

Abstract
Asthma is one of the most common chronic diseases globally and can be divided into presenting with or without an immune response. Current therapies have little effect on nonimmune disease, and the mechanisms that drive this type of asthma are poorly understood. Here, we have shown that loss of the transcription factors forkhead box P1 (Foxp1) and Foxp4, which are critical for lung epithelial development, in the adult airway epithelium evokes a non-Th2 asthma phenotype that is characterized by airway hyperresponsiveness (AHR) without eosinophilic inflammation. Transcriptome analysis revealed that loss of Foxp1 and Foxp4 expression induces ectopic expression of neuropeptide Y (Npy), which has been reported to be present in the airways of asthma patients, but whose importance in disease pathogenesis remains unclear. Treatment of human lung airway explants with recombinant NPY increased airway contractility. Conversely, loss of Npy in Foxp1- and Foxp4-mutant airway epithelium rescued the AHR phenotype. We determined that NPY promotes AHR through the induction of Rho kinase activity and phosphorylation of myosin light chain, which induces airway smooth muscle contraction. Together, these studies highlight the importance of paracrine signals from the airway epithelium to the underlying smooth muscle to induce AHR and suggest that therapies targeting epithelial induction of this phenotype may prove useful in treatment of noneosinophilic asthma.
Introduction
The contribution of the immune system and airway smooth muscle to asthma phenotype, including airway hyperresponsiveness (AHR), has been well studied, and most therapies are directed toward inhibiting the associated inflammatory response in this disease. Asthma can be divided into either disease involving a Th2 inflammatory/eosinophilic immune response or disease lacking eosinophilic involvement (1, 2). Current asthma therapies are not curative and have little effect on asthma lacking immune involvement. The role of epithelium in mediating the asthma diathesis has been studied, but the precise molecular mechanisms by which airway epithelium mediates AHR remain unknown. Importantly, whether paracrine signals originating in the airway epithelium can promote and exacerbate the asthmatic phenotype remains unclear. In this report, we show that the critical airway epithelial transcription factors forkhead box P1 (FOXP1) and FOXP4 act coordinately to restrict expression of neuropeptide Y (NPY) specifically in airway epithelium. Importantly, loss of FOXP1 and FOXP4 leads to increased NPY expression that can, in turn, induce airway hyperreactivity in a paracrine manner through activation of smooth muscle myosin light-chain phosphorylation. This phenotype can also be induced in mouse and human lung explants by treatment with recombinant NPY, revealing the importance of epithelial/smooth muscle paracrine signaling in promoting the asthma phenotype in both mouse and human lungs.
Results and Discussion
Loss of Foxp1 and Foxp4 in postnatal airway epithelium increases expression of goblet cell–associated proteins MUC5AC, MUC5B, CLCA3, and TFF2, whose expression is increased in asthmatic airways (3). To assess whether the disruption of airway epithelial homeostasis caused by loss of Foxp1 and Foxp4 evoked AHR, a defining phenotype of asthma, we performed lung resistance and airway compliance testing after exposure to methacholine on adult controls (Scgb1a1-Cre) and Scgb1a1-Cre Foxp1fl/fl Foxp4fl/fl (herein referred to as Foxp1/4DKO) mutants. The Scgb1a1 line results in a highly effective deletion of genes, including Foxp1 and Foxp4, in postnatal airway epithelium (3). While methacholine challenge increased airway resistance in a dose-dependent manner in control animals, the Foxp1/4DKO mutants responded with an increase in airway resistance (RL) and tissue damping (G), though there was no difference between control and mutants in Newtonian resistance (RN) (Figure 1, A and B, and Supplemental Figure 1, A–C; supplemental material available online with this article; doi:10.1172/JCI81389DS1). Additionally, at baseline, the Foxp1/4DKO mutants exhibited greater airway resistance, suggesting that an underlying increase in bronchomotor tone exists even in the absence of contractile stimulation of the airways (Figure 1, A and B). To eliminate the effect of baseline differences in lung-resistance testing, we compared the airway resistance response to methacholine using precision cut lung slices (PCLSs) from control and Foxp1/4DKO mutants (4–6). These experiments showed that Foxp1/4DKO mutants exhibited greater airway resistance both in vivo and in the PCLS system (Figure 1C). The AHR response in unchallenged Foxp1/4DKO mutants was similar to that of control mice challenged with Aspergillus fumigates (Supplemental Figure 1D). Moreover, the AHR response in Foxp1/4DKO mutants was further amplified after exposure to A. fumigates challenge (Supplemental Figure 1E). Our findings are consistent with those seen in asthma patients, who often manifest a diminished forced expiratory volume in 1 second (FEV1) (more obstruction) and greater methacholine sensitivity after challenge (7).
Figure 1. Loss of Foxp1 and Foxp4 in airway epithelial cells promotes AHR and induces ectopic expression of NPY.

(A and B) Lung resistance to cumulative doses of methacholine was measured. Deficiency in FOXP1 and FOXP4 in epithelial cells markedly enhances methacholine-induced lung resistance demonstrated in airway resistance (RL) (A) and dynamic elastance (B) (n = 6 for control and DKO, respectively) and in the mouse PCLS assay (C) (control n = 9, 30 slices; DKO n = 8, 19 slices). (D) Loss of Foxp1 and Foxp4 induced AHR without evoking airway eosinophilic inflammation, as demonstrated by no significant increase in IL-13, IL-17 (n = 11 for control, 8 for DKO), IL-6, and KC (n = 3 for WT and 4 for DKO, respectively) (E) Heat map from the microarray analyses showing that Npy expression was increased by in the secreted proteins category in Foxp1/4DKO mutants. (F) ISH showing increased Npy expression in the epithelium of Foxp1/4DKO mutants (arrowheads). Ai, airway. **P < 0.01. Scale bar: 200 μm.
Rodent models of AHR, including those that are sensitive to ovalbumin, house dust mite, and A. fumigatus challenge, have a significant inflammatory component (8, 9). We examined the lungs of Foxp1/4DKO mutants for expression of the eosinophilic inflammatory marker IL-13 and neutrophilic inflammatory markers IL-17, IL-6, and keratinocyte-derived chemokine (KC) to assess whether loss of Foxp1 and Foxp4 evoked an inflammatory response. Surprisingly, Foxp1/4DKO mutants did not exhibit increased levels of any of these inflammatory cytokines compared with control mice (Figure 1D). We did observe a modest increase in the numbers of neutrophils, but not macrophages, eosinophils, or lymphocytes in the bronchoalveolar lavage (BAL) fluid from Foxp1/4DKO mutants (Supplemental Figure 2A).
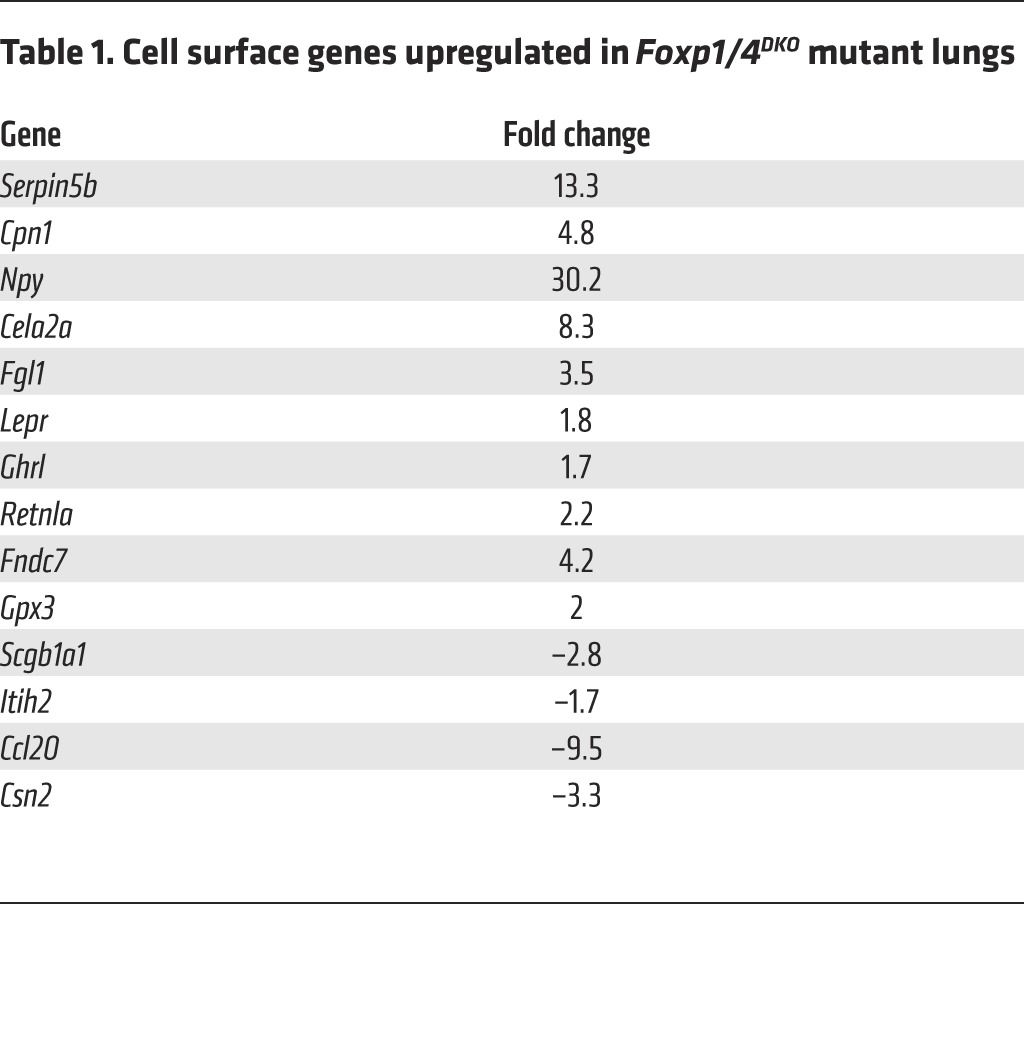
Transcriptome analysis revealed 109 genes significantly (P < 0.05) up- or downregulated upon genetic inactivation of Foxp1 and Foxp4. We focused our attention on those genes that were upregulated and that could act in a paracrine fashion upon airway smooth muscle cells to confer AHR. Among the 10 upregulated genes coding for secreted proteins, NPY expression was changed by the greatest extent (Figure 1E and Table 1). The increase in Npy expression was confirmed by ISH (Figure 1F) and quantitative real-time PCR (Q-PCR) (Supplemental Figure 2, B and C). NPY is a 36-aa polypeptide and is expressed in the central and peripheral nervous system and in other cell lineages, including endothelium, platelets, immune cells, and enteric neurons in the gut (10–15). Although NPY is one of the most abundant peptides in the peripheral nervous system, the role of NPY in asthma remains unclear (16, 17). Interestingly, increased Npy expression is associated with a mouse model of allergic airway inflammation, and polymorphisms in the Npy gene are associated with an increased risk for asthma (18, 19). Assessment of changes in the amount of airway smooth muscle in Foxp1/4DKO mutants did not reveal any changes compared with controls (Supplemental Figure 2, D–H). These data indicate that increased levels of Npy observed in the Foxp1/4DKO mutant airway epithelium generated a paracrine signal to promote airway smooth muscle contraction rather than increased mass.
Table 1. Cell surface genes upregulated in Foxp1/4DKO mutant lungs.
Two FOXP1 and FOXP4 DNA–binding sites are located in an evolutionarily conserved region approximately –0.8 kb to the transcriptional start of the mouse Npy gene (Supplemental Figure 2I). ChIP assays showed that FOXP1 and FOXP4 bind to these sites in chromatin from mouse lungs (Supplemental Figure 2J). Luciferase reporter constructs containing this –0.8 kb region were repressed by FOXP1, demonstrating that Npy is a transcriptional target of FOXP1 and FOXP4 (Supplemental Figure 2K).
To determine whether NPY potentiates airway smooth muscle contraction, we incubated mouse and human PCLSs (4–6) with recombinant NPY and characterized methacholine-mediated bronchoconstriction. These experiments demonstrated that NPY amplifies methacholine-induced bronchoconstriction (Figure 2, A and B, and Supplemental Figure 3).
Figure 2. NPY amplifies methacholine-induced bronchoconstriction in human airways.

(A) Human PCLSs were treated with 10 nM recombinant NPY for 18 hours, and cumulative dose curves to methacholine measuring luminal diameter narrowing were determined. (B) Pretreatment with NPY markedly enhances maximal bronchoconstriction induced by methacholine. (C) NPY treatment alone has little effect on pMLC; however, pretreatment with NPY markedly enhances methacholine-induced pMLC. Graphical data represent experiments performed in triplicate and are expressed as arbitrary densitometry units. Mch, methacholine. (D) NPY had little effect on peak calcium levels induced by contractile agonists BK or histamine, indicating that NPY does not act to induce calcium mobilization through these 2 pathways. (E) NPY promotes Rho kinase activity by itself and in cooperation with BK as measured by Western blot. *P < 0.01; **P < 0.005.
Phosphorylation of myosin light chain (pMLC) is a critical signaling event required for airway smooth muscle contraction (20, 21). We next examined the level of pMLC in primary human airway smooth muscle (HASM) cells. While methacholine increased pMLC as expected, Npy treatment in addition to methacholine significantly increased pMLC levels (Figure 2C). Activation of Rho kinase mediated sensitization of bronchoconstriction to contractile agonists in HASM. Bradykinin (BK) and histamine are known to increase peak calcium levels and bronchoconstriction in HASM cells (22). Treatment of HASM cells with NPY had little effect on peak cellular calcium levels over that induced by BK or histamine alone (Figure 2D). NPY alone, however, increased Rho kinase activity, and augmented BK induced Rho kinase activity above that induced by BK alone (Figure 2E).
To determine whether the phenotype observed in Foxp1/4DKO mutants is due to increased Npy expression, we genetically deleted Npy in Foxp1/4DKO mutants to determine whether this would attenuate the AHR phenotype. Foxp1/4DKO Npy–/– mutants (herein referred to as Foxp1/4 NpyTKO) were generated by mating Foxp1/4DKO mice with Npy–/– mice, which do not have a significant AHR phenotype (data not shown). Foxp1/4 NpyTKO mutants were grossly normal and had normal breeding habits. ISH and Q-PCR for Npy expression showed loss of Npy expression in Foxp1/4 NpyTKO mutants (Figure 3, A and B). Foxp1/4 NpyTKO mutants were challenged with methacholine, and the enhanced AHR observed in the Foxp1/4DKO mice was attenuated in the Foxp1/4 NpyTKO mutants (Figure 3C). BAL from Foxp1/4 NpyTKO mutants contained more neutrophils than that of Foxp1/4DKO mutants; however, the total cell counts from the BAL were not significantly different (Figure 3D). Expression of the proinflammatory cytokine IL-6 was not significantly altered in Foxp1/4 NpyTKO mutants compared with in Foxp1/4DKO mutants, whereas expression of KC was significantly increased (Supplemental Figure 4).
Figure 3. Loss of Npy expression rescues the AHR phenotype in Foxp1/4DKO mutants and implicates an epithelial/stromal signaling pathway regulating AHR.

(A) ISH for Npy expression in control, Foxp1/4DKO, and Foxp1/4 NpyTKO mutants. (B) Q-PCR for Npy expression in control, Foxp1/4DKO (DKO), and Foxp1/4 NpyTKO (TKO) mutants. (C) Methacholine challenge in control, Foxp1/4DKO, and Foxp1/4 NpyTKO mutants showing reduced AHR in Foxp1/4 NpyTKO mutants as compared with Foxp1/4DKO mutants. (D) BAL cell counts in control, Foxp1/4DKO, and Foxp1/4 NpyTKO mutants. *P < 0.01; **P < 0.005. Scale bar: 200 μm.
The airway epithelium of Foxp1/4DKO mutants contains large goblet-like cells with reduced expression of markers of differentiated club secretory cells, including Scgb1a1 (ref. 3 and Supplemental Figure 5). Interestingly, we found that expression of Scgb1a1 was partially rescued in the Foxp1/4 NpyTKO mutants (Supplemental Figure 6). In contrast, expression of multiple goblet cell markers that were elevated in the Foxp1/4DKO mutants was not altered by the additional loss of Npy expression (Supplemental Figure 6).
Current therapies for asthma target the inflammatory response or promote relaxation of airway smooth muscle shortening. While these treatments are effective in the short term, the underlying cause of asthma remains elusive, and many patients are insensitive to conventional therapy. Approximately half of asthma patients exhibit eosinophilic inflammation with increased levels of IL-4, IL-5, IL-13, and IgE (23, 24). In contrast, about half of asthma patients lack a strong eosinophilic inflammatory response, but exhibit a variable neutrophilic inflammatory response. Foxp1 and Foxp4 double mutants represent a model for noneosinophilic asthma, which remains poorly understood. In addition, patients with this disorder tend to be the least responsive to current therapies.
Previous studies have reported increased expression of Npy in animal models of allergen-induced AHR (19, 25). Moreover, Npy polymorphisms are associated with increased risk for asthma in overweight subjects (18, 25). Other reports have demonstrated that plasma levels of NPY were increased in asthmatics at rest and in acute exacerbations (26–28). Our study shows that increased expression of Npy in airway epithelium can induce an AHR phenotype in a paracrine manner (Supplemental Figure 6). This phenotype is recapitulated in a human lung explant assay, demonstrating that NPY can act on human small airways in a similar manner. Importantly, our studies are among the first, to our knowledge, to mechanistically link a critical transcription pathway in airway epithelium that activates a paracrine response to the promotion of AHR in the absence of a Th2-induced eosinophilic inflammatory response.
Methods
Animals.
Generation and genotyping of Foxp1fl/fl, Foxp4fl/fl, Scgb1a1-Cre, and Npy–/– mouse lines have been previously reported (3, 29). The mouse lines were kept on a mixed background consisting of C57BL/6 and 129SvJ. Control animals were derived from the same background and, when possible, the same litters as the mutant animals.
Microarray studies.
All original microarray data were deposited in the NCBI’s Gene Expression Omnibus (GEO GSE64774).
Statistics.
We used the R package lme4 (1, 30) to perform a linear mixed-effects analysis of the relationship between lung resistance and genotype. As fixed effects, we entered dose and genotype with an interaction term into the model. As random effects, we used a random intercept model for the subjects. P values were obtained by comparing the full model and reduced model without the genotype fixed effect using an F test with Kenward-Roger approximation (31). Other data are reported as mean ± SEM of at least 3 independent assays unless otherwise noted. Unpaired Student’s t test (2-tailed) was used for single comparisons and 1-way ANOVA for multiple comparisons. A P value of less than 0.05 was considered significant.
Study approval.
All animal procedures were approved by the Institute for Animal Care and Use Committee at the University of Pennsylvania.
See Supplemental Methods for additional information regarding these studies.
Author contributions
SL, RAP, and EEM designed and developed the assays and experiments. SL, CKW, JJ, MJ, HZ, GC, EY, WJ, SZ, YW, and MML performed experiments. MPM performed bioinformatics and statistical analysis. SL, RAP, and EEM wrote the manuscript.
Supplementary Material
Acknowledgments
These studies were supported by funding from the NIH to E.E. Morrisey (HL071589 and HL110942) and R.A. Panettieri Jr. (HL110942).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2016;126(5):1978–1982. doi:10.1172/JCI81389.
Contributor Information
Shanru Li, Email: shanruli@mail.med.upenn.edu.
Cynthia Koziol-White, Email: kcynthia@mail.med.upenn.edu.
Joseph Jude, Email: aantony@mail.med.upenn.edu.
Meiqi Jiang, Email: mjiang@mail.med.upenn.edu.
Hengjiang Zhao, Email: hezhao@mail.med.upenn.edu.
Gaoyuan Cao, Email: gaoyuan@mail.med.upenn.edu.
Edwin Yoo, Email: edwinyoo@mail.med.upenn.edu.
William Jester, Email: wjester@exchange.upenn.edu.
Su Zhou, Email: zhou5@mail.med.upenn.edu.
Yi Wang, Email: wangyi1@mail.med.upenn.edu.
Min Min Lu, Email: mlu2@mail.med.upenn.edu.
Edward E. Morrisey, Email: emorrise@mail.med.upenn.edu.
References
- 1.Douwes J, Gibson P, Pekkanen J, Pearce N. Non-eosinophilic asthma: importance and possible mechanisms. Thorax. 2002;57(7):643–648. doi: 10.1136/thorax.57.7.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fahy JV. Eosinophilic and neutrophilic inflammation in asthma: insights from clinical studies. Proc Am Thorac Soc. 2009;6(3):256–259. doi: 10.1513/pats.200808-087RM. [DOI] [PubMed] [Google Scholar]
- 3.Li S, et al. Foxp1/4 control epithelial cell fate during lung development and regeneration through regulation of anterior gradient 2. Development. 2012;139(14):2500–2509. doi: 10.1242/dev.079699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Amrani Y, et al. Expression and activation of the oxytocin receptor in airway smooth muscle cells: regulation by TNFα and IL-13. Respir Res. 2010;11: doi: 10.1186/1465-9921-11-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Banerjee A, et al. Trichostatin A abrogates airway constriction, but not inflammation, in murine and human asthma models. Am J Respir Cell Mol Biol. 2012;46(2):132–138. doi: 10.1165/rcmb.2010-0276OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cooper PR, Panettieri RA., Jr Steroids completely reverse albuterol-induced [beta]2-adrenergic receptor tolerance in human small airways. J Allergy Clin Immunol. 2008;122(4):734–740. doi: 10.1016/j.jaci.2008.07.040. [DOI] [PubMed] [Google Scholar]
- 7.Crapo RO, et al. Guidelines for methacholine and exercise challenge testing-1999. This official statement of the American Thoracic Society was adopted by the ATS Board of Directors, July 1999. Am J Respir Crit Care Med. 2000;161(1):309–329. doi: 10.1164/ajrccm.161.1.ats11-99. [DOI] [PubMed] [Google Scholar]
- 8.Mays LE, et al. Modified Foxp3 mRNA protects against asthma through an IL-10-dependent mechanism. J Clin Invest. 2013;123(3):1216–1228. doi: 10.1172/JCI65351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ghosh S, et al. B lymphocytes regulate airway granulocytic inflammation cytokine production in a murine model of fungal allergic asthma. Cell Mol Immunol. 2015;12(2):202–212. doi: 10.1038/cmi.2014.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tatemoto K, Carlquist M, Mutt V. Neuropeptide Y — a novel brain peptide with structural similarities to peptide YY and pancreatic polypeptide. Nature. 1982;296(5858):659–660. doi: 10.1038/296659a0. [DOI] [PubMed] [Google Scholar]
- 11.Higuchi H, Yang HY, Sabol SL. Rat neuropeptide Y precursor gene expression. mRNA structure, tissue distribution, and regulation by glucocorticoids, cyclic AMP, and phorbol ester. J Biol Chem. 1988;263(13):6288–6295. [PubMed] [Google Scholar]
- 12.Ericsson A, Hemsen A, Lundberg JM, Persson H. Detection of neuropeptide Y-like immunoreactivity and messenger RNA in rat platelets: the effects of vinblastine, reserpine, and dexamethasone on NPY expression in blood cells. Exp Cell Res. 1991;192(2):604–611. doi: 10.1016/0014-4827(91)90082-6. [DOI] [PubMed] [Google Scholar]
- 13.Zukowska-Grojec Z, et al. Neuropeptide Y: a novel angiogenic factor from the sympathetic nerves and endothelium. Circ Res. 1998;83(2):187–195. doi: 10.1161/01.RES.83.2.187. [DOI] [PubMed] [Google Scholar]
- 14.Dimitrijevic M, Stanojevic S. The intriguing mission of neuropeptide Y in the immune system. Amino Acids. 2013;45(1):41–53. doi: 10.1007/s00726-011-1185-7. [DOI] [PubMed] [Google Scholar]
- 15.Branchek TA, Gershon MD. Time course of expression of neuropeptide Y, calcitonin gene-related peptide, and NADPH diaphorase activity in neurons of the developing murine bowel and the appearance of 5-hydroxytryptamine in mucosal enterochromaffin cells. J Comp Neurol. 1989;285(2):262–273. doi: 10.1002/cne.902850208. [DOI] [PubMed] [Google Scholar]
- 16.Macia L, Rao PT, Wheway J, Sierro F, Mackay F, Herzog H. Y1 signalling has a critical role in allergic airway inflammation. Immunol Cell Biol. 2011;89(8):882–888. doi: 10.1038/icb.2011.6. [DOI] [PubMed] [Google Scholar]
- 17.Wheway J, Herzog H, Mackay F. NPY and receptors in immune and inflammatory diseases. Curr Top Med Chem. 2007;7(17):1743–1752. doi: 10.2174/156802607782341046. [DOI] [PubMed] [Google Scholar]
- 18.Jaakkola U, et al. Neuropeptide Y polymorphism increases the risk for asthma in overweight subjects; protection from atherosclerosis in asthmatic subjects — the cardiovascular risk in young Finns study. Neuropeptides. 2012;46(6):321–328. doi: 10.1016/j.npep.2012.09.005. [DOI] [PubMed] [Google Scholar]
- 19.Makinde TO, Steininger R, Agrawal DK. NPY and NPY receptors in airway structural and inflammatory cells in allergic asthma. Exp Mol Pathol. 2013;94(1):45–50. doi: 10.1016/j.yexmp.2012.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Erle DJ, Sheppard D. The cell biology of asthma. J Cell Biol. 2014;205(5):621–631. doi: 10.1083/jcb.201401050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kamm KE, Stull JT. The function of myosin and myosin light chain kinase phosphorylation in smooth muscle. Annu Rev Pharmacol Toxicol. 1985;25:593–620. doi: 10.1146/annurev.pa.25.040185.003113. [DOI] [PubMed] [Google Scholar]
- 22.Amrani Y, Da Silva A, Kassel O, Bronner C. Biphasic increase in cytosolic free calcium induced by bradykinin and histamine in cultured tracheal smooth muscle cells: is the sustained phase artifactual? Naunyn Schmiedebergs Arch Pharmacol. 1994;350(6):662–669. doi: 10.1007/BF00169372. [DOI] [PubMed] [Google Scholar]
- 23.Bradding P. Asthma: eosinophil disease, mast cell disease, or both? Allergy Asthma Clin Immunol. 2008;4(2):84–90. doi: 10.1186/1710-1492-4-2-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lambrecht BN, Hammad H. The immunology of asthma. Nat Immunol. 2015;16(1):45–56. doi: 10.1038/ni.3049. [DOI] [PubMed] [Google Scholar]
- 25.Wu ZX, Benders KB, Hunter DD, Dey RD. Early postnatal exposure of mice to side-steam tobacco smoke increases neuropeptide Y in lung. Am J Physiol Lung Cell Mol Physiol. 2012;302(1):L152–L159. doi: 10.1152/ajplung.00071.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Voedisch S, Rochlitzer S, Veres TZ, Spies E, Braun A. Neuropeptides control the dynamic behavior of airway mucosal dendritic cells. PLoS One. 2012;7(9): doi: 10.1371/journal.pone.0045951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dahlof C, Dahlof P, Lundberg JM, Strombom U. Elevated plasma concentration of neuropeptide Y and low level of circulating adrenaline in elderly asthmatics during rest and acute severe asthma. Pulm Pharmacol. 1988;1(1):3–6. doi: 10.1016/0952-0600(88)90003-8. [DOI] [PubMed] [Google Scholar]
- 28.Cardell LO, Uddman R, Edvinsson L. Low plasma concentrations of VIP and elevated levels of other neuropeptides during exacerbations of asthma. Eur Respir J. 1994;7(12):2169–2173. doi: 10.1183/09031936.94.07122169. [DOI] [PubMed] [Google Scholar]
- 29.Erickson JC, Clegg KE, Palmiter RD. Sensitivity to leptin and susceptibility to seizures of mice lacking neuropeptide Y. Nature. 1996;381(6581):415–421. doi: 10.1038/381415a0. [DOI] [PubMed] [Google Scholar]
- 30.Bates D, Machler M, Bolker BM, Walker SC. Fitting linear mixed-effects models using lme4. J Stat Softw. 2015;67(1):1–48. [Google Scholar]
- 31.Halekoh U, Hojsgaard S. Kenward-Roger approximation and parametric bootstrap methods for tests in linear mixed models — The R Package pbkrtest. J Stat Softw. 2014;59(9):1–32. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.