

Abstract
Although the use of antioxidants for the treatment of cancer and HIV/AIDS has been proposed for decades, new insights gained from redox research have suggested a very different scenario. These new data show that the major cellular antioxidant systems, the thioredoxin (Trx) and glutathione (GSH) systems, actually promote cancer growth and HIV infection, while suppressing an effective immune response. Mechanistically, these systems control both the redox- and NO-based pathways (nitroso-redox homeostasis), which subserve innate and cellular immune defenses. Dual inhibition of the Trx and GSH systems synergistically kills neoplastic cells in vitro and in mice and decreases resistance to anticancer therapy. Similarly, the population of HIV reservoir cells that constitutes the major barrier to a cure for AIDS is exquisitely redox sensitive and could be selectively targeted by Trx and GSH inhibitors. Trx and GSH inhibition may lead to a reprogramming of the immune response, tilting the balance between the immune system and cancer or HIV in favor of the former, allowing elimination of diseased cells. Thus, therapies based on silencing of the Trx and GSH pathways represent a promising approach for the cure of both cancer and AIDS and warrant further investigation.
Introduction
It is now well recognized that ROS and reactive nitrogen species (RNS) such as superoxide anion (O2–), H2O2, NO, and S-nitrosothiols (SNOs) are ubiquitously generated in vivo and influence a broad spectrum of physiological and pathological processes. ROS and RNS affect cellular function through redox-based modification of proteins, particularly S-oxidation and S-nitrosylation. Above all, reversible redox-based modifications of cysteine thiols, namely sulfenic acids (RSOH), disulfides (RSSR), and SNOs, regulate the activity of proteins to influence diverse cellular processes including cell division and differentiation, metabolism, and cell death (1–3).
The net cellular effects of ROS and RNS are coordinated by systems that generate these reactive species (NADPH oxidases and NO synthases), consume them (antioxidant systems), or reverse the protein modifications that subserve their activity (disulfide reductases and denitrosylases). In this regard, the antioxidant protein thioredoxin (Trx) and tripeptide glutathione (GSH) and their system components represent the major defenses against ROS and RNS (4–6). However, accumulating evidence indicates that Trx and GSH also play a homeostatic role in basic cellular processes including DNA synthesis, protein maturation, and cellular signaling (Figure 1). Accordingly, dysregulation of the Trx and GSH systems may contribute to a broad spectrum of diseases including cancer and various infectious and inflammatory disorders (4, 6, 7), and there is currently interest in targeting Trx and GSH for therapeutic gain. Here, we discuss recent progress regarding the potential benefits of dual targeting of the Trx and GSH systems for the management or treatment of cancer and HIV infection and outline the main molecular mechanisms that are the likely mediators of the antitumor and antiviral effects. Given the pathological and epidemiological overlap between HIV and several types of cancer, a shared therapeutic strategy to address both conditions could potentially have a marked impact on global health.
Figure 1. Role of the Trx and GSH systems in cellular nitroso-redox homeostasis and signaling.

ROS and RNS such as O2–, H2O2, and NO are generated in cells by the regulated activity of dedicated enzymes such as NADPH oxidases (NOX) and NOS and by additional sources such as mitochondria. NO is efficiently converted into low-molecular-weight SNOs (e.g., S-nitrosoglutathione [GSNOs]), which convey the large part of NO bioactivity. NO can react with O2– to produce ONOO–, a potent oxidant and nitrating agent. ROS and RNS play various roles in both homeostatic and pathological processes. The physiological roles of H2O2 and NO and SNO are principally mediated through the S-oxidation or S-nitrosylation of protein Cys residues (RSH) to form sulfenic acids (RSOH), disulfides (RSSR), and protein S-nitrosothiols (RSNO), thereby altering protein structure and function. GSH and Trx and associated redox enzymes, such as glutaredoxin (Grx), reverse these thiol modifications. Oxidized GSH (GSSG) and oxidized Trx (Trx-S2) are reduced by GSH reductase (GR) and TrxR, respectively, at the expense NADPH oxidation. The thiol-reducing activity of GSH and Trx is of key importance in maintaining the enzymatic activity of members of the peroxiredoxin (Prx) and GSH peroxidase (Gpx) enzymes, which remove peroxides. Additionally, GSH acts together with GSH S-transferases (GST enzymes) to detoxify various xenobiotics, and Trx acts with methionine sulfoxide reductases to repair oxidized methionines. GSNO is metabolized by the NADH-dependent GSNOR, which thereby shifts the cellular equilibrium to disfavor protein SNOs, while Trxs and TrxRs denitrosylate proteins directly. GSNHOH, N-hydroxy sulfenamide.
The Trx and GSH systems: redundancy and crosstalk
To understand the basis for therapies characterized by the inhibition of Trx and GSH, it is important to consider the relationships between the two systems in terms of redundancy and interrelated control mechanisms. Trx and GSH have both overlapping and nonredundant functions. ROS and RNS detoxification depends on both Trx and GSH, as exemplified by the removal of H2O2 by GSH- and Trx-dependent peroxidases (Figure 1). Functional redundancy is further supported by studies using cells deficient in Trx reductase 1 (TrxR1), a selenoenzyme that reduces the active site of the cytoplasmic isoform of Trx (Trx1). Conrad and colleagues demonstrated that, although cells from TrxR1-deficient mice have normal proliferative capacity, they exhibit heightened sensitivity to the GSH synthesis inhibitor buthionine sulfoximine (BSO) (8). In addition, Schmidt and colleagues showed that hepatocyte DNA replication requires either GSH or TrxR1 (9). This finding was recently confirmed in primary human T lymphocytes (10), where the two systems are required for optimal activity of ribonucleotide reductase. There is also evidence that the GSH and Trx redox systems back each other up in maintaining redox and cell homeostasis. Holmgren and colleagues have recently shown that GSH, together with the protein glutaredoxin, can reduce Trx1 when TrxR1 is inhibited (11). Moreover, it was found that the combined inhibition of TrxR1 and GSH had synergistic effects on Trx1 oxidation (11).
Notwithstanding the above observations, there is growing evidence that Trx and GSH target different cysteines in the proteome, supporting the existence of nonredundant functions (12, 13), as exemplified by the distinct mechanisms and roles of Trx and GSH in protein denitrosylation (7). Whereas both GSH and Trxs (Trx1 and -2 and Trx-related protein 14 [TRP14]) catalyze the direct removal of NO groups from proteins, the activity of GSH is dependent on a class of denitrosylases (S-nitrosoglutathione reductases [GSNORs]), which metabolize the GSNO formed in the denitrosylation reaction (7). The proteins that are denitrosylated by Trx- and GSH-dependent mechanisms are largely nonoverlapping (7, 14, 15). Even among the denitrosylating Trxs, substrate specificity is evident. This can also be understood by virtue of subcellular distribution: Trx1 is cytosolic and Trx2 is mitochondrial (16).
Given the above observations, it follows that perturbing one redox system — GSH or Trx — is likely to affect the other, even though these systems have nonoverlapping roles. In this regard, the nascent field of quantitative redox biology, incorporating in vitro data to guide in silico analysis, may help to provide a broader picture of these interconnections (17, 18). In particular, the quantitative association between the redox state of the cell and its biological state (such as quiescence, proliferation, or apoptosis) could help guide therapeutic manipulation of the antioxidant pathways (17). Moreover, these models may enhance our understanding of the interrelation between oxidative and nitrosative stress. For example, oxidative stress (e.g., localized production of H2O2) may tie up the activity of Trx-dependent defenses, increasing protein SNOs in the process (7). Oxidative stress thereby begets nitrosative stress (nitroso-redox imbalance). Under such circumstances, redox-based signaling and defense become increasingly reliant on GSH-dependent mechanisms, and, as illustrated throughout this Review, the GSH and Trx pathways can thus be targeted with synergistic consequences.
Oxidative and nitrosative stress in the immune system: the lymphocyte paradigm
Before considering the significance of ROS and RNS and the Trx and GSH systems in tumorigenesis and HIV infection, it is important to first discuss the involvement of these redox-active molecules in normal cellular function. For this purpose, we focus here on lymphocytes, which are critical regulators of the anticancer and antiviral immune responses, as a paradigm for thiol-based redox control.
ROS play opposing roles in lymphocytes. While ROS are necessary for lymphocyte activation and establishment of effective antiviral and antitumor immunity, they also promote lymphocyte senescence and cell death (for a recent review, see ref. 19). Lymphocyte activation involves the stimulation of cell-surface receptors (e.g., the T cell receptor [TCR] complex) by antigens bound to the MHC of specialized antigen-presenting cells (APCs) such as DCs and macrophages. This antigen presentation process is accompanied by a number of coordinated molecular events (e.g., CD80/86 interaction with CD28, and CD70 with CD27) that trigger an array of signals, leading to proliferation (clonal expansion) of the activated lymphocyte and differentiation into specialized subsets (20).
Oxidants play an early role in lymphocyte activation events. An oxidative burst occurs immediately upon the encounter with the antigen (recently reviewed in ref. 21). This early oxidant burst leads to thiol oxidation that enables signal transduction (22). Oxidants also act downstream of antigen sensing by promoting the activation of transcription factors such as NF-κB and nuclear factor of activated T cells (NFAT) that facilitate the G0/G1 transition (19, 23). However, the ROS burst may coincidentally activate apoptosis signal–regulating kinase 1 (ASK1), which propagates a proapoptotic signal (19); consequently, part of the activated lymphocyte population inevitably dies (19). Thus, an effective immune response also entails an antioxidant response that is needed for lymphocyte survival.
Trx and GSH contribute counterbalancing responses that promote lymphocyte proliferation and survival (19, 23), reflecting the context-specific nature of redox regulation. Trx upregulation is observed in murine splenic B lymphocytes following the initial oxidative burst (24). Conversely, inhibition of the Trx system impairs the mitogenic response of mouse splenocytes (25), favors ASK1-mediated cell death of CD4+ T lymphocytes (26), and downregulates costimulatory CD27 and CD28 molecules in T cells (26, 27). The GSH system is also necessary for efficient lymphocyte proliferation. Notably, cystine, the rate-limiting factor in GSH biosynthesis, is supplied to lymphocytes during their encounter with DCs (28, 29) and, upon activation, is taken up from the medium through the cystine/glutamate antiporter xc– (30). GSH facilitates the proliferation of T lymphocytes (23) and is necessary for their G1/S transition (31). GSH likely mediates its effects through posttranslational modification of proteins, particularly S-glutathionylation (32) and S-nitrosylation (33), and by protecting DNA from oxidative damage during replication (5, 7). Lymphocytes treated with inhibitors of both Trx and GSH systems exhibit impaired synthesis of DNA (10) and key proteins (e.g., α chain of the proliferation-related IL-2 receptor) and eventually die, while limited or incomplete effects are observed with either drug alone (34, 35). Thus, the Trx and GSH pathways play important and complementary roles during lymphocyte activation and proliferation.
Antioxidant defenses gradually decrease during lymphocyte differentiation, with memory T cells showing lower levels of intracellular GSH than do naive cells (26). Accordingly, the memory T cell subsets are most vulnerable to Trx inhibition and the resultant apoptosis (in this case, p38 MAP kinase–mediated apoptosis) (26); senescent CD27−CD28− T cells also demonstrate spontaneous p38 MAP kinase activation (36), which is an event linked to ROS production. As will be discussed below, the susceptibility to oxidative stress may represent an Achilles’ heel of memory T cell subsets.
Opposing actions of NO and S-nitrosylation have also been observed during lymphocyte activation and differentiation. NO is produced by the NOS family of enzymes, which include endothelial (eNOS), neuronal (nNOS), and inducible NOS (iNOS) (37). Classically, iNOS is responsible for the production of NO in a variety of immune cells, including macrophages and DCs, and was originally reported to suppress viral infection and cell death in human B lymphocytes (38). DC-derived NO can freely diffuse to bystander lymphocytes (37). However, while the exact roles of iNOS-derived NO remain controversial in lymphocytes (37), an emerging body of literature indicates that eNOS plays a central role at the immune synapse. Antigen presentation is coupled to NO production in Th cells via eNOS, which increases calcium and PI3K activity (39). Thus, eNOS regulates the coalescence of the T cell coreceptor CD3 at the T cell–APC synapse to induce IFN-γ synthesis and suppress IL-2 production. Perhaps through IFN-γ, NO promotes the differentiation of T lymphocytes into specific cell subsets (37) including Tregs and Th1 and Th9 cells (reviewed in ref. 37).
Whereas NO potentiates T cell signaling at the immune synapse, coincident S-nitrosylation of Ras consequent upon TCR engagement results in apoptosis (40). Thus, by direct analogy to ROS, the activation of NO-based signaling in T cells is required for cellular activation and proliferation but is also associated with cell death. NO-induced growth inhibition and apoptosis of lymphocytes has been well documented (reviewed in ref. 37). A proapoptotic effect of NO in lymphocytes may be a consequence of either direct activation of apoptotic signaling pathways (e.g., Ras/ERK, ref. 40; and Fas, ref. 41) or of nitrosative stress (accumulation of S-nitrosylated proteins to hazardous levels, ref. 42). In toto, these data set the stage for important contributions of redox- and NO-based signaling in immune responses against cancer and HIV, as discussed below.
The Trx and GSH systems in cancer
In recent years, there has been a growing appreciation that a cellular nitroso-redox imbalance can promote tumor growth and resistance to therapy. This imbalance involves not only the cancer cells themselves but also other cells that populate the tumor and its microenvironment including cancer stem cells, vascular cells, and immune cells (43–48). Increased cellular levels of ROS and RNS can damage DNA and thereby promote carcinogenesis (49, 50); consequently, antioxidant vitamins (C and E) and cellular reductants (Trx and GSH) have long been considered cancer-suppressing molecules (51–53). However, mounting evidence has shown that antioxidant supplementation fails to provide cancer protection and might even increase cancer risk (54–56). These observations are supported and rationalized by recent mechanistic, interdisciplinary studies demonstrating that oxidative stress can inhibit cancer progression and metastasis and that the GSH and Trx systems may promote tumorigenesis and resistance to therapy (51–53, 57–59).
Increased levels of Trx or TrxR have been detected in multiple tumors (e.g., lung, breast, colorectal, pancreatic, hepatocellular, gastric, myeloma, non-Hodgkin lymphoma, and acute lymphocytic leukemia) and in association with aggressive tumor behavior (60–63). A recent study showed that mitochondrial TrxR contributes to tumor growth through the HIF-1 pathway (64), supporting the idea that mitochondrial redox signaling is important for tumorigenesis. Conversely, the TrxR inhibitor auranofin partially inhibits the growth of leukemia and relapsed Hodgkin lymphoma cells in vitro (65, 66). TrxR inhibitors may generally promote apoptosis or differentiation. Indeed, auranofin was shown to induce apoptosis of acute promyelocytic leukemia cells in vitro and promote the differentiation of these cells when used in combination with retinoic acid (67) — effects reminiscent of arsenicals, which, apart from their main target, the promyelocytic leukemia protein, also inhibit TrxR (68). Inhibition of the Trx system may also sensitize cancer cells to ionizing radiation and chemotherapeutic drugs, which have pro-oxidant effects (69, 70), and to the antitumor effects of NO donor compounds, likely via S-nitrosylation (71, 72). Additionally, auranofin administered as a single agent to patients with chronic lymphocytic leukemia had a significant, albeit transient, cidal effect (73). A larger clinical trial has been completed, and its results await publication (ClinicalTrials.gov identifier: NCT01419691). Auranofin is also being administered along with several other chemotherapeutics as a salvage therapy for recurrent glioblastoma (74). A clinical trial of auranofin-based combination therapy is expected to start in Ulm, Germany, in 2016 (European Clinical Trials Database [EudraCT] number 2014-004197-42). Collectively, these studies support the viability of Trx and TrxR inhibition as an anticancer strategy.
Blockade of the GSH system represents another potentially important anticancer strategy (75). Like Trx, GSH content increases during malignant transformation of lymphocytes and other cell types (76, 77). Oncogenic transformation is associated with an increased ratio of reduced GSH to oxidized GSH (78). This change to a more reducing intracellular environment often involves the transcription factor nuclear factor, erythroid-derived 2, like 2 (NRF2), which positively regulates antioxidant and detoxification enzymes (79), including enzymes needed for synthesis and handling of GSH. Notable examples of enzymes associated with malignant transformation include γ-glutamylcysteine ligase (also referred to as γ-glutamylcysteine synthetase) (80), the target of BSO, and glutathione S-transferases (GSTs) (81), which catalyze the formation of thioether bonds between GSH and electrophilic groups in xenobiotics. This mechanism of detoxification can lead to the development of resistance against chemotherapeutic agents (81). Tumorigenic effects of GSTs may also be mediated by inhibition of phosphorylation of the transcription factor c-Jun, resulting in antiapoptotic effects (82). The important role of GSH and GSH-associated enzymes in tumor development and progression and drug resistance has been recently demonstrated in mouse models. Specifically, Harris and colleagues demonstrated that blocking GSH synthesis with BSO or through genetic approaches inhibits the spontaneous development of lymphomas, sarcomas, and breast tumors in mice (83). BSO was, however, ineffective when malignant transformation had already occurred. Further analyses supported the conclusion that the upregulation of Trx may have compensated for the inhibition of GSH. More generally, despite the lack of activity of BSO when used alone, inhibition of GSH synthesis may find a place in rationally designed combination therapy. For example, melphalan, which is detoxified by GST, showed synergistic effects with BSO in a number of tumor lines (84). Moreover, BSO and melphalan proved generally well tolerated in phase I clinical trials involving advanced cancers (85, 86), the one exception being in children with recurrent neuroblastoma (87). Of note, melphalan was shown to inhibit TrxR in vitro, although this is probably not the mechanism of action at physiological plasma concentrations, as melphalan is principally an alkylating agent (88).
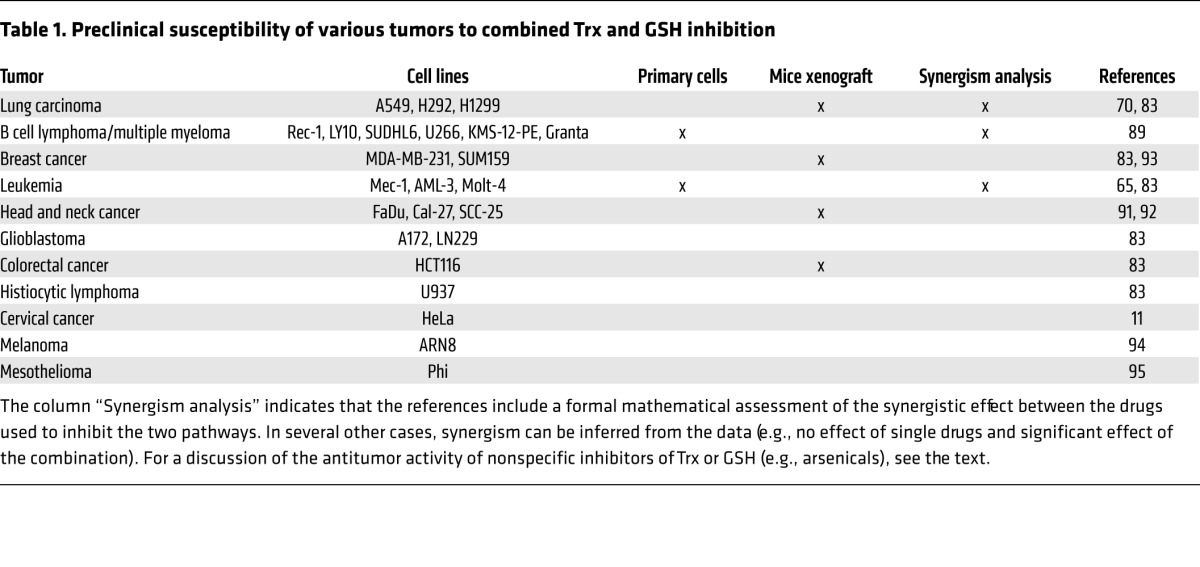
Several studies, including the study by Harris and colleagues (83), indicate that dual targeting of the GSH and Trx redox systems can result in powerful and synergistic antitumor effects. This conclusion is in line with studies showing that BSO markedly increased the effects of auranofin in leukemias and lymphomas, including chronic lymphocytic leukemia (65), large B cell lymphoma (an HIV-associated malignancy), mantle cell lymphoma, and in multiple myeloma (89). Potential redox-sensitive targets implicated in the antitumor effects of GSH and Trx silencing include ribonucleotide reductase (inhibition) and ASK1 (activation) (refs. 11, 90, and Figure 2). Recent evidence shows that glucose metabolism (e.g., GAPDH) is also inhibited by the induction of oxidative stress, which may impair the growth of tumors that are reliant on glycolysis (59). Involvement of additional oxidized or nitrosylated proteins is likely. The interrelation between the glycolytic pathway and Trx and GSH inhibition is also suggested by in vitro and in vivo data: the glycolysis inhibitor 2-deoxy-D-glucose (2DG) enhanced the antitumor effect of the combination of auranofin and BSO in a mouse xenograft model (70). Taken together, these and other studies support the idea that dual inhibition of the GSH and Trx pathways may represent an effective anticancer approach against tumors of multiple origins (refs. 11, 65, 70, 83, 89, 91–95, and Table 1). Notably, the combination of auranofin and BSO has been well tolerated in macaques and mice (70, 96), indicating that it may also be tolerated in patients.
Figure 2. Model of the anticancer and anti-HIV effects of targeting the Trx and GSH systems.

Left: Many cancer cells generate large amounts of ROS and RNS but also have elevated antioxidant defenses, largely through upregulation of the Trx and GSH systems. Inhibition of TrxR by auranofin can lead to elevated ROS and RNS levels, apoptosis, and decreased cell proliferation. However, when TrxR is inhibited, the GSH redox system often becomes upregulated, eliminating ROS and RNS and acting as a backup system to reduce Trx. Exposure of cancer cells to auranofin and BSO disables both Trx and GSH systems, severely compromising antioxidant capacity and leading to marked elevation of ROS and SNOs, which suppress cell proliferation and promote cell death. Cell death is in part mediated by inhibition of ribonucleotide reductase (RNR) and through activation of the proapoptotic kinase ASK1. Right: HIV-infected cells are characterized by elevated levels of ROS, coupled with decreased intracellular levels of antioxidant defenses (Trx, TrxR, and GSH). ART blocks HIV replication and partially counteracts the effects of oxidative stress, but is unable to target the latently infected cells. Inhibition of TrxR by auranofin increases oxidative (and potentially nitrosative) stress and apoptosis. TrxR inhibition also leads to a prodifferentiation effect on the latently HIV-infected cells, driving them toward short-lived phenotypes (i.e., effector memory) and, ultimately, decreasing the viral reservoir. Auranofin and BSO impair lymphocyte activation and increase HIV-infected cell death. ROS variations are documented, whereas RNS variations are predicted. Arrows beside the names of the molecules indicate the variation over time. SH, thiol.
Table 1. Preclinical susceptibility of various tumors to combined Trx and GSH inhibition.
Treating HIV/AIDS by increasing oxidative stress: results from in vitro and in vivo models
HIV and AIDS exhibit interesting similarities with cancer in terms of redox regulation and the history of its therapeutic exploitation, starting from the failure of antioxidant-based therapies and culminating in novel therapeutic strategies based on dual inhibition of the Trx and GSH systems. Classic studies conducted by the Dröge group and later confirmed by several independent investigators showed that intracellular levels of GSH were significantly reduced in HIV+ individuals and that this reduction was coupled with decreased levels of cystine in plasma (reviewed in ref. 97). The net result was oxidative stress that is linked to increased viral replication (98, 99). These findings paved the way for several clinical trials aimed at boosting antioxidant defenses. Numerous compounds were tested, including N-acetylcysteine (a thiol antioxidant, denitrosylating agent and a precursor of GSH); vitamins A, C, and E; and zinc, selenium, and several carotenoids. This trend paralleled the wave of studies of antioxidant therapies for cancer, with similarly disappointing effects on disease progression (in this case measured by viral load and CD4 counts) (100–102).
The subsequent success of combined antiretroviral therapy (ART) led to a decline in interest in alternative or complementary therapeutic approaches using agents such as antioxidants. However, while ART transformed a deadly disease into a manageable chronic condition, antiretroviral drugs proved unable to cure HIV infection, and the incidence of AIDS-related conditions, including cancer and heart disease, remains high in ART-treated individuals (103). The inability to cure HIV/AIDS resides in the fact that HIV persists in latently infected memory T cells (104), thus committing patients to lifelong therapy (105). In its dormant form, the virus, integrated within the host genome, is sheltered from both the immune system and antiretroviral drugs (104). Latent virus can survive for years in central and transitional memory T cells, and mathematical estimates suggest that viral reservoirs persist for more than 70 years (105). Latent proviral DNA may also spread through low-level antigenic activation and homeostatic proliferation of memory T cells, thus increasing the number of proviral DNA copies within the organism (104). Although not directly harmful, the HIV reservoir cell population does represent a danger for the host, as the virus can become reactivated following suspension of therapy (104). The population of latently infected cells living and proliferating within the organism may share an interesting similarity with cancer.
This paradigm of viral persistence has had important implications for the reevaluation of the role of oxidative and nitrosative stress in HIV infection (Figure 2). While increases in oxidative stress favor productive viral replication, antioxidant defenses favor the development and maintenance of latency (106–108). Under this model, inflammatory stimuli increase the activity of the transcriptional activators NF-κB and NFAT, which are required for the transcription of viral genes (109, 110) and p53, which coincidently mediates apoptosis (111). Antioxidants may therefore silence transcriptional activity (109, 110), while pro-oxidants, such as arsenic trioxide, may reactivate the viral reservoir (112). By the same token, both NO donors and endogenously generated NO may stimulate HIV reactivation (113). Relatedly, Kahn et al. (114) showed that anticancer conditioning agents (doxorubicin, etoposide, fludarabine phosphate, or vincristine) were able to induce HIV escape from latency and that this was associated with increased apoptosis rates (114). It is interesting to note that these drugs have been reported to induce oxidative stress (115–117), corroborating previous findings suggesting that the viral reservoir may be redox responsive and could potentially be eliminated by pro-oxidant drugs (118). The case of Mr. Timothy Brown (the “Berlin patient”), who was famously cured of HIV after a hematopoietic stem cell transplant to treat acute myeloid leukemia, may be instructive. The therapeutic approach was based on an initial conditioning regimen (which included the TrxR inhibitor cyclophosphamide), followed by heterologous stem cell transplantation from a donor carrying a mutation that confers resistance to HIV infection (119). Following suspension of ART, the virus was apparently eradicated (119).
That induction of oxidative or nitrosative stress is a promising therapeutic strategy for HIV/AIDS is shown by studies of ART-treated macaques that were chronically infected with the HIV homolog SIVmac251 (35, 96). In these studies, the combined inhibition of the Trx and GSH systems, obtained with auranofin and BSO coupled with a potent ART regimen, induced a functional cure, i.e., long-term, drug-free control in the absence of disease progression (120). This state has been maintained for at least two years following the suspension of all drugs, with follow-up still ongoing (35). The mechanism of action of auranofin and BSO is likely three-fold (Figures 2 and 3). First, auranofin causes a reduction in the viral reservoir as a direct consequence of the proapoptotic and prodifferentiating effects of auranofin on memory T cells (26, 27) harboring the bulk of latent virus (ref. 104 and Figure 2). Second, auranofin and BSO deplete the activated lymphocyte population that is the main target of HIV and whose levels are associated with a poor prognosis (recently reviewed in refs. 121, 122, and Figure 2). Third, and perhaps most important, the drugs boost immunity against the conserved sequences of the viral capsid Gag protein (Figure 3). Indeed, when this immunity was inhibited by temporary depletion of CD8+ cells, the control of the virus was lost, thereby establishing causality (35).
Figure 3. Hypothesized bottleneck mechanism for the enhancement of cell-mediated immunity following inhibition of the Trx and GSH systems.

A professional APC (DC) can phagocytize a foreign pathogen (HIV), degrade it into peptidic antigens, and present these antigens to naive lymphocytes. Because of high mutation rates, HIV antigens are characterized by high variability, with the exception of highly conserved, essential proteins, which are consequently well represented in a typical viral population. Following antigen presentation, the lymphocyte becomes activated and primed toward a specific antigen. During the progression of HIV infection (upper panel), the lymphocyte clones specific for conserved antigens (red halo in the figure) are reduced in frequency due to the accumulation of “nonfunctional clones” directed against increasingly mutated antigens (green and gold halos). When Trx and GSH systems are inhibited, lymphocyte activation is impaired, and the majority of activated lymphocyte clones undergo stochastic apoptosis. The more frequently represented clones have a selective advantage in survival as a result of their numbers. This leads to a bottleneck effect that can favor repopulation by those lymphocytes primed against the most frequently represented (conserved) antigens.
Modulation of immune activation by inhibition of Trx and GSH systems: anticancer and anti-HIV immune responses
The notion that inhibition of lymphocyte proliferation by auranofin and BSO enhances T cell–mediated immune responses against SIV and HIV may seem paradoxical at first glance. However, evidence that selective lymphocyte subset inhibition may improve cell-mediated immunity is supported by studies conducted in cancer patients in whom tumor-specific immunity is enhanced by cytotoxic agents that cause transient lymphopenia (reviewed in ref. 123). Notably, a number of these cytotoxic agents have redox-based activity. Some directly inhibit Trx and GSH systems (e.g., melphalan, arsenicals), others increase ROS (e.g., the histone deacetylase inhibitor vorinostat, ref. 124), while others mimic the effects of Trx and GSH inhibition on some key targets (e.g., ribonucleotide reductase inhibitors) (125).
How decreases in lymphocytes, although transient, may result in enhanced and more efficient immunity remains a matter of debate. Transient lymphodepletion has been observed to be associated with a lymphocyte recovery characterized by an “effector phenotype” (e.g., CD27–CD95+ T lymphocytes), i.e., a population of cells that can mount a specific immune response (reviewed in ref. 123). This may be due to the removal of immune inhibitory cell types such as Tregs (123).
A prodifferentiating effect of lymphocyte-depleting therapies (as seen with auranofin and BSO) is another possibility. In particular, a prodifferentiating effect of auranofin in T lymphocytes has recently been published by some of us (26). The mechanism can be decoupled from an H2O2-independent proapoptotic effect. Ingenuity pathway analysis has shown that one likely mediator of differentiation is TCR-associated ζ-chain protein kinase 70 kDa (ZAP70), a cysteine-rich protein that mediates the activation and differentiation of lymphocytes. Nitrosative stress can also mediate prodifferentiation effects. The cytokine IFN-γ may also be involved in this phenomena through NO-based mechanisms that favor the switch to a Th1 phenotype (i.e., a cytotoxic cell–mediated response) (126, 127).
GSH depletion does not increase the prodifferentiation effect induced by TrxR inhibition (35), but may alter the intrinsic immunogenic properties of target cells, potentially by acting on death receptors. In this regard, treatment of drug-resistant leukemia cells with BSO induces the formation of the CD95 death–inducing signaling complex (128). Other lymphocyte-depleting chemotherapeutic regimens, which interfere with GSH, increase the expression of death receptors (123).
Finally, mathematical modeling may aid in understanding the lymphodepletion and immune control paradox (129, 130), as mediated by auranofin and BSO. One insight relates to the removal of ineffective immune-dominant responses (131), which can be best understood in terms of a “bottleneck” effect on T cell survival following suspension of ART if lymphocyte activation is inhibited at the time of viral rebound. In this model, the temporary depletion of activated T lymphocytes, as seen with auranofin and BSO, would lead to the elimination of clones directed against HIV (Figure 3). This could spark a renewal of the lymphocyte populations that favor the development of immunity against antigens most frequently encountered by newly produced T cells (i.e., evolutionarily conserved antigens), which are least susceptible to escape mutations (Figure 3). The possibility of similar bottleneck effects on the boosting of cell-mediated immunity may also deserve consideration in the cancer setting.
Concluding remarks
The studies reviewed in this article support the premise that decreasing, rather than increasing, the cellular defenses against ROS and RNS results in an enhanced immune response and thus presents a viable anticancer and anti-HIV strategy. Oxidative and nitrosative stress–inducing drugs, of which auranofin, BSO, and arsenic oxides are prototypes, may represent a new frontier in cancer and HIV research. It should be emphasized that HIV/AIDS is associated with multiple types of cancer (103), and although the introduction of ART has led to a dramatic improvement in the prognosis of HIV infection, the incidence of cancer in HIV-infected patients has remained high. This is particularly evident in countries with a high incidence of HIV such as South Africa, where HIV and lymphoma comorbidity is a huge societal problem (132, 133). Clinical trials of Trx and GSH pathway inhibitors in individuals affected by HIV and lymphoma should be considered. The strategy should ideally be tested first in HIV-infected individuals, given the availability of safety and efficacy data in a phylogenetically close animal model, i.e., SIV-infected macaques, which closely recapitulate the disease progression and virus persistence seen during ART (134, 135). A phase II clinical trial that includes a treatment arm with ART and auranofin (but without BSO) is ongoing in Brazil (Ricardo Diaz, principal investigator; Fundação de Amparo à Pesquisa do Estado de São Paulo [FAPESP], grant no. 13/11323-5). This trial will help to estimate the effects of auranofin on the viral reservoir and may pave the way for a trial of combined Trx and GSH inhibition in HIV-infected individuals undergoing ART. The HIV trials will furnish important data to guide future trials in cancer patients. Further studies based on molecular markers and metabolic pathway analyses mapping the susceptibility of different tumors to Trx and GSH inhibition will be necessary in order to optimally combine this line of drugs with current chemotherapeutics.
Acknowledgments
We wish to thank the following organizations for grant support: the Israel Science Foundation (ISF) (1574/14) and the Israel Cancer Research Fund; the Italian Institute of Health (2013 intramural grant ONCOVR/1); and the NIH (P01HL075443 and R01GM099921).
Footnotes
Conflict of interest: M. Benhar, J.S. Stamler, and A. Savarino are inventors of patents covering some of the therapeutic drug combinations described in this article.
Reference information:J Clin Invest. 2016;126(5):1630–1639. doi:10.1172/JCI85339.
Contributor Information
Moran Benhar, Email: benhar@tx.technion.ac.il.
Iart Luca Shytaj, Email: luca.shytaj@gmail.com.
Jonathan S. Stamler, Email: jonathan.stamler@case.edu.
Andrea Savarino, Email: andrea.savarino@iss.it.
References
- 1.Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol. 2005;6(2):150–166. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- 2.Janssen-Heininger YM, et al. Redox-based regulation of signal transduction: principles, pitfalls, and promises. Free Radic Biol Med. 2008;45(1):1–17. doi: 10.1016/j.freeradbiomed.2008.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Go YM, Jones DP. The redox proteome. J Biol Chem. 2013;288(37):26512–26520. doi: 10.1074/jbc.R113.464131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lillig CH, Holmgren A. Thioredoxin and related molecules — from biology to health and disease. Antioxid Redox Signal. 2007;9(1):25–47. doi: 10.1089/ars.2007.9.25. [DOI] [PubMed] [Google Scholar]
- 5.Ghezzi P. Protein glutathionylation in health and disease. Biochim Biophys Acta. 2013;1830(5):3165–3172. doi: 10.1016/j.bbagen.2013.02.009. [DOI] [PubMed] [Google Scholar]
- 6.Zhang H, Forman HJ. Glutathione synthesis and its role in redox signaling. Semin Cell Dev Biol. 2012;23(7):722–728. doi: 10.1016/j.semcdb.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Benhar M, Forrester MT, Stamler JS. Protein denitrosylation: enzymatic mechanisms and cellular functions. Nat Rev Mol Cell Biol. 2009;10(10):721–732. doi: 10.1038/nrm2764. [DOI] [PubMed] [Google Scholar]
- 8.Mandal PK, et al. Loss of thioredoxin reductase 1 renders tumors highly susceptible to pharmacologic glutathione deprivation. Cancer Res. 2010;70(22):9505–9514. doi: 10.1158/0008-5472.CAN-10-1509. [DOI] [PubMed] [Google Scholar]
- 9.Prigge JR, et al. Hepatocyte DNA replication in growing liver requires either glutathione or a single allele of txnrd1. Free Radic Biol Med. 2012;52(4):803–810. doi: 10.1016/j.freeradbiomed.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Levring TB, et al. Human CD4+ T cells require exogenous cystine for glutathione and DNA synthesis. Oncotarget. 2015;6(26):21853–21864. doi: 10.18632/oncotarget.5213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Du Y, Zhang H, Lu J, Holmgren A. Glutathione and glutaredoxin act as a backup of human thioredoxin reductase 1 to reduce thioredoxin 1 preventing cell death by aurothioglucose. J Biol Chem. 2012;287(45):38210–38219. doi: 10.1074/jbc.M112.392225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jones DP, Go YM. Redox compartmentalization and cellular stress. Diabetes Obes Metab. 2010;12(suppl 2):116–125. doi: 10.1111/j.1463-1326.2010.01266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Go YM, et al. Selective targeting of the cysteine proteome by thioredoxin and glutathione redox systems. Mol Cell Proteomics. 2013;12(11):3285–3296. doi: 10.1074/mcp.M113.030437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pader I, et al. Thioredoxin-related protein of 14 kDa is an efficient L-cystine reductase and S-denitrosylase. Proc Natl Acad Sci U S A. 2014;111(19):6964–6969. doi: 10.1073/pnas.1317320111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Benhar M. Nitric oxide and the thioredoxin system: A complex interplay in redox regulation. Biochim Biophys Acta. 2015;1850(12):2476–2484. doi: 10.1016/j.bbagen.2015.09.010. [DOI] [PubMed] [Google Scholar]
- 16.Lu J, Holmgren A. The thioredoxin antioxidant system. Free Radic Biol Med. 2014;66:75–87. doi: 10.1016/j.freeradbiomed.2013.07.036. [DOI] [PubMed] [Google Scholar]
- 17.Buettner GR, Wagner BA, Rodgers VG. Quantitative redox biology: an approach to understand the role of reactive species in defining the cellular redox environment. Cell Biochem Biophys. 2013;67(2):477–483. doi: 10.1007/s12013-011-9320-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adimora NJ, Jones DP, Kemp ML. A model of redox kinetics implicates the thiol proteome in cellular hydrogen peroxide responses. Antioxid Redox Signal. 2010;13(6):731–743. doi: 10.1089/ars.2009.2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kesarwani P, Murali AK, Al-Khami AA, Mehrotra S. Redox regulation of T-cell function: from molecular mechanisms to significance in human health and disease. Antioxid Redox Signal. 2013;18(12):1497–1534. doi: 10.1089/ars.2011.4073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Malissen B, Grégoire C, Malissen M, Roncagalli R. Integrative biology of T cell activation. Nat Immunol. 2014;15(9):790–797. doi: 10.1038/ni.2959. [DOI] [PubMed] [Google Scholar]
- 21.Simeoni L, Bogeski I. Redox regulation of T-cell receptor signaling. Biol Chem. 2015;396(5):555–568. doi: 10.1515/hsz-2014-0312. [DOI] [PubMed] [Google Scholar]
- 22.Michalek RD, et al. The requirement of reversible cysteine sulfenic acid formation for T cell activation and function. J Immunol. 2007;179(10):6456–6467. doi: 10.4049/jimmunol.179.10.6456. [DOI] [PubMed] [Google Scholar]
- 23.Chiu J, Dawes IW. Redox control of cell proliferation. Trends Cell Biol. 2012;22(11):592–601. doi: 10.1016/j.tcb.2012.08.002. [DOI] [PubMed] [Google Scholar]
- 24.Vené R, et al. Redox remodeling allows and controls B-cell activation and differentiation. Antioxid Redox Signal. 2010;13(8):1145–1155. doi: 10.1089/ars.2009.3078. [DOI] [PubMed] [Google Scholar]
- 25.Ueno H, Kajihara H, Nakamura H, Yodoi J, Nakamuro K. Contribution of thioredoxin reductase to T-cell mitogenesis and NF-kappaB DNA-binding promoted by selenite. Antioxid Redox Signal. 2007;9(1):115–121. doi: 10.1089/ars.2007.9.115. [DOI] [PubMed] [Google Scholar]
- 26.Chirullo B, et al. A candidate anti-HIV reservoir compound, auranofin, exerts a selective ‘anti-memory’ effect by exploiting the baseline oxidative status of lymphocytes. Cell Death Dis. 2013;4: doi: 10.1038/cddis.2013.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lewis MG, et al. Gold drug auranofin restricts the viral reservoir in the monkey AIDS model and induces containment of viral load following ART suspension. AIDS. 2011;25(11):1347–1356. doi: 10.1097/QAD.0b013e328347bd77. [DOI] [PubMed] [Google Scholar]
- 28.Angelini G, et al. Antigen-presenting dendritic cells provide the reducing extracellular microenvironment required for T lymphocyte activation. Proc Natl Acad Sci U S A. 2002;99(3):1491–1496. doi: 10.1073/pnas.022630299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yan Z, Garg SK, Kipnis J, Banerjee R. Extracellular redox modulation by regulatory T cells. Nat Chem Biol. 2009;5(10):721–723. doi: 10.1038/nchembio.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Levring TB, et al. Activated human CD4+ T cells express transporters for both cysteine and cystine. Sci Rep. 2012;2: doi: 10.1038/srep00266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Messina JP, Lawrence DA. Cell cycle progression of glutathione-depleted human peripheral blood mononuclear cells is inhibited at S phase. J Immunol. 1989;143(6):1974–1981. [PubMed] [Google Scholar]
- 32.Grek CL, Zhang J, Manevich Y, Townsend DM, Tew KD. Causes and consequences of cysteine S-glutathionylation. J Biol Chem. 2013;288(37):26497–26504. doi: 10.1074/jbc.R113.461368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gould N, Doulias PT, Tenopoulou M, Raju K, Ischiropoulos H. Regulation of protein function and signaling by reversible cysteine S-nitrosylation. J Biol Chem. 2013;288(37):26473–26479. doi: 10.1074/jbc.R113.460261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vint IA, Chain BM, Foreman JC. The interaction of auranofin and buthionine sulfoximine blocks activation of human peripheral T lymphocytes. Cell Immunol. 1993;152(1):152–161. doi: 10.1006/cimm.1993.1275. [DOI] [PubMed] [Google Scholar]
- 35.Shytaj IL, et al. Two-year follow-up of macaques developing intermittent control of the human immunodeficiency virus homolog simian immunodeficiency virus SIVmac251 in the chronic phase of infection. J Virol. 2015;89(15):7521–7535. doi: 10.1128/JVI.00396-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lanna A, Henson SM, Escors D, Akbar AN. The kinase p38 activated by the metabolic regulator AMPK and scaffold TAB1 drives the senescence of human T cells. Nat Immunol. 2014;15(10):965–972. doi: 10.1038/ni.2981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bogdan C. Nitric oxide synthase in innate and adaptive immunity: an update. Trends Immunol. 2015;36(3):161–178. doi: 10.1016/j.it.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 38.Mannick JB, Asano K, Izumi K, Kieff E, Stamler JS. Nitric oxide produced by human B lymphocytes inhibits apoptosis and Epstein-Barr virus reactivation. Cell. 1994;79(7):1137–1146. doi: 10.1016/0092-8674(94)90005-1. [DOI] [PubMed] [Google Scholar]
- 39.Ibiza S, et al. Endothelial nitric oxide synthase regulates T cell receptor signaling at the immunological synapse. Immunity. 2006;24(6):753–765. doi: 10.1016/j.immuni.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 40.Ibiza S, et al. Endothelial nitric oxide synthase regulates N-Ras activation on the Golgi complex of antigen-stimulated T cells. Proc Natl Acad Sci U S A. 2008;105(30):10507–10512. doi: 10.1073/pnas.0711062105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Benhar M, Forrester MT, Hess DT, Stamler JS. Regulated protein denitrosylation by cytosolic and mitochondrial thioredoxins. Science. 2008;320(5879):1050–1054. doi: 10.1126/science.1158265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu L, et al. Essential roles of S-nitrosothiols in vascular homeostasis and endotoxic shock. Cell. 2004;116(4):617–628. doi: 10.1016/S0092-8674(04)00131-X. [DOI] [PubMed] [Google Scholar]
- 43.Benhar M, Engelberg D, Levitzki A. ROS, stress-activated kinases and stress signaling in cancer. EMBO Rep. 2002;3(5):420–425. doi: 10.1093/embo-reports/kvf094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grek CL, Tew KD. Redox metabolism and malignancy. Curr Opin Pharmacol. 2010;10(4):362–368. doi: 10.1016/j.coph.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Parri M, Chiarugi P. Redox molecular machines involved in tumor progression. Antioxid Redox Signal. 2013;19(15):1828–1845. doi: 10.1089/ars.2012.5040. [DOI] [PubMed] [Google Scholar]
- 46.Jorgenson TC, Zhong W, Oberley TD. Redox imbalance and biochemical changes in cancer. Cancer Res. 2013;73(20):6118–6123. doi: 10.1158/0008-5472.CAN-13-1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Castellani P, Balza E, Rubartelli A. Inflammation, DAMPs, tumor development, and progression: a vicious circle orchestrated by redox signaling. Antioxid Redox Signal. 2014;20(7):1086–1097. doi: 10.1089/ars.2012.5164. [DOI] [PubMed] [Google Scholar]
- 48.Burke AJ, Sullivan FJ, Giles FJ, Glynn SA. The yin and yang of nitric oxide in cancer progression. Carcinogenesis. 2013;34(3):503–512. doi: 10.1093/carcin/bgt034. [DOI] [PubMed] [Google Scholar]
- 49.Marnett LJ. Oxyradicals and DNA damage. Carcinogenesis. 2000;21(3):361–370. doi: 10.1093/carcin/21.3.361. [DOI] [PubMed] [Google Scholar]
- 50.Hussain SP, Hofseth LJ, Harris CC. Radical causes of cancer. Nat Rev Cancer. 2003;3(4):276–285. doi: 10.1038/nrc1046. [DOI] [PubMed] [Google Scholar]
- 51.Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8(7):579–591. doi: 10.1038/nrd2803. [DOI] [PubMed] [Google Scholar]
- 52.Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov. 2013;12(12):931–947. doi: 10.1038/nrd4002. [DOI] [PubMed] [Google Scholar]
- 53.Glasauer A, Chandel NS. Targeting antioxidants for cancer therapy. Biochem Pharmacol. 2014;92(1):90–101. doi: 10.1016/j.bcp.2014.07.017. [DOI] [PubMed] [Google Scholar]
- 54.Albanes D, et al. α-Tocopherol and β-carotene supplements and lung cancer incidence in the α-tocopherol, β-carotene cancer prevention study: effects of base-line characteristics and study compliance. J Natl Cancer Inst. 1996;88(21):1560–1570. doi: 10.1093/jnci/88.21.1560. [DOI] [PubMed] [Google Scholar]
- 55.Omenn GS, et al. Effects of a combination of beta carotene and vitamin A on lung cancer and cardiovascular disease. N Engl J Med. 1996;334(18):1150–1155. doi: 10.1056/NEJM199605023341802. [DOI] [PubMed] [Google Scholar]
- 56.Fortmann SP, Burda BU, Senger CA, Lin JS, Whitlock EP. Vitamin and mineral supplements in the primary prevention of cardiovascular disease and cancer: An updated systematic evidence review for the U.S. Preventive Services Task Force. Ann Intern Med. 2013;159(12):824–834. doi: 10.7326/0003-4819-159-12-201312170-00729. [DOI] [PubMed] [Google Scholar]
- 57.Piskounova E, et al. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature. 2015;5 27(7577):186–191. doi: 10.1038/nature15726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Harris IS, Brugge JS. Cancer: The enemy of my enemy is my friend. Nature. 2015;527(7577):170–171. doi: 10.1038/nature15644. [DOI] [PubMed] [Google Scholar]
- 59.Yun J, et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science. 2015;350(6266):1391–1396. doi: 10.1126/science.aaa5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lincoln DT, Ali Emadi EM, Tonissen KF, Clarke FM. The thioredoxin-thioredoxin reductase system: over-expression in human cancer. Anticancer Res. 2003;23(3B):2425–2433. [PubMed] [Google Scholar]
- 61.Powis G, Mustacich D, Coon A. The role of the redox protein thioredoxin in cell growth and cancer. Free Radic Biol Med. 2000;29(3–4):312–322. doi: 10.1016/s0891-5849(00)00313-0. [DOI] [PubMed] [Google Scholar]
- 62.Arnér ES, Holmgren A. The thioredoxin system in cancer. Semin Cancer Biol. 2006;16(6): doi: 10.1016/j.semcancer.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 63.Vance TM, et al. Thioredoxin 1 in prostate tissue is associated with gleason score, erythrocyte antioxidant enzyme activity, and dietary antioxidants. Prostate Cancer. 2015;2015: doi: 10.1155/2015/728046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hellfritsch J, et al. Knockout of mitochondrial thioredoxin reductase stabilizes prolyl hydroxylase 2 and inhibits tumor growth and tumor-derived angiogenesis. Antioxid Redox Signal. 2015;22(11):938–950. doi: 10.1089/ars.2014.5889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fiskus W, et al. Auranofin induces lethal oxidative and endoplasmic reticulum stress and exerts potent preclinical activity against chronic lymphocytic leukemia. Cancer Res. 2014;74(9):2520–2532. doi: 10.1158/0008-5472.CAN-13-2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Celegato M, et al. Preclinical activity of the repurposed drug auranofin in classical Hodgkin lymphoma. Blood. 2015;126(11):1394–1397. doi: 10.1182/blood-2015-07-660365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kim IS, Jin JY, Lee IH, Park SJ. Auranofin induces apoptosis and when combined with retinoic acid enhances differentiation of acute promyelocytic leukaemia cells in vitro. Br J Pharmacol. 2004;142(4):749–755. doi: 10.1038/sj.bjp.0705708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jungwirth U, Kowol CR, Keppler BK, Hartinger CG, Berger W, Heffeter P. Anticancer activity of metal complexes: involvement of redox processes. Antioxid Redox Signal. 2011;15(4):1085–1127. doi: 10.1089/ars.2010.3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Patwardhan RS, Sharma D, Checker R, Thoh M, Sandur SK. Spatio-temporal changes in glutathione and thioredoxin redox couples during ionizing radiation-induced oxidative stress regulate tumor radio-resistance. Free Radic Res. 2015;49(10):1218–1232. doi: 10.3109/10715762.2015.1056180. [DOI] [PubMed] [Google Scholar]
- 70.Fath MA, Ahmad IM, Smith CJ, Spence J, Spitz DR. Enhancement of carboplatin-mediated lung cancer cell killing by simultaneous disruption of glutathione and thioredoxin metabolism. Clin Cancer Res. 2011;17(19):6206–6217. doi: 10.1158/1078-0432.CCR-11-0736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Edes K, Cassidy P, Shami PJ, Moos PJ. JS-K, a nitric oxide prodrug, has enhanced cytotoxicity in colon cancer cells with knockdown of thioredoxin reductase 1. PLoS One. 2010;5(1): doi: 10.1371/journal.pone.0008786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cañas A, et al. Maintenance of S-nitrosothiol homeostasis plays an important role in growth suppression of estrogen receptor-positive breast tumors. Breast Cancer Res. 2012;14(6): doi: 10.1186/bcr3366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Saba NS, et al. Auranofin induces a reversible in-vivo stress response that correlates with a transient clinical effect in patients with chronic lymphocytic leukemia. Blood. 2013;122(21): [Google Scholar]
- 74.Kast RE, Karpel-Massler G, Halatsch ME. CUSP9* treatment protocol for recurrent glioblastoma: aprepitant, artesunate, auranofin, captopril, celecoxib, disulfiram, itraconazole, ritonavir, sertraline augmenting continuous low dose temozolomide. Oncotarget. 2014;5(18):8052–8082. doi: 10.18632/oncotarget.2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Traverso N, et al. Role of glutathione in cancer progression and chemoresistance. Oxid Med Cell Longev. 2013;2013: doi: 10.1155/2013/972913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Estrela JM, Ortega A, Obrador E. Glutathione in cancer biology and therapy. Crit Rev Clin Lab Sci. 2006;43(2):143–181. doi: 10.1080/10408360500523878. [DOI] [PubMed] [Google Scholar]
- 77.Zhang W, et al. Stromal control of cystine metabolism promotes cancer cell survival in chronic lymphocytic leukaemia. Nat Cell Biol. 2012;14(3):276–286. doi: 10.1038/ncb2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Balendiran GK, Dabur R, Fraser D. The role of glutathione in cancer. Cell Biochem Funct. 2004;22(6):343–352. doi: 10.1002/cbf.1149. [DOI] [PubMed] [Google Scholar]
- 79.DeNicola GM, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475(7354):106–109. doi: 10.1038/nature10189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yao KS, Godwin AK, Johnson SW, Ozols RF, O’Dwyer PJ, Hamilton TC. Evidence for altered regulation of gamma-glutamylcysteine synthetase gene expression among cisplatin-sensitive and cisplatin-resistant human ovarian cancer cell lines. Cancer Res. 1995;55(19):4367–4374. [PubMed] [Google Scholar]
- 81.Townsend DM, Tew KD. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene. 2003;22(47):7369–7375. doi: 10.1038/sj.onc.1206940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Laborde E. Glutathione transferases as mediators of signaling pathways involved in cell proliferation and cell death. Cell Death Differ. 2010;17(9):1373–1380. doi: 10.1038/cdd.2010.80. [DOI] [PubMed] [Google Scholar]
- 83.Harris IS, et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell. 2015;27(2):211–222. doi: 10.1016/j.ccell.2014.11.019. [DOI] [PubMed] [Google Scholar]
- 84.Wu JH, Batist G. Glutathione and glutathione analogues; therapeutic potentials. Biochim Biophys Acta. 2013;1830(5):3350–3353. doi: 10.1016/j.bbagen.2012.11.016. [DOI] [PubMed] [Google Scholar]
- 85.O’Dwyer PJ, et al. Phase I trial of buthionine sulfoximine in combination with melphalan in patients with cancer. J Clin Oncol. 1996;14(1):249–256. doi: 10.1200/JCO.1996.14.1.249. [DOI] [PubMed] [Google Scholar]
- 86.Bailey HH, et al. Phase I study of continuous-infusion L-S,R-buthionine sulfoximine with intravenous melphalan. J Natl Cancer Inst. 1997;89(23):1789–1796. doi: 10.1093/jnci/89.23.1789. [DOI] [PubMed] [Google Scholar]
- 87.Anderson CP, et al. Pilot study of intravenous melphalan combined with continuous infusion L-S,R-buthionine sulfoximine for children with recurrent neuroblastoma. Pediatr Blood Cancer. 2015;62(10):1739–1746. doi: 10.1002/pbc.25594. [DOI] [PubMed] [Google Scholar]
- 88.Witte AB, Anestål K, Jerremalm E, Ehrsson H, Arnér ES. Inhibition of thioredoxin reductase but not of glutathione reductase by the major classes of alkylating and platinum-containing anticancer compounds. Free Radic Biol Med. 2005;39(5):696–703. doi: 10.1016/j.freeradbiomed.2005.04.025. [DOI] [PubMed] [Google Scholar]
- 89.Kiebala M, et al. Dual targeting of the thioredoxin and glutathione antioxidant systems in malignant B cells: a novel synergistic therapeutic approach. Exp Hematol. 2015;43(2):89–99. doi: 10.1016/j.exphem.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zahedi Avval F, Holmgren A. Molecular mechanisms of thioredoxin and glutaredoxin as hydrogen donors for Mammalian s phase ribonucleotide reductase. J Biol Chem. 2009;284(13):8233–8240. doi: 10.1074/jbc.M809338200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Simons AL, Parsons AD, Foster KA, Orcutt KP, Fath MA, Spitz DR. Inhibition of glutathione and thioredoxin metabolism enhances sensitivity to perifosine in head and neck cancer cells. J Oncol. 2009;2009: doi: 10.1155/2009/519563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sobhakumari A, et al. Susceptibility of human head and neck cancer cells tocombined inhibition of glutathione and thioredoxin metabolism. PLoS One. 2012;7(10): doi: 10.1371/journal.pone.0048175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Scarbrough PM, Mapuskar KA, Mattson DM, Gius D, Watson WH, Spitz DR. Simultaneous inhibition of glutathione- and thioredoxin-dependent metabolism is necessary to potentiate 17AAG-induced cancer cell killing via oxidative stress. Free Radic Biol Med. 2012;52(2):436–443. doi: 10.1016/j.freeradbiomed.2011.10.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sachweh MC, et al. Redox effects and cytotoxic profiles of MJ25 and auranofin towards malignant melanoma cells. Oncotarget. 2015;6(18):16488–16506. doi: 10.18632/oncotarget.4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.You BR, Park WH. Auranofin induces mesothelioma cell death through oxidative stress and GSH depletion. Oncol Rep. 2015;35(1):546–551. doi: 10.3892/or.2015.4382. [DOI] [PubMed] [Google Scholar]
- 96.Shytaj IL, et al. Investigational treatment suspension and enhanced cell-mediated immunity at rebound followed by drug-free remission of simian AIDS. Retrovirology. 2013;10: doi: 10.1186/1742-4690-10-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Staal FJ. Glutathione and HIV infection: reduced, or increased oxidized? Eur J Clin Invest. 1998;28(3):194–196. doi: 10.1046/j.1365-2362.1998.00268.x. [DOI] [PubMed] [Google Scholar]
- 98.Schreck R, Rieber P, Baeuerle PA. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-kappa B transcription factor and HIV-1. EMBO J. 1991;10(8):2247–2258. doi: 10.1002/j.1460-2075.1991.tb07761.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Garaci E, et al. Intracellular GSH content and HIV replication in human macrophages. J Leukoc Biol. 1997;62(1):54–59. doi: 10.1002/jlb.62.1.54. [DOI] [PubMed] [Google Scholar]
- 100.Akerlund B, Jarstrand C, Lindeke B, Sönnerborg A, Akerblad AC, Rasool O. Effect of N-acetylcysteine(NAC) treatment on HIV-1 infection: a double-blind placebo-controlled trial. Eur J Clin Pharmacol. 1996;50(6):457–461. doi: 10.1007/s002280050140. [DOI] [PubMed] [Google Scholar]
- 101.Allard JP, et al. Effects of vitamin E and C supplementation on oxidative stress and viral load in HIV-infected subjects. AIDS. 1998;12(13):1653–1659. doi: 10.1097/00002030-199813000-00013. [DOI] [PubMed] [Google Scholar]
- 102.Kelly P, Musonda R, Kafwembe E, Kaetano L, Keane E, Farthing M. Micronutrient supplementation in the AIDS diarrhoea-wasting syndrome in Zambia: a randomized controlled trial. AIDS. 1999;13(4):495–500. doi: 10.1097/00002030-199903110-00008. [DOI] [PubMed] [Google Scholar]
- 103.Narayan KM, et al. HIV and noncommunicable disease comorbidities in the era of antiretroviral therapy: a vital agenda for research in low- and middle-income country settings. J Acquir Immune Defic Syndr. 2014;67(suppl 1):S2–S7. doi: 10.1097/QAI.0000000000000267. [DOI] [PubMed] [Google Scholar]
- 104.Chomont N, DaFonseca S, Vandergeeten C, Ancuta P, Sékaly RP. Maintenance of CD4+ T-cell memory and HIV persistence: keeping memory, keeping HIV. Curr Opin HIV AIDS. 2011;6(1):30–36. doi: 10.1097/COH.0b013e3283413775. [DOI] [PubMed] [Google Scholar]
- 105.Finzi D, et al. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat Med. 1999;5(5):512–517. doi: 10.1038/8394. [DOI] [PubMed] [Google Scholar]
- 106.Kazazi F, Koehler JK, Klebanoff SJ. Activation of the HIV long terminal repeat and viral production by H2O2-vanadate. Free Radic Biol Med. 1996;20(6):813–820. doi: 10.1016/0891-5849(95)02214-7. [DOI] [PubMed] [Google Scholar]
- 107.Kalebic T, Kinter A, Poli G, Anderson ME, Meister A, Fauci AS. Suppression of human immunodeficiency virus expression in chronically infected monocytic cells by glutathione, glutathione ester, and N-acetylcysteine. Proc Natl Acad Sci U S A. 1991;88(3):986–990. doi: 10.1073/pnas.88.3.986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Roederer M, Raju PA, Staal FJ, Herzenberg LA, Herzenberg LA. N-acetylcysteine inhibits latent HIV expression in chronically infected cells. AIDS Res Hum Retroviruses. 1991;7(6):563–567. doi: 10.1089/aid.1991.7.563. [DOI] [PubMed] [Google Scholar]
- 109.Piret B, Legrand-Poels S, Sappey C, Piette J. NF-κB transcription factor and human immunodeficiency virus type 1 (HIV-1) activation by methylene blue photosensitization. Eur J Biochem. 1995;228(2):447–455. doi: 10.1111/j.1432-1033.1995.tb20283.x. [DOI] [PubMed] [Google Scholar]
- 110.Bosque A, Planelles V. Induction of HIV-1 latency and reactivation in primary memory CD4+ T cells. Blood. 2009;113(1):58–65. doi: 10.1182/blood-2008-07-168393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Shinozaki S, et al. Inflammatory stimuli induce inhibitory S-nitrosylation of the deacetylase SIRT1 to increase acetylation and activation of p53 and p65. Sci Signal. 2014;7(351): doi: 10.1126/scisignal.2005375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lusic M, Marini B, Ali H, Lucic B, Luzzati R, Giacca M. Proximity to PML nuclear bodies regulates HIV-1 latency in CD4+ T cells. Cell Host Microbe. 2013;13(6):665–677. doi: 10.1016/j.chom.2013.05.006. [DOI] [PubMed] [Google Scholar]
- 113.Mannick JB, et al. Nitric oxide modulates HIV-1 replication. J Acquir Immune Defic Syndr. 1999;22(1):1–9. doi: 10.1097/00042560-199909010-00001. [DOI] [PubMed] [Google Scholar]
- 114.Khan SZ, Hand N, Zeichner SL. Apoptosis-induced activation of HIV-1 in latently infected cell lines. Retrovirology. 2015;12: doi: 10.1186/s12977-015-0169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kamio T, et al. B-cell-specific transcription factor BACH2 modifies the cytotoxic effects of anticancer drugs. Blood. 2003;102(9):3317–3322. doi: 10.1182/blood-2002-12-3656. [DOI] [PubMed] [Google Scholar]
- 116.Christopherson RI, Mactier S, Almazi JG, Kohnke PL, Best OG, Mulligan SP. Mechanisms of action of fludarabine nucleoside against human Raji lymphoma cells. Nucleosides Nucleotides Nucleic Acids. 2014;33(4-6):375–383. doi: 10.1080/15257770.2013.863334. [DOI] [PubMed] [Google Scholar]
- 117.Tsai SY, Sun NK, Lu HP, Cheng ML, Chao CC. Involvement of reactive oxygen species in multidrug resistance of a vincristine-selected lymphoblastoma. Cancer Sci. 2007;98(8):1206–1214. doi: 10.1111/j.1349-7006.2007.00513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Savarino A, et al. “Shock and kill” effects of class I-selective histone deacetylase inhibitors in combination with the glutathione synthesis inhibitor buthionine sulfoximine in cell line models for HIV-1 quiescence. Retrovirology. 2009;6: doi: 10.1186/1742-4690-6-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hütter G, et al. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N Engl J Med. 2009;360(7):692–698. doi: 10.1056/NEJMoa0802905. [DOI] [PubMed] [Google Scholar]
- 120.Trono D, et al. HIV persistence and the prospect of long-term drug-free remissions forHIV-infected individuals. Science. 2010;329(5988):174–180. doi: 10.1126/science.1191047. [DOI] [PubMed] [Google Scholar]
- 121.Lederman MM, Funderburg NT, Sekaly RP, Klatt NR, Hunt PW. Residual immune dysregulation syndrome in treated HIV infection. Adv Immunol. 2013;119:51–83. doi: 10.1016/B978-0-12-407707-2.00002-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Savarino A, Shytaj IL. Chloroquine and beyond: exploring anti-rheumatic drugs to reduce immune hyperactivation in HIV/AIDS. Retrovirology. 2015;12: doi: 10.1186/s12977-015-0178-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zitvogel L, Apetoh L, Ghiringhelli F, André F, Tesniere A, Kroemer G. The anticancer immune response: indispensable for therapeutic success? J Clin Invest. 2008;118(6):1991–2001. doi: 10.1172/JCI35180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Marks PA. Discovery and development of SAHA as an anticancer agent. Oncogene. 2007;26(9):1351–1356. doi: 10.1038/sj.onc.1210204. [DOI] [PubMed] [Google Scholar]
- 125.Cerqueira NM, Pereira S, Fernandes PA, Ramos MJ. Overview of ribonucleotide reductase inhibitors: an appealing target in anti-tumour therapy. Curr Med Chem. 2005;12(11):1283–1294. doi: 10.2174/0929867054020981. [DOI] [PubMed] [Google Scholar]
- 126.Niedbala W, Wei XQ, Piedrafita D, Xu D, Liew FY. Effects of nitric oxide on the induction and differentiation of Th1 cells. Eur J Immunol. 1999;29(8):2498–2505. doi: 10.1002/(SICI)1521-4141(199908)29:08<2498::AID-IMMU2498>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 127.Lee SW, Choi H, Eun SY, Fukuyama S, Croft M. Nitric oxide modulates TGF-β-directive signals to suppress Foxp3+ regulatory T cell differentiation and potentiate Th1 development. J Immunol. 2011;186(12):6972–6980. doi: 10.4049/jimmunol.1100485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Friesen C, Kiess Y, Debatin KM. A critical role of glutathione in determining apoptosis sensitivity and resistance in leukemia cells. Cell Death Differ. 2004;11(suppl 1):S73–S85. doi: 10.1038/sj.cdd.4401431. [DOI] [PubMed] [Google Scholar]
- 129.Conway JM, Perelson AS. Post-treatment control of HIV infection. Proc Natl Acad Sci U S A. 2015;112(17):5467–5472. doi: 10.1073/pnas.1419162112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Shytaj IL, Savarino A. Cell-mediated anti-Gag immunity in pharmacologically induced functional cure of simian AIDS: a ‘bottleneck effect’? J Med Primatol. 2015;44(5):227–240. doi: 10.1111/jmp.12176. [DOI] [PubMed] [Google Scholar]
- 131.Deng K, et al. Broad CTL response is required to clear latent HIV-1 due to dominance of escape mutations. Nature. 2015;517(7534):381–385. doi: 10.1038/nature14053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Patel M, Philip V, Fazel F. Human Immunodeficiency Virus Infection and Hodgkin’s Lymphoma in South Africa: an emerging problem. Adv Hematol. 2011;2011: doi: 10.1155/2011/578163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Davidson A, et al. Malignancies in South African children with HIV. J Pediatr Hematol Oncol. 2014;36(2):111–117. doi: 10.1097/MPH.0b013e31829cdd49. [DOI] [PubMed] [Google Scholar]
- 134.Shytaj IL, et al. A highly intensified ART regimen induces long-term viral suppression and restriction of the viral reservoir in a simian AIDS model. PLoS Pathog. 2012;8(6): doi: 10.1371/journal.ppat.1002774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Apetrei C, Pandrea I, Mellors JW. Nonhuman primate models for HIV cure research. PLoS Pathog. 2012;8(8): doi: 10.1371/journal.ppat.1002892. [DOI] [PMC free article] [PubMed] [Google Scholar]