

Abstract
The TALE-class homeoprotein MEIS1 specifically collaborates with HOXA9 to drive myeloid leukemogenesis. Although MEIS1 alone has only a moderate effect on cell proliferation in vitro, it is essential for the development of HOXA9-induced leukemia in vivo. Here, using murine models of leukemogenesis, we have shown that MEIS1 promotes leukemic cell homing and engraftment in bone marrow and enhances cell-cell interactions and cytokine-mediated cell migration. We analyzed global DNA binding of MEIS1 in leukemic cells as well as gene expression alterations in MEIS1-deficent cells and identified synaptotagmin-like 1 (Sytl1, also known as Slp1) as the MEIS1 target gene that cooperates with Hoxa9 in leukemogenesis. Replacement of SYTL1 in MEIS1-deficent cells restored both cell migration and engraftment. Further analysis revealed that SYTL1 promotes cell migration via activation of the CXCL12/CXCR4 axis, as SYTL1 determines intracellular trafficking of CXCR4. Together, our results reveal that MEIS1, through induction of SYTL1, promotes leukemogenesis and supports leukemic cell homing and engraftment, facilitating interactions between leukemic cells and bone marrow stroma.
Introduction
Genetic cooperation is important to expanding the capabilities of certain mutations of oncogenes and/or tumor suppressors. However, the functional significance of such genetic cooperation has not yet been clarified. Moreover, the role of oncogenic activation in the expansion of malignant cells in vivo is only partially understood. Abdominal B–like (AbdB-like) HOX genes play an important role both in normal hematopoiesis and leukemogenesis (1–4). HOXA9 is overexpressed in human acute myeloid leukemia (AML) of poor prognosis, and AbdB-like HOX is a downstream target of mixed-lineage leukemia (MLL) fusion oncoproteins (5–7). Furthermore, AbdB-like HOX is also found fused to NUP98 in human myeloid neoplasms (8). These HOX genes possess transforming activity for hematopoietic cells when the genes are overexpressed (9). However, HOX aberrations are frequently associated with alterations of other genes, such as Meis1, NPM1, and Trib1/2, in human and murine AML (10–13).
Meis1 was first identified as a common retroviral integration site in BXH2 mouse AML (14). Of greater significance to our study, Meis1 has been found to be cooperatively activated with Hoxa7/a9 in AML (10), and it indeed promotes leukemogenic activities of Hoxa9 as well as its chimeric mutant NUP98-HOXA9 (15, 16). Meis1 encodes a TALE-class homeodomain protein, and it is essential for both fetal and adult hematopoiesis (17–20). Loss of Meis1 results in severe impairment of hematopoietic stem cell (HSC) function, and HSCs with the Meis1-null mutation do not support long-term hematopoiesis due to stem cell exhaustion. Possible target genes of MEIS1 include Msi2 and Hif1a, which might be important for Meis1 function (20, 21). Moreover, several hematopoiesis/leukemia-related target genes, including Cd34, Flt3, Myb, and Erg, have been identified (21, 22). However, the molecular mechanisms of Meis1 cooperativity specific to Hoxa9 have not been clarified. It is very likely that Meis1’s cooperative activity with Hoxa9 is only effective in vivo, since hematopoietic cells can be transformed by overexpression of Hoxa9 alone (23).
Identification of the target genes downstream from MEIS1 that are responsible for the leukemogenic activity of Meis1 and Hox is therefore of great importance. Here, we determined that synaptotagmin-like 1 (Sytl1, also known as Slp1) was a direct target of MEIS1 and replaced MEIS1’s cooperative activity with HOXA9. By interacting with Rab27a/b, synaptotagmin-like proteins facilitated intracellular trafficking of cytokine and chemokine receptors as well as transportation and secretion of vesicles to extracellular spaces (24, 25). Prompt trafficking of receptors helps leukemic cells to access bone marrow niches that secrete specific ligands. Thus, MEIS1 coordinately orchestrates leukemic cell engraftment and growth expansion in vivo by regulating target gene transcription.
Results
MEIS1 is required for Hoxa9-induced leukemogenesis in vivo but is dispensable in vitro.
To evaluate the function of MEIS1 in Hoxa9-induced leukemia, an immortalized cell line (H9M1) was established by introducing pMYs-Hoxa9-IRES-mKO and pMYs-loxP-Meis1-IRES-GFP-loxP retroviruses into primary murine bone marrow cells derived from the Rosa26-Cre-ERT2 mouse (Supplemental Figure 1A; supplemental material available online with this article; doi:10.1172/JCI81516DS1). Cell growth and the colony-forming activity of H9M1 cells were mildly reduced by 4-hydroxy-tamoxifen–induced (4-OHT–induced) (as shown in Meis1-deleted H9M1 cells [HΔM cells], Figure 1A). Significant promotion of self-renewal by MEIS1 expression was reported previously (26), suggesting that MEIS1 might affect cellular growth in a lineage- and/or cytokine-dependent manner in vitro. There were no morphologically significant changes due to Meis1, although a decrease of cytoplasmic granules was detected (Supplemental Figure 1A). Slight myeloid differentiation was also induced, as detected by a reduced frequency of CD34-positive cells and a mild increase of Gr-1–positive cells (Supplemental Figure 1B).
Figure 1. Hoxa9 and Meis1 cooperation in leukemogenesis: leukemic cell engraftment is supported by MEIS1.

(A) Comparison of the proliferation and colony-forming activities of H9M1 cells expressing (+) or lacking (–) Meis1. MEIS1 expression in H9M1 and HΔM cells was confirmed by Western blotting; blots are representative of 2 experiments. Cumulative numbers of H9M1 cells in liquid culture were counted. Means of the natural logarithm of cell numbers from 3 independent experiments are shown (*P < 0.05, 2-tailed Student’s t test). Colony numbers per 1,000 H9M1 cells in methylcellulose culture were measured, and representative culture plates are shown (3 experiments). (B) Leukemia-free survival of sublethally irradiated animals transplanted with 1 × 106 H9M1 cells are shown for H9M1 cells with (red line) or without (blue line) Meis1. (C) H9M1 cells in bone marrow 48 hours after transplantation were detected as an mKO-positive fraction by flow cytometry. A significant reduction of H9M1 cells in bone marrow was observed in animals transplanted with Meis1 KO cells. The number was restored by reintroduction of Meis1. Frequencies of mKO-positive cells in bone marrow are indicated as mean ± SEM of 3 independent experiments (**P < 0.01, 1-way ANOVA with Dunnett’s multiple comparison test). (D) Representative images of H9M1 cells in frozen bone marrow sections (3 experiments). DiO-stained H9M1 cells were detected, though they were absent in Meis1 KO mice, and were observed after Meis1 reintroduction into HΔM cells. Gr-1 is indicated by red fluoro-dye, and nuclei were counterstained with DAPI. Scale bar: 20 μm. (E) Engraftment activities were assessed by flow cytometry, which detected H9M1 or HΔM cells in bone marrow 2 weeks after transplantation as mKO-positive fractions. Data are representative of 3 independent experiments. (F) Coculture of H9M1 cells with OP9 cells. Cobblestone areas were established by H9M1 cells, but not by Meis1 KOs, and were restored by Meis1 reintroduction. Scale bar: 100 μm. Numbers of cobblestone areas (CFAs) are indicated as mean ± SEM of 3 independent experiments (**P < 0.01, 1-way ANOVA with Dunnett’s multiple comparison test).
In contrast, Meis1 KO by 4-OHT treatment completely abolished leukemia development of transplanted H9M1 cells (Figure 1B). Similar results were obtained in primary bone marrow cells transduced with Hoxa9/Meis1-expressing retrovirus (12). Hoxa9 expression was sufficient to increase replating activities and immortalization, whereas Meis1 did not enhance self-renewal (Supplemental Figure 2A). However, Meis1 expression markedly accelerated Hoxa9-induced leukemogenesis (Supplemental Figure 2B). The effect of Meis1 expression was also tested in mixed-lineage leukemia/eleven-nineteen-leukemia–induced (MLL-ENL–induced) leukemia, since MLL fusion proteins are upstream regulators of Hoxa9 and Meis1, and Meis1 is upregulated in AML by MLL fusions (27, 28). Meis1 conditional KO by tamoxifen treatment in vivo was achieved with recipient mice transplanted with MLL-ENL-transduced bone marrow cells with a Meis1fl/fl Rosa26-Cre-ERT2 background (20). As expected, MLL/eleven nineteen leukemia–induced (MLL-ENL–induced) leukemogenesis was markedly suppressed, except when leukemic cells acquired a deletion of the Meis1 loxP site in one allele (Supplemental Figure 2C). Granulocytic and monocytic differentiation was induced by Meis1 KO in MLL-ENL–induced AML in vitro (Supplemental Figure 2D), although the effect was limited. These results clearly indicate that Meis1 must cooperate with Hoxa9 for myeloid leukemogenesis in vivo. In contrast, the growth promotion and differentiation effects of Meis1 are weak in vitro.
MEIS1 contributes to leukemia cell homing and engraftment in bone marrow.
Upon occupation of hematopoietic niches, leukemia-initiating cells can interact with bone marrow stromal cells, maintain dormancy, increase their self-renewal activity, and develop drug resistance (29). Promotion of leukemogenesis by MEIS1 in vivo suggests that MEIS1 may support leukemic cell homing and engraftment into bone marrow. To address this question, 1 × 107 H9M1 cells were injected into C57BL/6 mice irradiated with 4 Gy, and the efficiency of homing and engraftment was examined by flow cytometry as well as by fluorescent microscopy. Forty-eight hours after injection, H9M1 cells constituted approximately 1% of total bone marrow cells, whereas the fraction almost disappeared in Meis1 KO mice. The homing activity was restored by reinfection of HΔM cells with the Meis1 retrovirus (Figure 1, C and D). These findings were confirmed in bone marrow sections in which we detected H9M1 cells prestained with DiO (Figure 1D). The difference was continuously observed for 2 weeks (Figure 1E), indicating that engraftment activities are also supported by MEIS1. The number of transplanted leukemic cells was also significantly diminished in the spleen (Supplemental Figure 3, A and B). The same results were obtained with an independent cell line, H9M11, that expresses Hoxa9 and Meis1 in the same genetic background (Supplemental Figure 3, C and D).
To gain insight into the mechanisms of leukemic cell homing and engraftment, the interaction between leukemic cells and bone marrow–derived stromal cells was examined using cocultivation of H9M1 cells and OP9 cells. Cobblestone-like areas of H9M1 cells on OP9 cells (a signature of stem cell–like activity, ref. 30) were frequently observed in the coculture, and the number of cobblestone areas was greatly decreased by Meis1 KO and recovered by Meis1 reintroduction (Figure 1F). These data indicate that MEIS1 supports leukemic cell homing and engraftment into hematopoietic niches in bone marrow and spleen through its alteration of cell-cell interaction.
MEIS1 mediates increased migratory activity upon cytokine stimulation.
A number of molecules mediate the interaction between hematopoietic cells/leukemic cells and stromal cells in the bone marrow niche (29). These include cytokines, chemokines, adhesion molecules, extracellular matrix proteins, and their specific receptors (31–34). To clarify the biological processes that are responsible for MEIS1-induced promotion of AML homing and engraftment, we compared expression of adhesion molecules, receptor tyrosine kinases, and chemokine receptors between H9M1 cells and HΔM cells and between H9M1 cells before and after engraftment. Frequencies of leukemic cells positive for 12 different integrin α chains, 4 β chains, 3 integrin complexes, PECAM-1, PGP-1, VCAM-1, and CSF1R were not decreased by Meis1 KO (Supplemental Table 1). CXCR4-positive, KIT-positive, and FLT3-positive cells were slightly decreased. Meis1 KO also diminished expression of integrin α IIb (CD41).
Next, we examined cytokine-stimulated migration activity of H9M1 cells. FLT3L, CXCL12, CSF2 (also known as GM-CSF), IL-3, and CSF1 (also known as M-CSF) enhanced migration by H9M1 cells, whereas SCF and TPO had no such effect (Figure 2A). Because H9M1 is an IL-3–dependent cell line similar to several murine myeloid cell lines, the significant effect of CXCL12 was intriguing, given the important function of the CXCL12/CXCR4 axis in hematopoiesis and the hematopoietic niche (35). The existence of CXCL12-stimulated migration that is Meis1 dependent was confirmed in another cell line (H9M11) (Supplemental Figure 4A). Indeed, in vitro migration of H9M1 cells upon CXCL12 stimulation, homing of H9M1 cells, and the interaction between H9M1 and OP9 cells were significantly suppressed by anti-CXCR4 or CXCL12 knockdown in OP9 cells, respectively, similar to behavior of Meis1 KO cells (Figure 2B and Supplemental Figure 4, B and C).
Figure 2. Association of the cell migratory activity with MEIS1.

(A) Cell migration assay. CXCL12, FLT3L, GM-CSF, IL-3, and M-CSF significantly enhanced cell migration of H9M1 but not HΔM cells. Results represent mean frequencies of migratory cells per 5 × 105 cells ± SEM of 3 independent experiments (**P < 0.01, 2-tailed Student’s t test). (B) Inhibition of interaction between H9M1 and OP9 cells by CXCL12 knockdown in OP9 cells. A significant decrease of cobblestone areas was observed in OP9 cells treated with two independent CXCL12-targeting siRNAs. Scale bar: 100 μm. Numbers of cobblestone areas are indicated as the mean ± SEM of 3 independent experiments (**P < 0.01, 1-way ANOVA with Dunnett’s multiple comparison test). (C) Global gene expression changes of H9M1 cells in response to Meis1 KO. The data sets of gene expression differences resulting from Meis1 KO were used for GSEA. Enrichment plots are shown for selected sets identified by GSEA. Normalized enrichment scores (NES), nominal P values (calculated by an empirical phenotype-based permutation test), and FDR q values are indicated.
To examine signaling pathways that are important for cell migration, gene expression profiles were compared between H9M1 and HΔM cells. The microarray analysis showed that 551 genes were downregulated (<1.5) by Meis1 KO in H9M1 cells (accession number GSE50645). The 551 genes include those known to be downstream from Meis1 (Cd34, Flt3, and Msi2) as well as leukemia-associated genes, such as Erg, Gfi1b, Hhex, Myc, Nup98, and Trib1. Gene set enrichment analysis (GSEA) was carried out to identify important signaling pathways involved in transcriptional regulation by MEIS1. Interestingly, the pathways for chemokine/cytokine and receptor interaction, protein transport, and focal adhesion were enriched by altered Meis1 expression (Figure 2C and Supplemental Figure 5). The findings suggest that MEIS1-induced transcriptional regulation affected cell-cell interaction that promoted leukemic cell engraftment in vivo.
Identification of MEIS1 target genes that are responsible for HOXA9 cooperativity and leukemic cell homing/engraftment.
MEIS1 functions as a sequence-specific transcriptional activator (36, 37). Therefore, we tried to identify the target genes of MEIS1 that were involved in leukemic cell homing, engraftment, and/or cell-cell interaction. ChIP, followed by sequencing (ChIP-Seq) analyses, was carried out to identify genome-wide MEIS1-binding sites in H9M1 cells. In addition, HOXA9- and PBX-binding sites were assessed in the same cell line. This was done because clarification of cooperative DNA binding among MEIS1, HOXA9, and PBX is important to understanding how these homeodomain proteins regulate transcription in leukemia cells (21, 22). Using a FDR cutoff of 0.1, a total of 6,324 MEIS1-binding regions were identified (Figure 3A and Supplemental Table 2) (data are available at NCBI Gene Expression Omnibus, with accession number GSE48679). To validate the results, MEIS1 enrichment at representative peaks was confirmed by ChIP-qPCR for the Maf locus that contains a MEIS1 consensus binding sequence, and MEIS1 binding in hematopoietic cells was previously reported (ref. 38 and Supplemental Figure 6A). The ChIP-Seq analysis was performed in duplicate, and similar results were obtained in two experiments.
Figure 3. Sytl1 is a MEIS1 transcriptional target in leukemogenesis.

(A) Schematic diagram for target gene identification. (B) Sytl1 cooperated with Hoxa9 in leukemogenesis. Kaplan-Meier survival curves for recipient mice transplanted with Hoxa9-, Sytl1-, Hoxa9/Sytl1-, or Hoxa9/Meis1-transduced bone marrow cells. **P = 0.015, log-rank test. (C) ChIP-Seq analysis showed a MEIS1-binding peak 2 kb upstream of Sytl1. (D) Downregulation of Sytl1 by Meis1 KO in H9M1 cells. Representative gels of RT-PCR (3 experiments) and Q-RT-PCR (3 experiments) for Sytl1 mRNA expression and immunoblotting for the SYTL1 protein using α-SYTL1 in H9M1 (3 experiments). Immunoblotting for GAPDH and HA-MEIS1 proteins was performed on separate gels. Mean values of relative Sytl1 mRNA expression were normalized to Gapdh mRNA (**P < 0.01, 1-way ANOVA with Dunnett’s multiple comparison test). (E) Sytl1 expression was upregulated by Meis1 overexpression in 32Dcl3 cells. Sytl1 mRNA expression relative to Gapdh mRNA based upon Q-RT-PCR (n = 3, **P < 0.01, 2-tailed Student’s t test). (F) Luciferase reporter assay. A 2-kb fragment upstream of Sytl1 was introduced upstream of the luciferase cDNA. The mutant clone was created by introducing TGACAG to TGTTAG mutations into 3 putative MEIS1-binding sequences (MBS). Relative luciferase activities are shown in the presence or absence of MEIS1. Data represent mean ± SEM of 3 independent experiments (**P < 0.01, 2-tailed Student’s t test).
The ChIP-Seq analyses for HOXA9 and total PBX revealed 770 and 3,198 peaks, respectively. The numbers of HOXA9 and PBX peaks were increased to 2,892 and 4,163 upon Meis1 KO, respectively (Supplemental Table 2). The exact cause of the increased HOXA9 and PBX peaks in the absence of MEIS1 is unclear; however, the components of DNA-binding complexes might be affected by depletion of MEIS1. Within 6,324 MEIS1-binding peaks, PBX binding was associated with 1,966 loci, whereas 564 peaks were associated with HOXA9 binding. While we observed 543 HOXA9/PBX peaks, there were 465 MEIS1/PBX/HOXA9 trimeric peaks, indicating that the majority of the HOXA9/MEIS1- and HOXA9/PBX-binding complexes were in the HOXA9/PBX/MEIS1 complex (Supplemental Figure 6B). The data suggest that MEIS1 upregulation affects not only its own target genes, but also PBX/HOXA9-regulated genes. Approximately 30.1% of the binding sites were located in the introns of known genes and less than 4% were located in the promoter regions within 1 kb upstream of known transcription start sites (Supplemental Figure 6C). Most of the MEIS1-binding peaks (84%) and the HOXA9-binding peaks (70.9%) that were reported in the ChIP-Seq analysis of Huang et al. (22) are also identified in our current analysis. These results suggest that our ChIP-Seq analysis provided reliable information in spite of the low number of experimental repetitions.
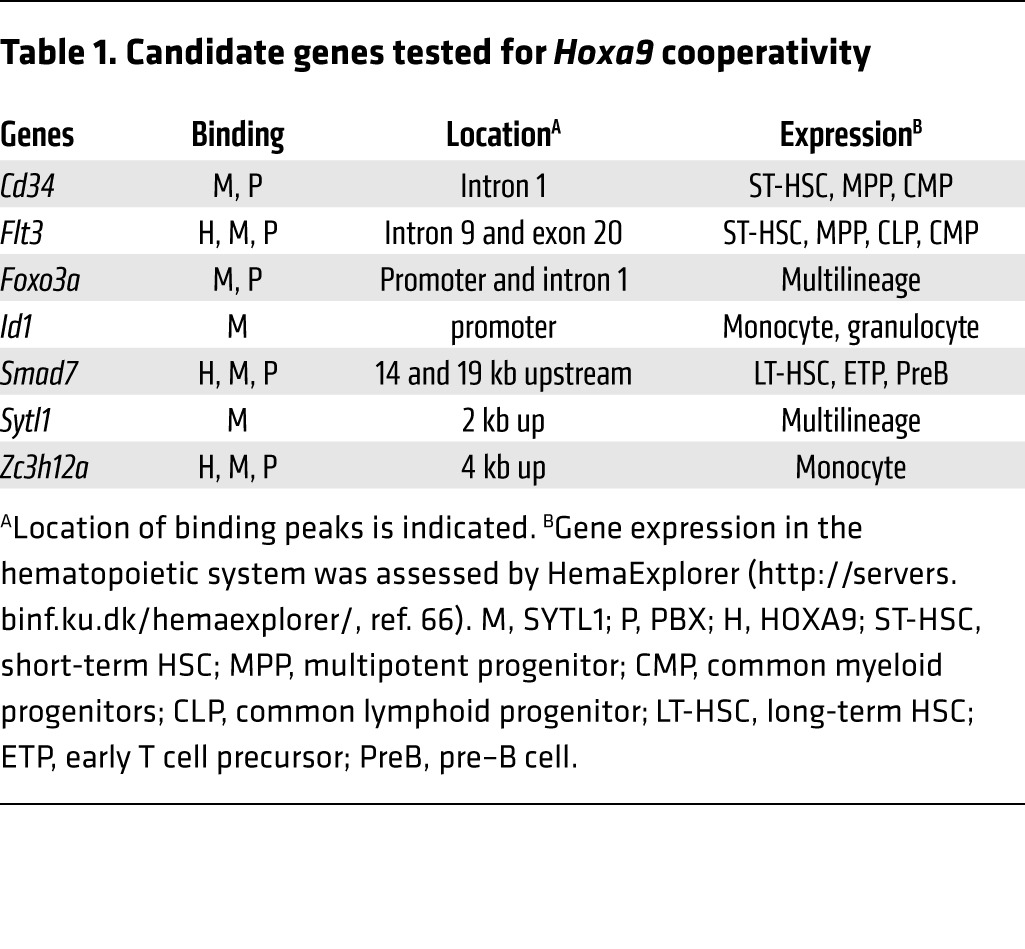
A total of 2,644 genes that have MEIS1 peaks within 30 kb of known gene bodies have been annotated (Supplemental Table 3). The results of gene expression profiles were compared with the ChIP-Seq data, and expression of 281 genes containing MEIS1-binding peaks was found to be downregulated by Meis1 KO (Figure 3A and Supplemental Table 3). Evaluation of the known functions of those 281 genes indicated that 32 of the 281 genes are implicated in hematopoiesis and/or malignancy (Figure 3A and Supplemental Table 4). Seven genes (Cd34, Flt3, Foxo3a, Id1, Smad7, Sytl1, and Zc3h12a) were selected to identify the MEIS1 target gene responsible for MEIS1’s HOXA9 cooperativity in vivo (Table 1).
Table 1. Candidate genes tested for Hoxa9 cooperativity.
Sytl1 is a transcriptional target of MEIS1 that cooperated with HOXA9 in leukemogenesis.
We examined the 7 aforementioned genes for their oncogenic cooperation with HOXA9 using cotransduction of retroviruses bearing the candidate genes with Hoxa9 into mouse primary bone marrow cells followed by bone marrow transplantation. Sytl1 is 1 of 2 genes that significantly accelerated Hoxa9-induced leukemogenesis (Figure 3B). Another cooperative gene that accelerated Hoxa9-induced leukemogenesis is Smad7. The others failed to show such cooperative activities with Hoxa9. Like Meis1, Sytl1 by itself does not show any leukemogenic activity (Figure 3B). Sytl1 belongs to the synaptotagmin-like or Slp gene family, which encodes a Rab27-binding protein with a 58-kDa molecular weight (39). In association with Rab27, SYTL1 family proteins facilitate transportation and secretion of cytoplasmic granules, such as platelet granules and zymogen granules, to cellular surfaces and transportation of membrane proteins such as receptor tyrosine kinases (24, 25, 40).
The MEIS1-binding peak is located 2 kb upstream from the transcriptional start site of Sytl1 (Figure 3C), and MEIS1 binding at the locus was validated by quantitative ChIP-PCR (Supplemental Figure 6D). Meis1 KO in H9M1 cells showed downregulated Sytl1 mRNA as well as protein (Figure 3D). In MLL-ENL–induced AML, 4-OHT–mediated KO of Meis1 downregulated Sytl1 (Supplemental Figure 6E). This was also observed in H9M11 cells (Supplemental Figure 6E). Furthermore, Meis1 overexpression in 32Dcl3 myeloid cells increased Sytl1 expression (Figure 3E). A reporter assay using a 2-kb fragment containing the Sytl1 promoter showed that MEIS1 expression increased the luciferase activity that was abolished by introducing mutations into putative MEIS1-binding sites (Figure 3F). These results indicate that Sytl1 cooperates with Hoxa9 in myeloid leukemogenesis as a direct transcriptional target of MEIS1.
SYTL1 supports homing and engraftment of leukemia.
To clarify the role of SYTL1 in its cooperation with HOXA9 in leukemogenesis and leukemic cell homing and engraftment, Sytl1 was retrovirally introduced into HΔM cells. As expected, Sytl1 introduction restored leukemic cell homing and engraftment in bone marrow and spleens (Figure 4, A and B, and Supplemental Figure 7, A–C). In addition, cobblestone formation of leukemic cells in OP9 coculture and CXCL12-induced cellular migration were recovered by Sytl1 expression (Figure 4, C and D). These results strongly indicate that SYTL1, a molecule downstream from MEIS1, initiates MEIS1’s function by cooperating with HOXA9. That is, it promotes leukemic cell engraftment and cell-cell interaction with stromal cells. Furthermore, bone marrow cells derived from Sytl1 homozygous KO mice (41) transduced with the Meis-Hoxa9 retrovirus failed to show AML development, and AML development was recovered by exogenous introduction of Sytl1 (Figure 4E). Coexpression of Hoxa9 and Meis1 in Sytl1 KO bone marrow cells supported neither bone marrow homing nor cellular migration upon CXCL12 stimulation, and these activities were rescued by Sytl1 introduction (Figure 4, F and G). These observations are in contrast to the results obtained in wild-type bone marrow cells in which both MEIS1 and SYTL1 promoted homing activity (Supplemental Figure 7D). The expression level of exogenous SYTL1 was comparable with endogenous expression in the presence of MEIS1 (Figure 4H). Moreover, knockdown of Sytl1 or Rab27b inhibited cellular migration of 32Dcl3 cells upon CXCL12 stimulation (Figure 4I). Similar findings were obtained by direct injection of H9M1 cells into bone marrow (Supplemental Figure 7E), indicating that the phenomena were not caused by artificial experimental conditions, i.e., bone marrow transplantation via the tail vein. Collectively, these data indicate that Sytl1 is essential for Meis1-related leukemogenesis. On the other hand, suppression of Sytl1 expression (by doxycycline-induced expression of shRNA) inhibited homing, but it did not reduce in vivo expansion of leukemic cells (Supplemental Figure 8). These findings suggest that Sytl1 activation might be very effective in the early stage of leukemogenesis, at which time genetic and/or epigenetic abnormalities have not accumulated.
Figure 4. SYTL1 supports leukemic cell engraftment.

(A) Sytl1 was retrovirally introduced into HΔM cells. Forty-eight hours after transplantation, bone marrow samples were analyzed for mKO-positive fractions by flow cytometry. Data are representative of 3 experiments. (B) Representative flow cytometric analysis (same as A) was performed 2 weeks after transplantation (3 experiments). (A and B) Frequencies of mKO-positive cells in bone marrow are indicated. (C) Representative images of OP9 coculture experiments (3 experiments). Cobblestone areas were recovered after SYTL1 was introduced into HΔM cells. The numbers of cobblestone areas are indicated. Scale bar: 100 μm. (D) Cell migration assay. H9M1, HΔM, and HΔM/SYTL1 cells were loaded onto chamber inserts. Cells migrated to the lower chamber containing CXCL12. Mean values of migratory cells per 5 × 105 cells ± SEM. (E) Sytl1 deletion abrogated HOXA9/MEIS1-induced leukemogenesis. Kaplan-Meier survival curves are shown for recipients transplanted with Hoxa9/Meis1-transduced bone marrow cells on a wild-type or Sytl1–/– background and Sytl1/Hoxa9/Meis1-transduced bone marrow cells on a Sytl1–/– background. (F) Homing of Hoxa9-transformed bone marrow cells of Sytl1–/– background was enhanced by Sytl1 but not by Meis1. Frequencies of mKO-positive cells in bone marrow were measured 48 hours after transplantation. (G) Migration upon CXCL12 stimulation of Hoxa9-transformed bone marrow cells of Sytl1–/– background was enhanced by Sytl1 but not by Meis1. All the experiments were performed in triplicate. (H) The expression levels of both endogenous and exogenous SYTL1 proteins were assessed by immunoblotting; blots are representative of 2 independent experiments. (I) Knockdown of Sytl1 or Rab27b inhibited cellular migration of 32Dcl3 cells after CXCL12 stimulation. (A–D, F, and G–I) n = 3, **P < 0.01, 1-way ANOVA with Dunnett’s multiple comparison test. Mean ± SEM is shown throughout.
SYTL1 is expressed in human AML and has key functions.
The important role of SYTL1 in human AML was also examined by knocking down SYTL1 with shRNA. When NOD/SCID mice underwent bone marrow transplantation with RS4;11 human AML cells with a fused MLL-AF4 gene, we observed suppression of homing and engraftment (Figure 5, A and B). As a result, SYTL1 knockdown extended the survival of recipient NOD/SCID mice transplanted with RS4;11 by 14 days, and during this time period, there were no detectable leukemic cells in peripheral blood (Figure 5C). Moreover, cellular migration upon CXCL12 stimulation was decreased (Figure 5D). We then analyzed the gene expression profile of 526 human AML cases. The analysis revealed that SYTL1 upregulation was markedly associated with that of MEIS1, HOXA7, and HOXA9 (Figure 5E). Coexpression of SYTL1 and MEIS1 is also remarkable in an AML subgroup characterized by having NPM1 mutations that are frequently associated with HOX gene overexpression (11). Interestingly, high levels of SYTL1 expression were observed in MLL-associated AML, with BREloEVI1hi expression but not BREhiEVI1lo expression (refs. 42, 43, and Figure 5E), suggesting that SYTL1 upregulation might be associated with poor prognosis in MLL-related AMLs. Collectively, these findings suggest that SYTL1 upregulation is closely associated with MEIS1 expression and that SYTL1 may be a key factor in HOX/MEIS1-related human AML.
Figure 5. SYTL1 functions in human AML.

(A) SYTL1 silencing inhibits homing of RS4;11 AML cells in NOD/SCID mice. Bone marrow samples were analyzed for the human CD45-positive fraction by flow cytometry (representative of 3 experiments). Data are compared between shSYTL1-treated (shSytl1 #4 and #5) and control shRNA (negative control) RS4;11 cells. Efficiencies of SYTL1 silencing were confirmed by RT-PCR. GAPDH, which was run on a separate gel, is shown as a loading control of cDNA. (B) SYTL1 silencing inhibits engraftment of RS4;11 AML cells in NOD/SCID mice. Bone marrow samples were analyzed for the human CD45-positive fraction by flow cytometry (n = 3). SYTL1 knockdown by shSYTL1 was confirmed by immunoblotting; blots are representative of 3 independent experiments. (A and B) Frequencies of human CD45-positive cells in bone marrow are indicated (mean ± SEM). (C) Survival curve of NOD/SCID mice transplanted with RS4;11 cells with shSYTL1 lentiviral vectors or empty vector. (D) Suppression of cell migration by SYTL1 knockdown. RS4;11 cells were transduced with lentiviral vectors bearing shSYTL1, and frequencies of cell migration were examined in the presence or absence of CXCL12. Data represent mean ± SEM of 3 independent experiments. (E) Pairwise correlations between gene expression profiles of 526 AML samples hybridized to the Affymetrix HGU 133Plus 2.0 GeneChips identified subsets with high SYTL1, HOXA9, HOXA7, and MEIS1 expression levels. The bar next to each sample indicates AML with the following genetic mutations in red: A: t(8;21), B: t(15;17), C: inv(16), D: CEBPA double mutation, E: CEBPA single mutation, F: NPM1 mutation, and G: 11q23 abnormalities/MLL fusions. Histograms next to the bar indicate expression levels of the following: 1: SYTL1, 2: HOXA9, 3: HOXA7, 4: MEIS1. Correlation coefficients were 0.33, 0.32, and 0.40 between SYTL1 and HOXA9, HOXA7, and MEIS1, respectively. An inset is enlarged to show two subsets of MLL-associated AML. I: BREhiEVI1lo, II: BREloEVI1hi. (A, B, and D) **P < 0.01, 1-way ANOVA with Dunnett’s multiple comparison test.
Sytl1 facilitates cellular responses to CXCL12 stimulation and promotes membrane trafficking of CXCR4.
Previous studies reported that the synaptotagmin family of proteins modulated cellular migration mediated by CXCL12 and CXCR4 signaling (44, 45). Since Meis1 KO suppressed the migratory activity induced by CXCL12, the role of SYTL1 in cell migration and CXCR4 status was further examined. SYTL1 overexpression in 32Dcl3 cells significantly enhanced CXCL12-induced cell motility (Figure 6, A and B, and Supplemental Videos 1 and 2). Moreover, Meis1 KO in H9M1 cells significantly suppressed the incorporation of CXCL12-coated beads, and the suppression was restored by Sytl1 expression (Figure 6C). Prolonged CXCR4 expression on the cellular surface was observed in H9M1 and HΔM/SYTL1 cells, compared with that in HΔM cells (Figure 7, A and B), and similar results were obtained in Sytl1-expressing 32Dcl3 cells (Supplemental Figure 9, A and B). Prompt internalization of CXCR4 was observed in Hoxa9-expressing cells, regardless of Meis1 or Sytl1 expression (Figure 7B), indicating that SYTL1 promotes membrane trafficking of CXCR4 but does not inhibit internalization of the receptor. Increased and prolonged phosphorylation of ERK1/2 and AKT upon CXCL12 stimulation was exhibited in cells expressing SYTL1 (Figure 7C and Supplemental Figure 9C). In addition, a close association between H9M1 or HΔM/SYTL1 cells and CXCL12-abundant reticular (CAR) cells (46) was frequently observed in the bone marrow of recipient mice (Figure 7D and Supplemental Figure 9D). Collectively, these results suggest that SYTL1 promotes trafficking of CXCR4 from cytoplasmic vesicles to the plasma membrane following the binding to CXCL12 and internalization (Figure 7E). Since the CXCR4/CXCL12 axis also plays a role in the maintenance of HSCs, we analyzed hematopoiesis in Sytl1 KO mice. The colony-forming activity of Sytl1 KO bone marrow cells was mildly increased for multilineage progenitors (Supplemental Figure 10A). However, comparing Sytl1 KO and wild-type bone marrow, there were no significant differences in the frequencies of SLAM cells (CD34- and CD48-negative and CD150-positive LSK cells) or LSK cells (Supplemental Figure 10B). These results suggest possible redundant functions by other SYT/SYTL1 family proteins in normal hematopoiesis.
Figure 6. Sytl1 increases CXCL12/CXCR4 signaling.

(A) Motility of 32Dcl3 cells with or without Sytl1 overexpression was observed during a 30-minute period of stimulation by CXCL12-coated beads. The original position of each cell is indicated as a white mark at 10, 20, or 30 minutes. See Supplemental Videos 1 and 2 for videos of the same cells. Representative images of 3 independent experiments are shown. Scale bar: 20 μm. (B) CXCL12-induced chemotaxis was quantified by measuring the accumulated distance in 40 cells each for SYTL1-expressing and -nonexpressing 32Dcl3 cells. Average accumulated distances (in number of 280-nm units) are indicated in boxes (n = 3, *P < 0.05; **P < 0.01, Mann-Whitney test). (C) Frequent incorporation of CXCL12-coated beads in H9M1 and HΔM/SYTL1 cells. Cells were seeded into VCAM1-coated chambers. The cells were fixed with 4% paraformaldehyde 5 minutes after addition of CXCL12-coated beads. Representative images of 3 independent experiments (arrows) are shown. The number of cells incorporating CXCL12-coated beads was measured. Frequencies of positive cells are indicated as the mean ± SEM of 3 independent experiments (**P < 0.01, 2-tailed Student’s t test). Scale bar: 20 μm.
Figure 7. SYTL1 promotes membrane trafficking of CXCR4 and activates downstream signaling.

(A) Representative immunofluorescence of H9M1, HΔM, or HΔM/SYTL1 cells after the indicated incubation periods with CXCL12 (3 experiments). Alexa Fluor 350–conjugated wheat germ agglutinin (WGA) was used to indicate plasma membrane. Scale bar: 20 μm. (B) The expression of CXCR4 on the surfaces of H9M1, HΔM, or HΔM/SYTL1 cells was analyzed by flow cytometry at the indicated times after CXCL12 stimulation. The cells were incubated with the anti-CXCR4 antibody after (same staining condition as A) or before CXCL12 stimulation. The data are representative of 3 independent experiments. (C) ERK and AKT phosphorylation after CXCL12 stimulation in the indicated cell types; blots are representative of 3 independent experiments. (D) Juxtaposition of H9M1 or HΔM/SYTL1 cells and CAR cells in bone marrow. CAR cells are stained with anti-S100. Representative images of 3 independent experiments. Quantification with statistical analysis is shown in Supplemental Figure 9D. Scale bar: 20 μm. (E) Proposed model showing enhanced membrane trafficking of CXCR4 after SYTL1 upregulation. CXCR4 is internalized and ubiquitinated upon binding to CXCL12. However, CXCR4 is rapidly provided from its reservoir on the limiting membrane of multivesicular bodies in the presence of SYTL1 and Rab27b.
Discussion
Here, we demonstrate that Meis1 and Sytl1 cooperate with Hoxa9 in vivo (but not in vitro) in promoting myeloid leukemogenesis. Leukemic cell homing and engraftment in bone marrow niches is an important early event in MEIS1/HOXA9 cooperation. It has been difficult to clarify the fundamental mechanisms for in vivo activity of transcription factors in the leukemogenic process. Identification of MEIS1-binding sites in the whole genome, evaluation of binding using microarray analysis, and cotransduction experiments enabled us to identify Sytl1 as a MEIS1 target gene responsible for cooperativity with HOXA9. Coexpression of SYTL1, HOXA7/A9, and MEIS1 in a subset of human AML supports the functional association of SYTL1 with these homeodomain proteins.
SYTL1 is a cytoplasmic protein that belongs to the SYTL1/JFC1 family that interacts with Rab family proteins to facilitate membrane trafficking of cytoplasmic granules and proteins (47–49). SYTL1 family proteins associate with the plasma membrane using C-terminal C2 motifs, and they also recognize Rab family proteins through their N-terminal regions (25, 50). SYTL1- and Rab-mediated vesicular trafficking plays an important role in a variety of cellular processes, including cellular migration, secretion of bioactive structures/substances, cell adhesion, and luminal formation (24, 25, 51, 52).
This study demonstrates for the first time to our knowledge that SYTL1 is involved in leukemogenesis and in the modulation of CXCR4 trafficking and signaling. The enhanced response to CXCL12 stimulation is mediated by SYTL1 expression. The results strongly suggest that the trafficking of CXCR4 helps leukemic cells obtain chemokine signals produced by bone marrow microenvironment CAR cells, resulting in prompt homing in bone marrow niches. Suppression of leukemogenic activity as well as homing and engraftment of Hoxa9/Meis1 in Sytl1 KO hematopoietic cells support this possibility. In addition, KO of Meis1 decreased cytoplasmic granules, suggesting that trafficking of cytoplasmic granules might be affected by SYTL1 expression.
Modification of receptors and cytokine signaling pathways other than the CXCR4/CXCL12 axis by SYTL1 should also be noted. FLT3 is one such receptor. Migration of H9M1 cells stimulated by FLT3L was inhibited by Meis1 KO and restored by Sytl1 expression (Figure 2A). Although a previous study showed that Flt3 gene expression was dispensable for Meis1/Hoxa9-induced leukemogenesis (53), additional effects of enhanced FLT3 signaling might contribute to MEIS1-related leukemogenesis. Furthermore, Flt3 is a transcriptional target of MEIS1 (Supplemental Tables 3 and 4), suggesting that MEIS1 orchestrates its function from Flt3’s transcription to FLT3’s transportation.
Previous ChIP-Seq studies of MEIS1-binding sites in hematopoietic cells revealed that the majority of MEIS1-binding loci were located outside of promoters (22, 38), a finding that was confirmed by this study. PBX proteins are TALE-class homeodomain proteins, as is MEIS1. They are DNA-binding cofactors of MEIS1, with a strong affinity for a bipartite DNA sequence (36). The close correlation between MEIS1 and PBX was highlighted by recent ChIP-Seq analyses of E11.5 mouse embryo trunks (54). In our analyses, a close interaction between MEIS1 and PBX in DNA binding was confirmed in vivo, and the presence of the MEIS1/PBX/HOXA9 trimeric complexes appeared to be common. Given the proposed role of MEIS1 as a transcriptional activator (37, 55), the current microarray and ChIP-Seq analyses indicate that target genes of PBX and/or HOXA9 are actively regulated by MEIS1. Sytl1 was not identified as a target of MEIS1 in mouse HSCs (38). The differences in DNA binding of transcription factors as well as differences in transcriptional regulation between normal and neoplastic cells might be caused by alterations in chromatin conditions at target loci (56). In contrast, known target genes of MEIS1, such as Msi2 (20) and Hif1a (21), were also identified in the present study (Supplemental Table 3). These data suggest that MEIS1 transcriptional regulation may have important roles in the musashi/numb signaling axis, asymmetric cell division, and leukemic cell maintenance in hypoxic niches (21, 57).
In conclusion, the present study directly demonstrates that MEIS1 abnormally affects transcriptional regulation and plays important roles in HOXA9-induced oncogenic events in vivo. In addition, we showed the role of the SYTL1 protein family in leukemogenesis and intracellular trafficking of CXCR4. The onset of leukemia in animals transplanted with HOXA9/SYTL1-expressing bone marrow cells was slightly delayed, compared with that in those transplanted with HOXA9/MEIS1-expressing bone marrow cells, suggesting that other downstream genes, such as Smad7, which was also identified as a MEIS1 downstream gene, might reinforce MEIS1-associated leukemogenic activity. Nevertheless, the fact that SYTL1 facilitated interaction between leukemic cells and the bone marrow niche suggests new therapeutic approaches to eliminate leukemic stem cells that occupy hematopoietic niches and are otherwise resistant to chemotherapy.
Methods
Plasmids.
Hoxa9, Meis1, Sytl1, and MLL-ENL (a gift from Tetsuya Nosaka, Mie University, Mie, Japan) cDNAs were cloned into pMYs retroviral vectors. The loxP sites were inserted to encompass MEIS1-IRES-GFP in the pMYs vector.
Cells.
Bone marrow cells were prepared from 8-week-old female Rosa26-Cre-ERT2 mice (58) 5 days after injection of 150 mg 5-fluorouracil/kg body weight (Kyowa Hakko Kogyo). Bone marrow cells were cultured for 24 hours in IMDM (Invitrogen) supplemented with 10% FBS (HyClone) and 10 ng/ml IL-6, 10 ng/ml IL-3, and 100 ng/ml SCF (R&D Systems). Retroviral stocks of pMYs-HOXA9-IRES-mKO and pMYs-loxP-MEIS1-IRES-EGFP-loxP were added to the medium containing bone marrow cells with 6 μg/ml Polybrene (Sigma-Aldrich) and then spun at 1,400 g for 2 hours. The cells were plated on Petri dishes containing Methocult M3534 methylcellulose medium (Stem Cell Technologies) at 1 × 104 cells per plate, and the H9M1 cells were established by replating the same number of cells 3 times. Expression of HOXA9 and MEIS1 was confirmed by detecting kusabira orange (mKO) and EGFP by flow cytometry as well as immunoblotting. For conditional KO of exogenous Meis1, 2 μM 4-OHT (Sigma-Aldrich) was added to the medium, and Meis1 deletion was confirmed by genomic PCR. 32Dcl3 cells were maintained in RPMI-1640 medium containing 10% FBS and 10 ng/ml IL-3. Human RS4;11 AML cells were obtained from ATCC and maintained in IMDM supplemented with 10% FBS.
Antibodies.
The following antibodies were obtained from eBioscience and were used for flow cytometry: rat anti-mouse CD11b (M1/70), rat anti-mouse Ly-6G/Gr-1 (RB6-8C5), rat anti-mouse CD117 (2B8), rat anti-mouse Ly-6A/E/Sca-1 (D7), rat anti-mouse CD34 (RAM34), mouse anti-human CD45 (HI30), rat anti-mouse CXCR4 (2B11), hamster anti-mouse CD3 (145-2C11), rat anti-mouse CD4 (RM4-5), rat anti-mouse CD8 (53-6.7), rat anti-mouse B220 (RA3-6B2), rat anti-mouse Ter119 (TER-199), rat anti-mouse c-kit (2B8), rat anti-mouse FCγR (clone 93), hamster anti-mouse CD48 (HM48-1), rat anti-mouse CD150 (mShad150), and mouse anti-human CD45 (HI30). Antibodies used for immunofluorescence staining were as follows: rat anti-mouse Ly6H/Gr-1 (RB-8C5, eBioscience), mouse anti-CD31 (390, BioLegend), and rabbit anti-S100 (Dako). Antibodies used for Western blotting, co-IP, and ChIP were as follows: rabbit anti-MEIS1 (ab19867, Abcam), rabbit anti-FLAG (F7425, Sigma-Aldrich), rabbit anti-PBX1/2/3 (C-20, Santa Cruz Biotechnology), rabbit anti-JFC1/SYTL1 (H-80, Santa Cruz Biotechnology), mouse anti-GAPDH (9GHT, HyTest), mouse anti-HA (ab-hatag, Invivogen), mouse anti-c-myc (9E10, Roche), rabbit anti-phospho p44/42 MAPK T202/Y204 (9101, Cell Signaling), rabbit anti-p44/42 MAPK (9102, Cell Signaling), rabbit anti-phospho AKT (S473, Cell Signaling), and mouse anti-AKT (2H10, Cell Signaling). Antibodies used for adhesion molecule analysis by flow cytometry are shown in Supplemental Table 1.
Replating assay.
H9M1 cells or retrovirally transduced bone marrow cells were plated in Petri dishes in Methocult M3534 (STEMCELL Technologies), and colonies were counted.
Animals and bone marrow transfer.
The Meis1 conditional KO mouse was generated by inserting loxP sequences into introns 7 and 8 (20). The Rosa26-Cre-ERT2 mouse was provided by Tyler Jacks (Massachusetts Institute of Technology, Cambridge, Massachusetts, USA), and the Sytl1 KO mouse was provided by Mitsunori Fukuda (Tohoku University, Sendai, Japan). Both Meis1 and Sytl1 KO mice are on the C57BL/6 background. C57BL/6J recipient mice were irradiated (8.5-Gy X-rays for leukemogenesis assays with primary bone marrow cells or 4-Gy X-rays for assays with cell lines; MBR-1520A, Hitachi Medico) and injected with primary bone marrow or leukemic cells. For transplantation of primary bone marrow cells, hematopoietic stem/progenitor cells were enriched by 5-FU administration 5 days before preparation and cotransduced with the indicated retroviruses, and 1 × 106 unsorted cells were injected into each mouse. For transplantation of RS4;11 human AML cells, 1 × 106 cells were injected into (2-Gy X-ray) NOD/SCID mice. Bone marrow cells and leukemic cells were injected via tail vein or directly into femoral bone. Mice were monitored daily for evidence of disease, and smears of peripheral blood were examined every week. The onset of AML was determined by detecting when immature myeloid cells constituted at least 20% of the leukocytes in peripheral blood (59). All of the diseased mice were subjected to autopsy and analyzed morphologically and by flow cytometry using a FACSCalibur flow cytometer (BD Biosciences).
OP9/H9M1 coculture assay.
Bone marrow–derived OP9 stromal cells were cultured in 6-well plates. At confluence H9M1 cells were added to the wells. The number of cobblestone areas was compared between MEIS1-positive and -negative or between SYTL1-expressing and -nonexpressing cells after a 48-hour incubation.
Leukemic cell homing and engraftment.
Mice were subjected to 4.0-Gy irradiation and injected intravenously with 1 × 107 H9M1 cells labeled with DiO. The frequencies of mKO-positive H9M1 cells in bone marrow and spleen were examined by flow cytometry, and cellular localization was evaluated using DiO signals by fluorescence microscopy. For CXCR4 inhibition, 1 × 107 H9M1 cells were injected after a 30-minute incubation with 10 μg/ml anti-CXCR4 in vitro, and 75 mg/kg anti-CXCR4 was intraperitoneally administered to recipient mice 24 hours after injection of H9M1 cells. A rat anti-CXCR4 antibody (RM0366-30L4) and its isotype control IgG2b were purchased from Abcam.
Flow cytometric analysis.
Immunophenotypic analysis was performed by flow cytometric analysis using fluorochrome- or biotin-conjugated monoclonal antibodies described above and a FACSAria flow cytometer (BD Biosciences).
ChIP and ChIP-Seq.
A total of 5 × 107 cells per immunoprecipitation were cross-linked with 1% formaldehyde for 10 minutes at room temperature. Chromatin was sheared in lysis buffer containing 1% Triton X-100, 0.1% sodium deoxycholate, and 0.1% SDS to an average size of 300 to 500 bp through sonication on ice in a Bioruptor (UCD-200T, Diagenode) for 20 minutes (30 seconds on, 30 seconds off; high-power load). ChIP was then performed with anti-MEIS1 (Abcam), anti-FLAG (Sigma-Aldrich), or anti-total PBX (Santa Cruz Biotechnology) antibodies preconjugated to protein G magnetic beads. Immunoprecipitated DNA was purified and subjected to next-generation sequencing. Libraries were prepared according to instructions accompanying Illumina’s DNA sample kit (part 0801-0303). Briefly, DNA was end repaired using a combination of T4 DNA polymerase, E. coli DNA Pol I large fragment (Klenow polymerase), and T4 polynucleotide kinase. The blunt, phosphorylated ends were treated with a Klenow fragment (32 to 52 Exo-minus) and dATP to yield a protruding 3-“A” base for ligation of Illumina’s adapters that have a single “T” base overhang at the 3′ end. After adapter ligation, DNA was PCR amplified with Illumina primers for 15 cycles, and library fragments of approximately 250 bp (insert plus adaptor and PCR primer sequences) were band isolated from an agarose gel. The purified DNA was captured on an Illumina flow cell for cluster generation. Libraries were sequenced on the Genome Analyzer following the manufacturer’s protocols. Base calls were performed using CASAVA version 1.4. ChIP-Seq reads were aligned to the mm9 genome assembly using ELAND (Illumina). Peaks were called using PeaksFind version 2.2 with the following setting: ChIP threshold of 0.2, enrichment fold of 2.5, and rescue fold of 3. The enriched genomic regions were determined using CisGenome (60). Overlapping of MEIS1, HOXA9, or PBX peaks was determined using an intersect function of BEDtools (61).
Microarray analysis.
GeneChip analysis was conducted to determine gene expression profiles. Total RNA was extracted using the RNeasy Mini Kit (QIAGEN). The murine Genome 430 PM Array (Affymetrix) was hybridized with aRNA probes generated from H9M1 cells. After staining with streptavidin-phycoerythrin conjugates, arrays were scanned using an Affymetrix GeneAtlas Scanner and analyzed using Affymetrix GeneChip Command Console Software (AGCC, Affymetrix) and GeneSpring GX 11.0.2 (Agilent Technologies). GSEA was performed using GSEA-P 2.0 software (62).
Western blotting.
Western blot analysis was performed using lysates of whole-tumor tissues with specific antibodies as described above.
RT-PCR and real-time quantitative RT-PCR.
Total RNA extraction, reverse transcription, and RNA quantification were performed by standard methods. Conventional RT-PCR and real-time quantitative RT-PCR (Q-RT-PCR) were performed with a Gene Amp 9700 thermal cycler (Applied Biosystems) and a 7500 Fast Real-Time PCR System (Applied Biosystems), respectively. The sequences of the oligonucleotide primers are shown in Supplemental Table 5.
Luciferase assay.
A 2.0-kbp genomic DNA fragment upstream of murine Sytl1 exon 1 was amplified by PCR using the forward (5′-AAGTGCTGGGATTTAGAGGTG-3′) and reverse (5′-GACGACAACTTCAAAGGCGAC-3′) primers. The fragment was inserted into the pGL4.10 vector (Promega). Mutations of putative MEIS1-binding sites were introduced by in vitro mutagenesis using the KOD-Plus Mutagenesis Kit (Toyobo). Reporter plasmids and the expression plasmid of Meis1 were nucleofected into 32Dcl3 cells using Nucleofector Kit V (Lonza). Luciferase assays were performed by a standard method.
Gene expression profiling of human patients with AML.
Gene expression profiling of 526 AML cases was carried out on Affymetrix HGU133A Plus2.0 GeneChips as previously described (63). The validation of the scaling/normalization factors of the GeneChips was less than 3-fold. Mutational analyses to detect recurrent mutations in AML were performed as previously described (64). All supervised class prediction analyses were performed with Prediction Analysis for Microarrays software version 1.28 in R version 2.1.0.
Cell migration assay.
The cell migration assay was performed as described previously with modification (65). Ten ng/ml of CXCL12, FLT3L, or IL-3 or 100 ng/ml of M-CSF, GM-CSF, SCF, or TPO was added to the lower chambers. All the chemokines and cytokines above were purchased from R&D Systems. To block CXCR4, 10 μg/ml anti-CXCR4 was added to the upper chambers.
RNA interference studies.
For knockdown of Sytl1 and Cxcl12, siRNAs were purchased from QIAGEN (GS269589 and GS20315). siRNAs were introduced into 32Dcl3 cells by nucleofection as described above or into OP9 cells using Lipofectamine 2000 (Life Technologies). Knockdown efficiencies were confirmed by Western blotting using anti-JFC1 (H-80, Santa Cruz Biotechnology) or RT-PCR for Cxcl12. SYTL1 knockdown in RS4;11 cells was performed by infecting the cells with the pLKO.1-puro lentiviral vector bearing shSYTL1 (Sigma-Aldrich). Tetracycline-inducible silencing of Sytl1 was performed using the BLOCK-IT Inducible H1 Lentiviral RNAi System (Invitrogen). For induction of shSytl1 expression, mice were given 0.5 mg/ml doxycycline in their drinking water supplemented with 2 mg/ml sucrose. For cell culture, doxycycline was used at a concentration of 2 μg/ml.
Confocal laser microscopy.
Murine bone marrow and spleen samples were fixed with 4% paraformaldehyde and embedded in OCT compound. Frozen sections were subjected to immunofluorescence using the antibodies described above. H9M1 or 32Dcl3 cells stimulated with CXCL12 were incubated on ice with a biotin-conjugated anti-CXCR4 antibody, streptavidin rhodamine (Jackson ImmunoResearch), and Alexa Fluor 350–conjugated wheat germ agglutinin (Life Technology), fixed with 1% paraformaldehyde for 10 minutes and washed with PBS twice. Lysosomes were detected with LysoTracker Green DND-26 (Molecular Probes). Immunofluorescent images were photographed with a Zeiss LSM 710 laser scanning microscope with a ×60 objective (Zeiss) and LSM Software ZEN 2009 (Zeiss).
Live-cell imaging.
The murine myeloid cell line 32Dcl3 was transfected with an expression vector bearing Cxcr4-GFP fusion and Sytl1-DsRed fusion cDNAs as well as control siRNAs with Nucleofector Kit V. GFP- and/or DsRed-positive cells were collected with a FACSAria cell sorter 24 hours after transfection, and 2 × 104 sorted 32Dcl3 cells were cultured on 35-mm glass-based dishes (Iwaki) coated with 2 μg/ml recombinant mouse VCAM-1/CD106 Fc chimera (R&D Systems), with growth medium containing 5 ng/ml IL-3. CXCL12-coated microbeads were made by incubating sulfate latex beads (4% w/v, 6 μm, Molecular Probes) with 100 ng/ml CXCL12 and 2 μg/ml fibronectin at 4°C overnight. After 24 hours of cultivation, 2 × 104 CXCL12-coated beads were added to the dishes. After addition of CXCL12-coated beads, GFP and DsRed images were obtained in a 5% CO2 humidified atmosphere at 37°C using a CellVoyager CV1000 confocal microscope (Yokogawa) with an Olympus UPLSApo ×60 objective (Olympus). Time-lapse images were taken at 30-second intervals over 30 minutes using an EMCCD camera. Acquired images were processed and analyzed with Photoshop (Adobe) and ImageJ. Cell tracking for migration was performed using the Manual Tracking and Chemotaxis Tool of the MTrackJ plugin of ImageJ.
Accession numbers.
The microarray and ChIP-Seq data sets are accessible through the NCBI Gene Expression Omnibus database (http://www.ncbi.nlm.nih.gov/geo), with accession numbers GSE50645 and GSE48679, respectively.
Statistics.
All in vitro experiments were performed at least in triplicate. The numbers of mice used in each experiment are presented in the text or figure legends. Values are expressed as mean ± SEM, and statistical significances were compared with 2-tailed Student’s t test for comparison between 2 groups, 1-way ANOVA with Dunnett’s test for multiple comparisons, Mann-Whitney test, or χ2 test. Survival analysis was performed using the Kaplan-Meier life-table method, and survival between groups was compared with the log-rank test. All P values were 2 sided, and a P value of less than 0.05 was considered significant.
Study approval.
Animals were housed, observed daily, and handled in accordance with the guidelines reviewed and approved by the Institutional Animal Care Committee at the Japanese Foundation for Cancer Research.
Author contributions
TY, MN, TK, AC, and RG performed and interpreted experiments. ST, HA, PJMV, and RD performed genomic and bioinformatic analyses. TN supervised the entire study and wrote the paper.
Supplementary Material
Acknowledgments
We would like to thank Mitsunori Fukuda, Tohoku University, Sendai, Japan, for providing Sytl1 KO mice; Toshio Kitamura, Institute of Medical Science, University of Tokyo, Tokyo, Japan, for Flt3 cDNA, pMYs vector, and PLAT-E cells; and Tetsuya Nosaka, Mie University, Mie, Japan, for MLL-ENL cDNA. We also thank Yohei Kanno, Yukari Yamazaki, Tomoko Takahara, and Yoshiko Yamazaki for technical assistance and Masaru Ushijima for helping with statistical analyses. This work was supported by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (23240125).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2016;126(5):1664–1678. doi:10.1172/JCI81516.
Contributor Information
Takashi Yokoyama, Email: takashi.yokoyama@jfcr.or.jp.
Mayuka Nakatake, Email: nakatake@mext.go.jp.
Takeshi Kuwata, Email: tkuwata@east.ncc.go.jp.
Ryo Goitsuka, Email: ryogoi@rs.noda.tus.ac.jp.
Shuichi Tsutsumi, Email: tsutsumi@ja2.so-net.ne.jp.
Hiroyuki Aburatani, Email: haburata-tky@umin.ac.jp.
Ruud Delwel, Email: h.delwel@erasmusmc.nl.
Takuro Nakamura, Email: takuro-ind@umin.net.
References
- 1.Lawrence HJ, Sauvageau G, Humphries RK, Largman C. The role of HOX homeobox genes in normal and leukemic hematopoiesis. Stem Cells. 1996;14(3):281–291. doi: 10.1002/stem.140281. [DOI] [PubMed] [Google Scholar]
- 2.Magli MC, Largman C, Lawrence HJ. Effects of HOX homeobox genes in blood cell differentiation. J Cell Physiol. 1997;173(2):168–177. doi: 10.1002/(SICI)1097-4652(199711)173:2<168::AID-JCP16>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 3.Alharbi RA, Pettengell R, Pandha HS, Morgan R. The role of HOX genes in normal hematopoiesis and acute leukemia. Leukemia. 2013;27(5):1000–1008. doi: 10.1038/leu.2012.356. [DOI] [PubMed] [Google Scholar]
- 4.Heuser M, et al. Cell of origin in AML: susceptibility to MN1-induced transformation is regulated by the MEIS1/AbdB-like HOX protein complex. Cancer Cell. 2011;20(1):39–52. doi: 10.1016/j.ccr.2011.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Golub TR, et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286(5439):531–537. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- 6.Yokoyama A, Somervaille TC, Smith KS, Rozenblatt-Rosen O, Meyerson M, Cleary M. The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell. 2005;123(2):207–218. doi: 10.1016/j.cell.2005.09.025. [DOI] [PubMed] [Google Scholar]
- 7.Gough SM, Slape CI, Aplan PD. NUP98 gene fusions and hematopoietic malignancies: common themes and new biologic insights. Blood. 2011;118(24):6247–6257. doi: 10.1182/blood-2011-07-328880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nakamura T. NUP98 fusion in human leukemia: dysregulation of the nuclear pore and homeodomain proteins. Int J Hematol. 2005;82(1):21–27. doi: 10.1532/IJH97.04160. [DOI] [PubMed] [Google Scholar]
- 9.Breitlinger C, Maethner E, Garcia-Cuellar MP, Slany RK. The homeodomain region controls the phenotype of HOX-induced murine leukemia. Blood. 2012;120(19):4018–4027. doi: 10.1182/blood-2011-10-384685. [DOI] [PubMed] [Google Scholar]
- 10.Nakamura T, Largaespada DA, Shaughnessy JD, Jenkins NA, Copeland NG. Cooperative activation of Hoxa and Pbx1-related genes in murine myeloid leukaemias. Nat Genet. 1996;12(2):149–153. doi: 10.1038/ng0296-149. [DOI] [PubMed] [Google Scholar]
- 11.Mullighan CG, et al. Pediatric acute myeloid leukemia with NPM1 mutations is characterized by a gene expression profile with dysregulated HOX gene expression distinct from MLL-rearranged leukemias. Leukemia. 2007;21(9):2000–2009. doi: 10.1038/sj.leu.2404808. [DOI] [PubMed] [Google Scholar]
- 12.Jin G, et al. Trib1 and Evi1 cooperate with Hoxa and Meis1 in myeloid leukemogenesis. Blood. 2007;109(9):3998–4005. doi: 10.1182/blood-2006-08-041202. [DOI] [PubMed] [Google Scholar]
- 13.Keeshan K, Shestova O, Ussin L, Pear WS. Tribbles homolog 2 (Trib2) and HoxA9 cooperate to accelerate acute myelogenous leukemia. Blood Cells Mol Dis. 2008;40(1):119–121. doi: 10.1016/j.bcmd.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 14.Moskow JJ, Bullrich F, Huebner K, Darr IO, Buchberg AM. Meis1, a PBX1-related homeobox gene involved in myeloid leukemia in BXH-2 mice. Mol Cell Biol. 1995;15(10):5434–5443. doi: 10.1128/MCB.15.10.5434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kroon E, Krosl J, Thorsteinsdottir U, Baban S, Buchberg AM, Sauvageau G. Hoxa9 transforms primary bone marrow cells through specific collaboration with Meis1a but not Pbx1b. EMBO J. 1998;17(13):3714–3725. doi: 10.1093/emboj/17.13.3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kroon E, Thorsteinsdottir U, Mayotte N, Nakamura T, Sauvageau G. NUP98-HOXA9 expression in hemopoietic stem cells induces chronic and acute myeloid leukemias in mice. EMBO J. 2001;20(3):350–361. doi: 10.1093/emboj/20.3.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hisa T, et al. Hematopoietic, angiogenic and eye defects in Meis1 mutant animals. EMBO J. 2004;23(2):450–459. doi: 10.1038/sj.emboj.7600038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kocabas F, et al. Meis1 regulates the metabolic phenotype and oxidant defense of hematopoietic stem cells. Blood. 2012;120(25):4963–4972. doi: 10.1182/blood-2012-05-432260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Unnisa Z, et al. Meis1 preserves hematopoietic stem cells in mice by limiting oxidative stress. Blood. 2012;120(25):4973–4981. doi: 10.1182/blood-2012-06-435800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ariki R, et al. Homeodomain transcription factor Meis1 is a critical regulator of adult bone marrow hematopoiesis. PLoS One. 2014;9(2): doi: 10.1371/journal.pone.0087646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Simsek T, et al. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell. 2010;7(3):380–390. doi: 10.1016/j.stem.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang Y, et al. Identification and characterization of Hoxa9 binding sites in hematopoietic cells. Blood. 2012;119(2):388–398. doi: 10.1182/blood-2011-03-341081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bach C, et al. Leukemogenic transformation by HOXA cluster genes. Blood. 2010;115(14):2910–2918. doi: 10.1182/blood-2009-04-216606. [DOI] [PubMed] [Google Scholar]
- 24.Arimura N, et al. Anterograde transport of TrkB in axons is mediated by direct interaction with Slp1 and Rab27. Dev Cell. 2009;16(5):675–686. doi: 10.1016/j.devcel.2009.03.005. [DOI] [PubMed] [Google Scholar]
- 25.Galvez-Santisteban M, et al. Synaptotagmin-like proteins control the formation of a single apical membrane domain in epithelial cells. Nat Cell Biol. 2012;14(8):838–849. doi: 10.1038/ncb2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Calvo KR, Knoepfler PS, Sykes DB, Pasillas MP, Kamps MP. Meis1a suppresses differentiation by G-CSF and promotes proliferation by SCF: Potential mechanisms of cooperativity with Hoxa9 in myeloid leukemia. Proc Natl Acad Sci USA. 2001;98(23):13120–13125. doi: 10.1073/pnas.231115398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kumar AR, et al. A role for MEIS1 in MLL-fusion gene leukemia. Blood. 2009;113(8):1756–1758. doi: 10.1182/blood-2008-06-163287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yokoyama A, Lin M, Naresh A, Kitabayashi I, Cleary ML. A higher-order complex containing AF4 and ENL family proteins with P-TEFb facilitates oncogenic and physiologic MLL-dependent transcription. Cancer Cell. 2010;17(2):198–212. doi: 10.1016/j.ccr.2009.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lane SW, Scadden DT, Gilliland DG. The leukemic stem cell niche: current concepts and therapeutic opportunities. Blood. 2009;114(6):1150–1157. doi: 10.1182/blood-2009-01-202606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schuringa JJ, Morrone G, Moore MA. Enforced activation of STAT5A facilitates the generation of embryonic stem-derived hematopoietic stem cells that contribute to hematopoiesis in vivo. Stem Cells. 2004;22(7):1191–1204. doi: 10.1634/stemcells.2004-0033. [DOI] [PubMed] [Google Scholar]
- 31.Ding L, Saunders TL, Enikolopov G, Morrison SJ. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature. 2012;481(7382):457–462. doi: 10.1038/nature10783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frenette PS, Pinho S, Lucas D, Scheiermann C. Mesenchymal stem cell: keystone of the hematopoietic stem cell niche and a stepping-stone for regenerative medicine. Annu Rev Immunol. 2013;31:285–316. doi: 10.1146/annurev-immunol-032712-095919. [DOI] [PubMed] [Google Scholar]
- 33.Grassinger J, et al. Thrombin-cleaved osteopontin regulates hemopoietic stem and progenitor cell functions through interactions with α9β1 and α4β1 integrins. Blood. 2009;114(1):49–59. doi: 10.1182/blood-2009-01-197988. [DOI] [PubMed] [Google Scholar]
- 34.Nakamura-Ishizu A, et al. Extracellular matrix protein tenascin-C is required in the bone marrow microenvironment primed for hematopoietic regeneration. Blood. 2012;119(23):5429–5437. doi: 10.1182/blood-2011-11-393645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity. 2006;25(6):977–988. doi: 10.1016/j.immuni.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 36.Chang CP, et al. Meis proteins are major in vivo DNA binding partners for wild-type but not chimeric Pbx proteins. Mol Cell Biol. 1997;17(10):5679–5687. doi: 10.1128/MCB.17.10.5679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goh SL, et al. Transcriptional activation by MEIS1A in response to protein kinase A signaling requires the transducers of regulated CREB family of CREB co-activators. J Biol Chem. 2009;284(28):18904–18912. doi: 10.1074/jbc.M109.005090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wilson NK, et al. Combinatorial transcriptional control in blood stem/progenitor cells: genome-wide analysis of ten major transcriptional regulators. Cell Stem Cell. 2010;7(4):532–544. doi: 10.1016/j.stem.2010.07.016. [DOI] [PubMed] [Google Scholar]
- 39.Holt O, et al. Slp1 and Slp2-a localize to the plasma membrane of CTL and contribute to secretion from the immunological synapse. Traffic. 2008;9(4):446–457. doi: 10.1111/j.1600-0854.2008.00714.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Neumuller O, Hoffmeister M, Babica J, Prelle C, Gegenbauer K, Smolenski AP. Synaptotagmin-like protein 1 interacts with the GTPase-activating protein Rap1GAP2 and regulates dense granule secretion in platelets. Blood. 2009;114(7):1396–1404. doi: 10.1182/blood-2008-05-155234. [DOI] [PubMed] [Google Scholar]
- 41.Saegusa C, Kanno E, Itohara S, Fukuda M. Expression of Rab27B-binding protein Slp1 in pancreatic acinar cells and its involvement in amylase secretion. Arch Biochem Biophys. 2008;475(1):87–92. doi: 10.1016/j.abb.2008.04.031. [DOI] [PubMed] [Google Scholar]
- 42.Noordermeer SM, et al. High BRE expression predicts favorable outcome in adult acute myeloid leukemia, in particular among MLL-AF9-positive patients. Blood. 2011;118(20):5613–5621. doi: 10.1182/blood-2011-06-359182. [DOI] [PubMed] [Google Scholar]
- 43.Groschel S, et al. Deregulated expression of EVI1 defines a poor prognostic subset of MLL-rearranged acute myeloid leukemias: a study of the German-Austrian Acute Myeloid Leukemia Study Group and the Dutch-Belgian-Swiss HOVON/SAKK Cooperative Group. J Clin Oncol. 2013;31(1):95–103. doi: 10.1200/JCO.2011.41.5505. [DOI] [PubMed] [Google Scholar]
- 44.Masztalerz A, et al. Synaptotagmin 3 deficiency in T cells impairs recycling of the chemokine receptor CXCR4 and thereby inhibits CXCL12 chemokine-induced migration. J Cell Sci. 2007;120(pt 2):219–228. doi: 10.1242/jcs.03328. [DOI] [PubMed] [Google Scholar]
- 45.Colvin RA, et al. Synaptotagmin-mediated vesicle fusion regulates cell migration. Nat Immunol. 2010;11(6):495–502. doi: 10.1038/ni.1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Omatsu Y, Seike M, Sugiyama T, Kume T, Nagasawa T. Foxc1 is a critical regulator of haematopoietic stem/progenitor cell niche formation. Nature. 2014;508(7497):536–540. doi: 10.1038/nature13071. [DOI] [PubMed] [Google Scholar]
- 47.Hokanson DE, Bretscher AP. EPI64 interacts with Slp1/JFC1 to coordinate Rab8a and Arf6 membrane trafficking. Mol Biol Cell. 2012;23(4):701–715. doi: 10.1091/mbc.E11-06-0521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kurowska M, et al. Terminal transport of lytic granules to the immune synapse is mediated by the kinesin-1/Slp3/Rab27a complex. Blood. 2012;119(17):3879–3889. doi: 10.1182/blood-2011-09-382556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Johnson JL, Monfregola J, Napolitano G, Kiosses WB, Catz SD. Vesicular trafficking through cortical actin during exocytosis is regulated by the Rab27a effect of JCR1/Slp1 and the RhoA-GTPase-activating protein Gem-interacting protein. Mol Biol Cell. 2012;23(10):1902–1916. doi: 10.1091/mbc.E11-12-1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fukuda M. Versatile role of Rab27 in membrane trafficking: focus on the Rab27 effector families. J Biochem. 2005;137(1):9–16. doi: 10.1093/jb/mvi002. [DOI] [PubMed] [Google Scholar]
- 51.Subramani D, Alahari SK. Integrin-mediated function of Rab GTPases in cancer progression. Mol Cancer. 2010;9: doi: 10.1186/1476-4598-9-312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bobrie A, et al. Rab27a supports exosome-dependent and –independent mechanisms that modify the tumor microenvironment and can promote tumor progression. Cancer Res. 2012;72(19):4920–4930. doi: 10.1158/0008-5472.CAN-12-0925. [DOI] [PubMed] [Google Scholar]
- 53.Morgado E, Albouhair S, Lavau C. Flt3 is dispensable to the Hoxa9/Meis1 leukemogenic cooperation. Blood. 2007;109(9):4020–4022. doi: 10.1182/blood-2006-01-039586. [DOI] [PubMed] [Google Scholar]
- 54.Penkov D, et al. Analysis of the DNA-binding profile and function of TALE homeoproteins reveals their specialization and specific interactions with Hox genes/proteins. Cell Reports. 2013;3(4):1321–1333. doi: 10.1016/j.celrep.2013.03.029. [DOI] [PubMed] [Google Scholar]
- 55.Wang Z, et al. GSK-3 promotes conditional association of CREB and its coactivators with MEIS1 to facilitate HOX-mediated transcription and oncogenesis. Cancer Cell. 2010;17(6):597–608. doi: 10.1016/j.ccr.2010.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tanaka M, et al. Ewing’s sarcoma precursors are highly enriched in embryonic osteochondrogenic progenitors. J Clin Invest. 2014;124(7):3061–3074. doi: 10.1172/JCI72399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ito T, et al. Regulation of myeloid leukaemia by the cell-fate determinant Musashi. Nature. 2010;466(7307):765–768. doi: 10.1038/nature09171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ventura A, et al. Restoration of p53 function leads to tumour regression in vivo. Nature. 2007;445(7128):661–665. doi: 10.1038/nature05541. [DOI] [PubMed] [Google Scholar]
- 59.Kogan S, et al. Bethesda proposals for classification of nonlymphoid hematopoietic neoplasms in mice. Blood. 2002;100(1):238–245. doi: 10.1182/blood.V100.1.238. [DOI] [PubMed] [Google Scholar]
- 60.Ji H, Jiang H, Ma W, Johnson DS, Myers RM, Wong WH. An integrated software system for analyzing ChIP-chip and ChIP-seq data. Nat Biotechnol. 2008;26(11):1293–1300. doi: 10.1038/nbt.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26(6):841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Subramanian A, Kuehn H, Gould J, Tamayo P, Mesirov JP. GSEA-P: a desktop application for gene set enrichment analysis. Bioinformatics. 2007;23(23):3251–3253. doi: 10.1093/bioinformatics/btm369. [DOI] [PubMed] [Google Scholar]
- 63.Alberich-Jorda M, et al. C/EBPγ deregulation results in differentiation arrest in acute myeloid leukemia. J Clin Invest. 2012;122(12):4490–4504. doi: 10.1172/JCI65102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Verhaak RGW, et al. Prediction of molecular subtypes in acute myeloid leukemia based on gene expression profiling. Haematologica. 2009;94(1):131–134. doi: 10.3324/haematol.13299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fujino T, et al. Function of EWS-POU5F1 in sarcomagenesis and tumor cell maintenance. Am J Pathol. 2010;176(4):1973–1982. doi: 10.2353/ajpath.2010.090486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bagger FO, et al. HemaExploler: a database of mRNA expression profiles in normal and malignant haematopoiesis. Nucl Acids Res. 2013;41(Database issue):D1034–D1039. doi: 10.1093/nar/gks1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.