematoxylin QS counterstaing (Vector Laboratories). Light microscopy was performed using an Olympus BX40 fitted with UplanFL 10×/0.30, 20×/0.50, 40×/0.65, and 60×/1.25 objectives. Antibodies for immunofluorescent microscopy were visualized with affinity purified, whole donkey IgG absorbed against alternate species conjugated to DyLight 488, 594, and 649 (Jackson Immunoresearch Laboratories Inc.). Immunofluorescent confocal microscopy was performed using an Olympus IX70 microscope with a UPlanAPO 60×/1.40 objective, one 30 mW argon laser exciting at 488 nm, one 1 mW helium-neon laser exciting at 543 nm, and one 5 mW helium-neon laser exciting at 633 nm. Confocal microscopy data were collected using proprietary image acquisition software and images edited for optimal color contrast using Adobe Creative Suite 2 premium (Adobe Systems).
Measurement of secreted Ig.
Plasma IgM and IgG were measured by ELISA. Ninety-six-well flat-bottom, high-binding microplates (Corning) were coated overnight at 4°C with 100 μl/well of 5 μg/ml goat F(ab)′2 anti-human IgM or IgG polyclonal antibodies (pAbs; Southern Biotech), washed 3 times with 0.1% Tween 20 (Fisher Scientific), blocked 1 hour with 100 μl/well of 10% FBS, washed 3 times, followed by 2 hours incubation with 80 μl/well dilutions of samples and human IgM or IgG standards (Sigma-Aldrich). After extensive washing, plates were incubated 1 hour with 100 μl/well of a 1:8000 dilution of a 1:1 mixture of goat anti-human Igκ and Igλ pAbs conjugated with horseradish peroxidase (Southern Biotech), washed, and incubated with 60 μl/well of TMB Sure Blue (KPL). After color reaction deepened sufficiently, 60 μl/well of 1 N HCl was added to stop reactions, and absorption at 450 nm measured with an ELx808 absorbance microplate reader (BioTek Instruments, Inc). Sample absorbance units were compared with the 4-parameter logistic curve fit of the human IgM or IgG standards to determine Ig concentrations using KCjunior (v1.22) software. The limit of human IgM and IgG detection varied from 0.3 to 37.8 μg/ml, depending on lowest detectable standard concentration and starting sample dilution.
Assessment of T cell–derived cytokines.
At euthanasia, PB was collected and plasma stored at –80°C. Samples were analyzed for IL-2, IL-4, IL-5, IL-10, IL-21, TNF, and IFNγ levels by cytometric bead array (BD Biosciences) as suggested. Results were correlated with the sum of the number of human CD45+CD4+CD5+, CD45+CD8+CD5+, and CD45+CD4+CD8+CD5+ cells expressed as a ratio over the number of mCD45+hCD45– cells obtained using identical forward scatter (FSC) and side scatter (SSC) gates from FACS analysis of single cell splenic suspensions obtained at the same timepoint.
RT-PCR for IGHV-D-J rearrangements.
hCD45+CD5+CD19+CD38–/loCD138– and hCD45+CD5+CD19+CD38++CD138+ cells were sorted (Figure 1A) with a BD FACSAria (BD Immunocytometry Systems) and washed, pelleted, and stored at –80°C. Total RNA from these fractions and from unsorted splenic suspensions was prepared with RLT (Qiagen). cDNA (1 μl), prepared from 1 μg RNA by reverse transcription with AccuScript High Fidelity RT (Stratagene) and random primers (300 ng/μl) (Invitrogen) at 42°C for 1 hour, was used to amplify IGHV-D-J-IGHC μ, γ, and α regions with IGHC-specific and IGHV family–specific leader and framework 1 primers (5) using PfuTurbo Hotstart DNA Polymerase (Stratagene) in 30 cycles of 5 seconds at 94°C, 30 seconds at 65°C, and 30 seconds at 72°C. Resultant IGHV-D-J-IGHC PCR products were purified using QIAquick Spin Columns (Qiagen) followed by Sanger sequencing with the same primers (Genewiz Inc.).
Deep sequencing analyses of IGHV-D-J rearrangements.
CLL clonal IGHV-D-J rearrangements (GenBank KU674888–KU674894) were amplified from cDNA using described high-fidelity methods (57) with IGHM-specific and IGHV family–specific framework 1 primers (5). Amplicons were magnified to prepare libraries for ultra-deep sequencing using the 454 FLX system (Roche Diagnostics) pyrosequencing technology (57). The resulting 3,260,173 sequence reads, deposited in GenBank (Sequence Read Archive [SRA] accession SRP069209), were processed and analyzed by custom bioinformatics computation (57), and summarized in Supplemental Table 1. Phylogenetic trees of new IGHV-D-J sequences were calculated using K80 pairwise distances (82) and neighbor-joining estimation (83) as implemented in the phangorn R package (84). These were illustrated using a polar tree layout with the Figtree software package (http://tree.bio.ed.ac.uk/software/figtree/).
Statistics.
Statistical tests were performed using Prism v5 (GraphPad Software) with normality assessed using D’Agostino and Pearson omnibus tests. The following tests were used: 1-way ANOVA test for analysis of time of appearance of CD5+CD19+CD38++ cells; Spearman rank tests for assessing correlation of plasma levels of IgM and IgG with % CD5+CD19+CD38++ cells and correlation of plasma IFNγ and IL-5 with numbers of spleen-residing T cells at euthanasia; 2-tailed, Mann-Whitney tests for expression of AID protein by undivided and multiply divided B cells and for comparison of M-CLL and U-CLL sequences; 2-tailed paired t-test for comparisons of IgM with IgG/A mutation frequencies; 2-tailed, unpaired t tests for comparison of intracellular IFNγ expression before and after transfer and comparison of %T-bet+ cells in the presence or absence of B-CLL maturation; 2-tailed matched Wilcoxon tests for analysis of AID hotspot and coldspot differences (following transformation by log2 of the fold mutation frequency) and for the analysis of T-bet expression in CD4, CD8, and CD5/CD19 cells before and after transfer. P < 0.05 was considered significant for all tests.
Study approval.
In accordance with the Helsinki Declaration and as approved by the IRB of Northwell Health (Great Neck, New York, USA), written informed consent was received from participants prior to enrollment in the study. The Institutional Animal Care and Use Committee of Northwell Health approved xenograft studies.
Author contributions
PEMP, CCC, and NC conceived and designed study. PEMP, TM, and CCC developed methodology. PEMP, GF, SSC, RS, SM, XJY, ZG, CY, TM, and CCC acquired data. PEMP, GF, SSC, RS, SM, XJY, ZG, CY, TM, CCC, and NC performed data analysis and interpretation. JEK, JB, SLA, and KRR collected patient samples for xenografting. PEMP, GF, SSC, TM, CCC, and NC wrote manuscript.
Supplementary Material
Acknowledgments
This work was supported in part by an R01 grant from the National Cancer Institute/NIH (CA081554) and by philanthropic contributions from The Karches Foundation, Marks Foundation, Nash Family Foundation, Jerome Levy Foundation, Leon Levy Foundation, Frank and Mildred Feinberg Foundation, Mona and Edward Albert Foundation, and Jean Walton Fund for Leukemia, Lymphoma, and Myeloma Research. Bloodwise, United Kingdom, supported PEMP.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:JCI Insight. 2016;1(4):e86288. doi:10.1172/jci.insight.86288.
Contributor Information
Gerardo Ferrer, Email: GFerrer1@NSHS.edu.
Shih-Shih Chen, Email: schen9@nshs.edu.
Rita Simone, Email: Rita.Simone@hotmail.it.
Sonia Marsilio, Email: MarsilioSonia@Gmail.com.
Xiao-Jie Yan, Email: xyan@nshs.edu.
Zachary Gitto, Email: zachary.gitto@med.einstein.yu.edu.
Chaohui Yuan, Email: yuanch87@gmail.com.
Jacqueline Barrientos, Email: JBarrientos@NSHS.edu.
Thomas MacCarthy, Email: thomas.maccarthy@stonybrook.edu.
Nicholas Chiorazzi, Email: nchizzi@nshs.edu.
References
- 1.Chiorazzi N, Rai KR, Ferrarini M. Chronic lymphocytic leukemia. N Engl J Med. 2005;352(8):804–815. doi: 10.1056/NEJMra041720. [DOI] [PubMed] [Google Scholar]
- 2.Klein U, et al. Gene expression profiling of B cell chronic lymphocytic leukemia reveals a homogeneous phenotype related to memory B cells. J Exp Med. 2001;194(11):1625–1638. doi: 10.1084/jem.194.11.1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rosenwald A, et al. Relation of gene expression phenotype to immunoglobulin mutation genotype in B cell chronic lymphocytic leukemia. J Exp Med. 2001;194(11):1639–1647. doi: 10.1084/jem.194.11.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Damle RN, et al. B-cell chronic lymphocytic leukemia cells express a surface membrane phenotype of activated, antigen-experienced B lymphocytes. Blood. 2002;99(11):4087–4093. doi: 10.1182/blood.V99.11.4087. [DOI] [PubMed] [Google Scholar]
- 5.Fais F, et al. Chronic lymphocytic leukemia B cells express restricted sets of mutated and unmutated antigen receptors. J Clin Invest. 1998;102(8):1515–1525. doi: 10.1172/JCI3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Agathangelidis A, et al. Stereotyped B-cell receptors in one-third of chronic lymphocytic leukemia: a molecular classification with implications for targeted therapies. Blood. 2012;119(19):4467–4475. doi: 10.1182/blood-2011-11-393694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Messmer BT, et al. Multiple distinct sets of stereotyped antigen receptors indicate a role for antigen in promoting chronic lymphocytic leukemia. J Exp Med. 2004;200(4):519–525. doi: 10.1084/jem.20040544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bulian P, et al. CD49d is the strongest flow cytometry-based predictor of overall survival in chronic lymphocytic leukemia. J Clin Oncol. 2014;32(9):897–904. doi: 10.1200/JCO.2013.50.8515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wiestner A, et al. ZAP-70 expression identifies a chronic lymphocytic leukemia subtype with unmutated immunoglobulin genes, inferior clinical outcome, and distinct gene expression profile. Blood. 2003;101(12):4944–4951. doi: 10.1182/blood-2002-10-3306. [DOI] [PubMed] [Google Scholar]
- 10.Dohner H, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med. 2000;343(26):1910–1916. doi: 10.1056/NEJM200012283432602. [DOI] [PubMed] [Google Scholar]
- 11.Landau DA, et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell. 2013;152(4):714–726. doi: 10.1016/j.cell.2013.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Griffin DO, Holodick NE, Rothstein TL. Human B1 cells in umbilical cord and adult peripheral blood express the novel phenotype CD20+ CD27+ CD43+ CD70. J Exp Med. 2011;208(1):67–80. doi: 10.1084/jem.20101499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seifert M, et al. Cellular origin and pathophysiology of chronic lymphocytic leukemia. J Exp Med. 2012;209(12):2183–2198. doi: 10.1084/jem.20120833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chiorazzi N, Ferrarini M. Cellular origin(s) of chronic lymphocytic leukemia: cautionary notes and additional considerations and possibilities. Blood. 2011;117(6):1781–1791. doi: 10.1182/blood-2010-07-155663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burger JA, Ghia P, Rosenwald A, Caligaris-Cappio F. The microenvironment in mature B-cell malignancies: a target for new treatment strategies. Blood. 2009;114(16):3367–3375. doi: 10.1182/blood-2009-06-225326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Messmer BT, et al. In vivo measurements document the dynamic cellular kinetics of chronic lymphocytic leukemia B cells. J Clin Invest. 2005;115(3):755–764. doi: 10.1172/JCI23409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Soma LA, Craig FE, Swerdlow SH. The proliferation center microenvironment and prognostic markers in chronic lymphocytic leukemia/small lymphocytic lymphoma. Hum Pathol. 2006;37(2):152–159. doi: 10.1016/j.humpath.2005.09.029. [DOI] [PubMed] [Google Scholar]
- 18.Chiorazzi N, Ferrarini M. B cell chronic lymphocytic leukemia: lessons learned from studies of the B cell antigen receptor. Annu Rev Immunol. 2003;21:841–894. doi: 10.1146/annurev.immunol.21.120601.141018. [DOI] [PubMed] [Google Scholar]
- 19.Ghia P, et al. Chronic lymphocytic leukemia B cells are endowed with the capacity to attract CD4+, CD40L+ T cells by producing CCL22. Eur J Immunol. 2002;32(5):1403–1413. doi: 10.1002/1521-4141(200205)32:5<1403::AID-IMMU1403>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 20.Patten PE, et al. CD38 expression in chronic lymphocytic leukemia is regulated by the tumor microenvironment. Blood. 2008;111(10):5173–5181. doi: 10.1182/blood-2007-08-108605. [DOI] [PubMed] [Google Scholar]
- 21.Bagnara D, et al. A novel adoptive transfer model of chronic lymphocytic leukemia suggests a key role for T lymphocytes in the disease. Blood. 2011;117(20):5463–5472. doi: 10.1182/blood-2010-12-324210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Patten P, et al. Engraftment of CLL-derived T cells in NSG mice is feasible, can support CLL cell proliferation, and eliminates the need for third party antigen presenting cells. Blood. 2011;118(4):e86288 [Google Scholar]
- 23.Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102(5):553–563. doi: 10.1016/S0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- 24.Fais F, et al. Examples of in vivo isotype class switching in IgM+ chronic lymphocytic leukemia B cells. J Clin Invest. 1996;98(7):1659–1666. doi: 10.1172/JCI118961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pham P, Bransteitter R, Petruska J, Goodman MF. Processive AID-catalysed cytosine deamination on single-stranded DNA simulates somatic hypermutation. Nature. 2003;424(6944):103–107. doi: 10.1038/nature01760. [DOI] [PubMed] [Google Scholar]
- 26.Jacob J, Kelsoe G, Rajewsky K, Weiss U. Intraclonal generation of antibody mutants in germinal centres. Nature. 1991;354(6352):389–392. doi: 10.1038/354389a0. [DOI] [PubMed] [Google Scholar]
- 27.Kelsoe G. Life and death in germinal centers (redux) Immunity. 1996;4(2):107–111. doi: 10.1016/S1074-7613(00)80675-5. [DOI] [PubMed] [Google Scholar]
- 28.Victora GD, Wilson PC. Germinal center selection and the antibody response to influenza. Cell. 2015;163(3):545–548. doi: 10.1016/j.cell.2015.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cattoretti G, Shaknovich R, Smith PM, Jäck HM, Murty VV, Alobeid B. Stages of germinal center transit are defined by B cell transcription factor coexpression and relative abundance. J Immunol. 2006;177(10):6930–6939. doi: 10.4049/jimmunol.177.10.6930. [DOI] [PubMed] [Google Scholar]
- 30.Phan RT, Dalla-Favera R. The BCL6 proto-oncogene suppresses p53 expression in germinal-centre B cells. Nature. 2004;432(7017):635–639. doi: 10.1038/nature03147. [DOI] [PubMed] [Google Scholar]
- 31.Ye BH, et al. The BCL-6 proto-oncogene controls germinal-centre formation and Th2-type inflammation. Nat Genet. 1997;16(2):161–170. doi: 10.1038/ng0697-161. [DOI] [PubMed] [Google Scholar]
- 32.Crotty S. T follicular helper cell differentiation, function, and roles in disease. Immunity. 2014;41(4):529–542. doi: 10.1016/j.immuni.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lazarevic V, Glimcher LH. T-bet in disease. Nat Immunol. 2011;12(7):597–606. doi: 10.1038/ni.2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fu SM, Winchester RJ, Feizi T, Walzer PD, Kunkel HG. Idiotypic specificity of surface immunoglobulin and the maturation of leukemic bone-marrow-derived lymphocytes. Proc Natl Acad Sci U S A. 1974;71(11):4487–4490. doi: 10.1073/pnas.71.11.4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fu SM, Chiorazzi N, Kunkel HG, Halper JP, Harris SR. Induction of in vitro differentiation and immunoglobulin synthesis of human leukemic B lymphocytes. J Exp Med. 1978;148(6):1570–1578. doi: 10.1084/jem.148.6.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tötterman TH, Nilsson K, Sundström C. Phorbol ester-induced differentiation of chronic lymphocytic leukaemia cells. Nature. 1980;288(5787):176–178. doi: 10.1038/288176a0. [DOI] [PubMed] [Google Scholar]
- 37.Gutierrez A, Jr, Arendt BK, Tschumper RC, Kay NE, Zent CS, Jelinek DF. Differentiation of chronic lymphocytic leukemia B cells into immunoglobulin secreting cells decreases LEF-1 expression. PLoS One. 2011;6(10):e86288. doi: 10.1371/journal.pone.0026056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klein U, et al. Transcription factor IRF4 controls plasma cell differentiation and class-switch recombination. Nat Immunol. 2006;7(7):773–782. doi: 10.1038/ni1357. [DOI] [PubMed] [Google Scholar]
- 39.Sciammas R, Shaffer AL, Schatz JH, Zhao H, Staudt LM, Singh H. Graded expression of interferon regulatory factor-4 coordinates isotype switching with plasma cell differentiation. Immunity. 2006;25(2):225–236. doi: 10.1016/j.immuni.2006.07.009. [DOI] [PubMed] [Google Scholar]
- 40.Capello D, et al. Identification of three subgroups of B cell chronic lymphocytic leukemia based upon mutations of BCL-6 and IgV genes. Leukemia. 2000;14(5):811–815. doi: 10.1038/sj.leu.2401766. [DOI] [PubMed] [Google Scholar]
- 41.William J, Euler C, Christensen S, Shlomchik MJ. Evolution of autoantibody responses via somatic hypermutation outside of germinal centers. Science. 2002;297(5589):2066–2070. doi: 10.1126/science.1073924. [DOI] [PubMed] [Google Scholar]
- 42.Berek C, Kim HJ. B-cell activation and development within chronically inflamed synovium in rheumatoid and reactive arthritis. Semin Immunol. 1997;9(4):261–268. doi: 10.1006/smim.1997.0076. [DOI] [PubMed] [Google Scholar]
- 43.Di Niro R, et al. Salmonella infection drives promiscuous B cell activation followed by extrafollicular affinity maturation. Immunity. 2015;43(1):120–131. doi: 10.1016/j.immuni.2015.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sweet RA, Cullen JL, Shlomchik MJ. Rheumatoid factor B cell memory leads to rapid, switched antibody-forming cell responses. J Immunol. 2013;190(5):1974–1981. doi: 10.4049/jimmunol.1202816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cunningham AF, et al. Salmonella induces a switched antibody response without germinal centers that impedes the extracellular spread of infection. J Immunol. 2007;178(10):6200–6207. doi: 10.4049/jimmunol.178.10.6200. [DOI] [PubMed] [Google Scholar]
- 46.Chiorazzi N, Fu SM, Montazeri G, Kunkel HG, Rai K, Gee T. T cell helper defect in patients with chronic lymphocytic leukemia. J Immunol. 1979;122(3):1087–1090. [PubMed] [Google Scholar]
- 47.Cantwell M, Hua T, Pappas J, Kipps TJ. Acquired CD40-ligand deficiency in chronic lymphocytic leukemia. Nat Med. 1997;3(9):984–989. doi: 10.1038/nm0997-984. [DOI] [PubMed] [Google Scholar]
- 48.Lang J, et al. Studies of lymphocyte reconstitution in a humanized mouse model reveal a requirement of T cells for human B cell maturation. J Immunol. 2013;190(5):2090–2101. doi: 10.4049/jimmunol.1202810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bürgler S, et al. Chronic lymphocytic leukemia cells express CD38 in response to Th1 cell-derived IFN-γ by a T-bet-dependent mechanism. J Immunol. 2015;194(2):827–835. doi: 10.4049/jimmunol.1401350. [DOI] [PubMed] [Google Scholar]
- 50.Onoe T, Kalscheuer H, Chittenden M, Zhao G, Yang YG, Sykes M. Homeostatic expansion and phenotypic conversion of human T cells depend on peripheral interactions with APCs. J Immunol. 2010;184(12):6756–6765. doi: 10.4049/jimmunol.0901711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hatzi K, et al. BCL6 orchestrates Tfh cell differentiation via multiple distinct mechanisms. J Exp Med. 2015;212(4):539–553. doi: 10.1084/jem.20141380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Riches JC, et al. T cells from CLL patients exhibit features of T-cell exhaustion but retain capacity for cytokine production. Blood. 2013;121(9):1612–1621. doi: 10.1182/blood-2012-09-457531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Oppezzo P, et al. Chronic lymphocytic leukemia B cells expressing AID display dissociation between class switch recombination and somatic hypermutation. Blood. 2003;101(10):4029–4032. doi: 10.1182/blood-2002-10-3175. [DOI] [PubMed] [Google Scholar]
- 54.McCarthy H, et al. High expression of activation-induced cytidine deaminase (AID) and splice variants is a distinctive feature of poor-prognosis chronic lymphocytic leukemia. Blood. 2003;101(12):4903–4908. doi: 10.1182/blood-2002-09-2906. [DOI] [PubMed] [Google Scholar]
- 55.Albesiano E, Messmer BT, Damle RN, Allen SL, Rai KR, Chiorazzi N. Activation induced cytidine deaminase in chronic lymphocytic leukemia B cells: expression as multiple forms in a dynamic, variably sized fraction of the clone. Blood. 2003;102(9):3333–3339. doi: 10.1182/blood-2003-05-1585. [DOI] [PubMed] [Google Scholar]
- 56.Palacios F, et al. High expression of AID and active class switch recombination might account for a more aggressive disease in unmutated CLL patients: link with an activated microenvironment in CLL disease. Blood. 2010;115(22):4488–4496. doi: 10.1182/blood-2009-12-257758. [DOI] [PubMed] [Google Scholar]
- 57.Patten PEM, et al. IGHV-unmutated and IGHV-mutated chronic lymphocytic leukemia cells produce activation-induced deaminase protein with a full range of biologic functions. Blood. 2012;120(24):4802–4811. doi: 10.1182/blood-2012-08-449744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gurrieri C, et al. Chronic lymphocytic leukemia B cells can undergo somatic hypermutation and intraclonal immunoglobulin V(H)DJ(H) gene diversification. J Exp Med. 2002;196(5):629–639. doi: 10.1084/jem.20011693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cols M, et al. Stromal endothelial cells establish a bidirectional crosstalk with chronic lymphocytic leukemia cells through the TNF-related factors BAFF, APRIL, and CD40L. J Immunol. 2012;188(12):6071–6083. doi: 10.4049/jimmunol.1102066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Leuenberger M, et al. AID protein expression in chronic lymphocytic leukemia/small lymphocytic lymphoma is associated with poor prognosis and complex genetic alterations. Mod Pathol. 2010;23(2):177–186. doi: 10.1038/modpathol.2009.156. [DOI] [PubMed] [Google Scholar]
- 61.Falini B, et al. A monoclonal antibody (MUM1p) detects expression of the MUM1/IRF4 protein in a subset of germinal center B cells, plasma cells, and activated T cells. Blood. 2000;95(6):2084–2092. [PubMed] [Google Scholar]
- 62.Pileri SA, et al. The pathologist’s view point. Part I — indolent lymphomas. Haematologica. 2000;85(12):1291–1307. [PubMed] [Google Scholar]
- 63.Ito M, et al. MUM1/IRF4 expression is an unfavorable prognostic factor in B-cell chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL) Jpn J Cancer Res. 2002;93(6):685–694. doi: 10.1111/j.1349-7006.2002.tb01307.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dorfman DM, Hwang ES, Shahsafaei A, Glimcher LH. T-bet, a T-cell-associated transcription factor, is expressed in a subset of B-cell lymphoproliferative disorders. Am J Clin Pathol. 2004;122(2):292–297. doi: 10.1309/AQQ2DVM75DVY0PWP. [DOI] [PubMed] [Google Scholar]
- 65.Herman SE, et al. Modeling tumor-host interactions of chronic lymphocytic leukemia in xenografted mice to study tumor biology and evaluate targeted therapy. Leukemia. 2013;27(12):2311–2321. doi: 10.1038/leu.2013.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Traggiai E, et al. Development of a human adaptive immune system in cord blood cell-transplanted mice. Science. 2004;304(5667):104–107. doi: 10.1126/science.1093933. [DOI] [PubMed] [Google Scholar]
- 67.Riches JC, Ramsay AG, Gribben JG. T-cell function in chronic lymphocytic leukaemia. Semin Cancer Biol. 2010;20(6):431–438. doi: 10.1016/j.semcancer.2010.09.006. [DOI] [PubMed] [Google Scholar]
- 68.Ramsay AG, et al. Chronic lymphocytic leukemia T cells show impaired immunological synapse formation that can be reversed with an immunomodulating drug. J Clin Invest. 2008;118(7):2427–2437. doi: 10.1172/JCI35017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ramsay AG, Clear AJ, Fatah R, Gribben JG. Multiple inhibitory ligands induce impaired T-cell immunologic synapse function in chronic lymphocytic leukemia that can be blocked with lenalidomide: establishing a reversible immune evasion mechanism in human cancer. Blood. 2012;120(7):1412–1421. doi: 10.1182/blood-2012-02-411678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McWhirter JR, et al. Antibodies selected from combinatorial libraries block a tumor antigen that plays a key role in immunomodulation. Proc Natl Acad Sci U S A. 2006;103(4):1041–1046. doi: 10.1073/pnas.0510081103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wong KK, Brenneman F, Chesney A, Spaner DE, Gorczynski RM. Soluble CD200 is critical to engraft chronic lymphocytic leukemia cells in immunocompromised mice. Cancer Res. 2012;72(19):4931–4943. doi: 10.1158/0008-5472.CAN-12-1390. [DOI] [PubMed] [Google Scholar]
- 72.Os A, et al. Chronic lymphocytic leukemia cells are activated and proliferate in response to specific T helper cells. Cell Rep. 2013;4(3):566–577. doi: 10.1016/j.celrep.2013.07.011. [DOI] [PubMed] [Google Scholar]
- 73.Oestreich KJ, Mohn SE, Weinmann AS. Molecular mechanisms that control the expression and activity of Bcl-6 in TH1 cells to regulate flexibility with a TFH-like gene profile. Nat Immunol. 2012;13(4):405–411. doi: 10.1038/ni.2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ueda Y, Liao D, Yang K, Patel A, Kelsoe G. T-independent activation-induced cytidine deaminase expression, class-switch recombination, and antibody production by immature/transitional 1 B cells. J Immunol. 2007;178(6):3593–3601. doi: 10.4049/jimmunol.178.6.3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pillai S, Cariappa A, Moran ST. Marginal zone B cells. Annu Rev Immunol. 2005;23:161–196. doi: 10.1146/annurev.immunol.23.021704.115728. [DOI] [PubMed] [Google Scholar]
- 76.Dogan I, et al. Multiple layers of B cell memory with different effector functions. Nat Immunol. 2009;10(12):1292–1299. doi: 10.1038/ni.1814. [DOI] [PubMed] [Google Scholar]
- 77.Taylor JJ, Pape KA, Jenkins MK. A germinal center-independent pathway generates unswitched memory B cells early in the primary response. J Exp Med. 2012;209(3):597–606. doi: 10.1084/jem.20111696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nowak D, Stewart D, Koeffler HP. Differentiation therapy of leukemia: 3 decades of development. Blood. 2009;113(16):3655–3665. doi: 10.1182/blood-2009-01-198911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Alexandrov LB, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dubovsky JA, et al. Ibrutinib is an irreversible molecular inhibitor of ITK driving a Th1-selective pressure in T lymphocytes. Blood. 2013;122(15):2539–2549. doi: 10.1182/blood-2013-06-507947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Corral LG, Kaplan G. Immunomodulation by thalidomide and thalidomide analogues. Ann Rheum Dis. 1999;58(suppl 1):I107–I113. doi: 10.1136/ard.58.2008.i107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kimura M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 1980;16(2):111–120. doi: 10.1007/BF01731581. [DOI] [PubMed] [Google Scholar]
- 83.Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4(4):406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 84.Schliep KP. phangorn: phylogenetic analysis in R. Bioinformatics. 2011;27(4):592–593. doi: 10.1093/bioinformatics/btq706. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.