

Abstract
The nervous and immune systems interact in complex ways to maintain homeostasis and respond to stress or injury, and rapid nerve conduction can provide instantaneous input for modulating inflammation. The inflammatory reflex referred to as the cholinergic antiinflammatory pathway regulates innate and adaptive immunity, and modulation of this reflex by vagus nerve stimulation (VNS) is effective in various inflammatory disease models, such as rheumatoid arthritis and inflammatory bowel disease. Effectiveness of VNS in these models necessitates the integration of neural signals and α7 nicotinic acetylcholine receptors (α7nAChRs) on splenic macrophages. Here, we sought to determine whether electrical stimulation of the vagus nerve attenuates kidney ischemia-reperfusion injury (IRI), which promotes the release of proinflammatory molecules. Stimulation of vagal afferents or efferents in mice 24 hours before IRI markedly attenuated acute kidney injury (AKI) and decreased plasma TNF. Furthermore, this protection was abolished in animals in which splenectomy was performed 7 days before VNS and IRI. In mice lacking α7nAChR, prior VNS did not prevent IRI. Conversely, adoptive transfer of VNS-conditioned α7nAChR splenocytes conferred protection to recipient mice subjected to IRI. Together, these results demonstrate that VNS-mediated attenuation of AKI and systemic inflammation depends on α7nAChR-positive splenocytes.
Introduction
Acute kidney injury (AKI) is associated with high mortality and morbidity, has an expanding incidence, and is a strong risk factor for chronic kidney disease (CKD) and end stage renal disease (ESRD) (1–3). Despite our understanding of the causes and the mechanisms of AKI, few specific preventive and therapeutic options exist (4); thus, new approaches are needed. Less is known about early extrarenal factors that are key modulators of AKI (5). Very early in the course of AKI (within 1 minute), various proinflammatory molecules are released into the arterial inflow and venous outflow of the kidney (6). Therefore, proinflammatory mediators entering into the kidney early in the course of AKI could modulate kidney injury.
As a promising preventative option, we showed that pulsed ultrasound (US) treatment protects the kidney from ischemia-reperfusion injury (IRI), a common AKI animal model, in a spleen-dependent manner (7, 8). Our previous work revealed mechanistic insight on the protective effect of US and demonstrated that the spleen is capable of blocking inflammation and tissue injury in kidney IRI. In addition, hematopoietic α7 nicotinic acetylcholine receptors (α7nAChR, encoded by Chrna7) and sympathetic innervation of the spleen are required for US-induced protection (8). These facts are consistent with stimulation of the inflammatory reflex called the cholinergic antiinflammatory pathway (CAP) (9, 10). This pathway modulates innate and adaptive immunity, and modulation of the reflex is effective in various inflammatory disease models (11).
In the inflammatory reflex, mediators of inflammation are sensed by the peripheral and central nervous system, and CAP signaling is initiated in brain stem nuclei of the vagus nerve and can be activated by vagus nerve stimulation (VNS) (12). The spleen is a critical site in an inflammatory reflex for the neural control of distant organ inflammation and provides a potential therapeutic target for immune-mediated diseases (9). As part of the CAP (13–15), acetylcholine-producing (ACh-producing) lymphocytes might have an important role. Although the intervening steps between VNS and splenic activation are not clearly defined (10, 16), norepinephrine (NE) that is released from the splenic sympathetic nerve binds to β2-adrenergic receptors (β2AR) on nearby choline acetyltransferase–expressing (ChAT-expressing) splenic memory T cells (CD4+CD44hiCD62Llo) and B cells (17). This subset of splenic ACh-producing T cells likely activates adjacent splenic macrophages via the α7nAChR (15), which leads to suppression of proinflammatory cytokines.
The FDA approved the use of VNS as a therapy for medically refractory partial-onset epilepsy in 1997 and for treatment-resistant depression (18, 19) in 2005. Besides these therapeutic roles, VNS may be an interesting tool for activating the CAP in the treatment of inflammatory disorders, such as rheumatoid arthritis (RA) and inflammatory bowel disease (IBD). Indeed, at the present time, 64 clinical studies on VNS are registered on ClinicalTrials.gov, a service of the NIH, mostly focused on epilepsy and depression, but also on studies of RA and IBD.
In animal models, VNS has been evaluated as a therapy in various diseases, such as sepsis, hemorrhagic shock, IBD, IRI in brain and heart, and arthritis (11, 20). In rats, α7nAChR agonists reduce kidney IRI (21), but the effect of VNS on kidney injury and the underlying mechanisms have not been examined. Considering our previous studies regarding US, the similarities between putative protective mechanisms of VNS and US, and the protective effects of VNS on other disorders, we hypothesized that VNS can protect the kidney from IRI. In the current studies, we explored the importance of the spleen and the splenic CAP in modulating kidney IRI. We now report that the effect of VNS requires the spleen, especially α7nAChR-expressing splenocytes, for protection from IRI.
Results
VNS activates both afferent and efferent axons, elicits a vagovagal reflex, and attenuates LPS-induced TNF-α production.
Stimulation of the intact left vagus nerve with the selected parameters (1 ms, 50 μA, 5 Hz) produced a small but reliable bradycardia (6 mice; –9.2 ± 2.1 bpm, range of resting heart rate [HR] 504.3–525.6 bpm; P < 0.05) without BP reduction (Figure 1, A and B). The response threshold was consistently between 10 and 20 μA. Stimulation of the central end of the cut left vagus nerve with the same parameters (1 ms, 50 μA, 5 Hz) elicited a robust evoked response in the contralateral efferent vagal nerve (Figure 1, C and D).
Figure 1. Optimization of VNS.

BP and HR were recorded while mice underwent left or right VNS at constant frequency (5 Hz), but with varied current (10, 30, and 50 μA). Average change in mean arterial BP (A) and HR (B) during vagal stimulation compared with conditions without vagal stimulation. 50 μA stimulation decreased HR significantly. (C and D) Recording of the right vagus efferent nerve during the left VNS. (C) Representative example of right vagus efferent nerve activity (VNA) during left VNS (5 Hz, 1 ms, 50 μA, 10 minutes). Representative data of 3 independent experiments. rVNA, rectified vagus efferent nerve activity. (D) Stimulus-triggered rectified vagus efferent nerve activity was averaged (3,000 sweeps). Arrow indicates stimulation. The latency of the evoked potential is about 20 ms. (E) Mice underwent VNS or sham stimulation (no VNS) surgery 24 hours prior to LPS (10 μg/ml infused at the rate of 10 μl/h for 3 hours) or saline administration, and blood was collected at the end of the infusion period. VNS treatment 24 hours before LPS administration suppressed the LPS-induced increase in circulating TNF. n = 6 each in A and B and n = 5 in E. Data in A and B were analyzed using 2-way ANOVA, and data in E were analyzed using 1-way ANOVA. Means were compared by post hoc multiple-comparison test (Tukey’s). ***P < 0.001.
Stimulation of the intact left vagal nerve (1 ms, 50 μA, 5 Hz) applied 24 hours before LPS infusion greatly reduced the plasma concentration of TNF (5 mice each; no VNS 173.6 ± 25.6 and VNS 43.3 ± 9.2 pg/ml; P < 0.001; Figure 1E). The above evidence shows that vagal stimulation with the chosen parameters activated both efferent (bradycardic response) and afferent vagal axons (vagovagal response). The fact that afferent VNS activated contralateral vagal efferents implies that parasympathetic preganglionic neurons were activated by all 3 modes of stimulation of the vagus nerve (intact, afferent, or efferent nerve). Finally, we demonstrated that VNS with the selected parameters produced the expected antiinflammatory response (22) to an i.v. injection of LPS.
VNS protects kidney from IRI and suppresses proinflammatory cytokines.
Since VNS can provide protection from sepsis when applied before or after endotoxin administration (23, 24), we first tested whether VNS stimulation performed only 10 minutes before renal ischemia attenuated the signs of injury observed 24 hours later. VNS did not change the rise in plasma creatinine (Figure 2A), nor did it attenuate the acute tubular necrosis (ATN) (Figure 2, B and C) produced by IRI; thus, VNS performed 10 minutes before ischemia did not protect the kidney from injury.
Figure 2. Kidneys are not protected from IRI when VNS is applied 10 minutes before IRI.

Mice underwent left VNS (5 Hz, 1 ms, 50 μA for 10 minutes) or sham stimulation surgery 10 minutes prior to IRI (26 minutes ischemia, 24 hours reperfusion) or sham IRI surgery. VNS applied 10 minutes before IRI did not protect kidneys from IRI, as shown by plasma creatinine (A), tissue morphology (B; representative H&E staining of kidney sections), and ATN (C; scored from H&E samples). n = 4 each. Data were analyzed using 1-way ANOVA. Means were compared by post hoc multiple-comparison test (Tukey’s). ***P < 0.001. Scale bars: 100 μm; 50 μm (inset).
Ischemic kidney injury is significantly attenuated when mice are exposed to a single US treatment, but the benefit requires that US be delivered 1 or 2 days before IRI (7). Given that US treatment benefits kidney IRI by activating CAP (7), we reasoned that VNS stimulation might be effective against IRI if delivered 24 hours prior to the injury. We indeed found marked attenuation of AKI in all mice subjected to VNS 24 hours prior to renal ischemia (Figure 3, A–D). Kim1 (also known as Havcr1) expression in whole kidney after IRI was also reduced by VNS (sham-IRI 4037.7 ± 624.5 and intact vagus nerve [VNS(i)]-IRI 1157.3 ± 537.6-fold change compared with sham-sham), providing another indicator of reduced kidney injury (Figure 3B). VNS was also effective in protecting the kidney when applied 48 hours before IRI (plasma creatinine: 1.24 ± 0.18 and 0.13 ± 0.01 mg/dl for sham-IRI and VNS-IRI, respectively; n = 4; P < 0.001).
Figure 3. VNS 24 hours before IRI protects kidneys from injury.

Mice underwent either VNS(i), VNS(a), VNS(e), or sham stimulation surgery (all on the left vagus nerve) 24 hours before IRI or sham IRI surgery. Prior VNS protected kidneys and reduced the IRI-induced increase in plasma creatinine (A), Kim1 expression (RNA from whole kidney) (B), and ATN (C, representative H&E staining of kidney sections; D, scored from H&E samples). n = 5–11. Data were analyzed using 1-way ANOVA. Means were compared by post hoc multiple-comparison test (Tukey’s) *P < 0.05; **P < 0.01; ***P < 0.001. Scale bars: 100 μm; 50 μm (inset).
Stimulating the VNS(i) had the same beneficial effect as stimulating the peripheral end (VNS[e], efferent) or the central end (VNS[a], afferent) of the cut nerve (Figure 3). Based on these results, we stimulated the intact nerve in all subsequent experiments.
Inflammation is a key component of IRI, and stimulation of the CAP reduces systemic cytokine production (25). Therefore, the protective effect of VNS in IRI may involve modulation of systemic inflammatory pathways. Consistent with this view, from a panel of 32 cytokines evaluated, IRI-induced increases in circulating IL-6, IL-10, IL-15, and TNF concentrations were reduced by VNS applied 24 hours prior to IRI (Table 1). Confirming the cytokine screening data, the IRI-induced increase in circulating TNF (measured with ELISA) was significantly reduced by VNS (Figure 4). In addition, the cytokine profile was evaluated in the kidney. RNA was isolated from the whole kidney, and quantitative PCR (qPCR) was performed. Relative expressions compared with the sham-sham group were calculated, and cluster analysis was performed to generate a heat map (Figure 5; raw data in Supplemental Figure 1; supplemental material available online with this article; doi:10.1172/JCI83658DS1). Among evaluated cytokine mRNA, more than half were upregulated by IRI, including Tnf, Lif, Csf1, Tgfb1, and Il1b, and the IRI-induced increase in Il1b was significantly reduced by VNS applied 24 hours prior to IRI. A significant increase in Vegfa produced by VNS was reversed by subsequent IRI, while an IRI-induced decrease in Cxcl9 was not altered by prior VNS (Supplemental Figure 1).
Table 1. IRI-induced increases in plasma cytokine concentrations are reduced by prior VNS.
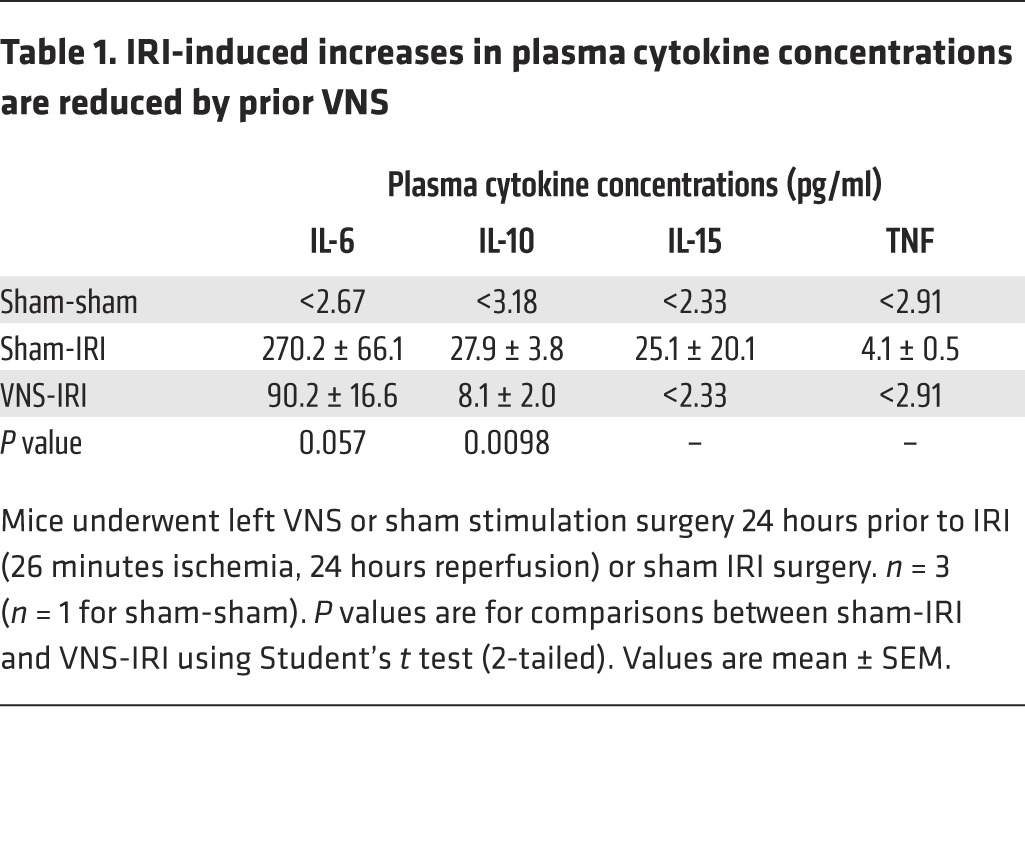
Figure 4. VNS 24 hours before IRI suppresses IRI-induced increases in circulating TNF-α.

Mice underwent VNS or sham stimulation surgery 24 hours prior to IRI or sham IRI surgery. VNS applied 24 hours before IRI suppressed IRI-induced increases in circulating TNF. n = 6. Data were analyzed using 1-way ANOVA. Means were compared by post hoc multiple-comparison test (Tukey’s). *P < 0.05.
Figure 5. More than half of the cytokines evaluated are upregulated in kidney by IRI and suppressed by prior VNS.

Mice underwent VNS or sham stimulation surgery 24 hours prior to IRI or sham IRI surgery. RNA was isolated from whole kidneys, and qPCR was performed. Relative gene expressions compared with sham-sham group were calculated (raw data in Supplemental Figure 1), and clustering was performed to generate a heat map. n = 5–11.
VNS protects against kidney IRI by activating the CAP.
The spleen is a key component of the CAP. Therefore we removed this organ 7 days before VNS and subjected the mice to IRI 24 hours after VNS. Splenectomy had no effect on the rise in plasma creatinine or the increase in ATN elicited by IRI; however, splenectomy prevented the beneficial effect of VNS on IRI (Figure 6).
Figure 6. Protection against IRI by VNS requires the spleen.

Splenectomy (SPLX) or sham surgery was performed 7 days before VNS or sham VNS treatment. Twenty-four hours after VNS treatment, mice were subjected to IRI or sham IRI operation. The protective effect by VNS was eliminated or reduced by prior splenectomy, as demonstrated by plasma creatinine (A) and ATN (B, representative H&E staining of kidney sections; C, scored from H&E samples). n = 6 each. Data were analyzed using 1-way ANOVA. Means were compared by post hoc multiple-comparison test (Tukey’s). *P < 0.05; **P < 0.01. Scale bars: 100 μm; 50 μm (inset).
Next, we performed adoptive transfer experiments. Donor mice were subjected to VNS or sham stimulation, splenocytes were isolated 1 day later, and the recipient mice received splenocytes (105 or 106) from donor mice; 24 hours later, IRI was performed. In a preliminary study, adoptive transfer of splenocytes from VNS-treated mice provided greater protection than those from sham-stimulated mice, especially with 1 million cells (Figure 7A). In a more thorough evaluation, adoptive transfer of 1 million splenocytes from VNS-treated mice significantly protected recipient mice from IRI compared with treatment with PBS or splenocytes from sham-treated mice (Figure 7B). Thus, adoptive transfer of splenocytes from a VNS-treated mouse is sufficient to induce protection from IRI in a recipient mouse. α7nAChR-positive macrophages are another critical component of the CAP (26). Therefore, we tested to determine whether VNS is capable of reducing IRI in Chrna7–/– (α7KO) mice. VNS applied 24 hours before IRI significantly reduced AKI in WT (Chrna7+/+) mice, but this protection was absent in α7nAChR+/– and α7KO mice (Figure 8, A–C). We further investigated the role of α7nAChR-positive cells in spleen using adoptive transfer studies. WT or α7KO donor mice underwent VNS or sham VNS treatment, and 24 hours later, 1 million splenocytes (based on results from Figure 7) from donor mice were injected i.v. into naive WT recipient mice. The mice that received splenocytes from VNS-treated WT mice were protected from IRI, but the protection was abolished when splenocytes from VNS-treated α7KO mice were used (Figure 9). Thus, α7nAChR-positive splenocytes are required for the protection from kidney injury induced by adoptively transferred splenocytes from VNS-treated mice.
Figure 7. Adoptive transfer of splenocytes from VNS-treated mice confers protection from kidney injury after IRI in naive recipient mice.

Donor mice underwent VNS or sham VNS treatment, and 24 hours later, splenocytes isolated from donor mice were injected i.v. into recipient mice. The recipient mice were subjected to IRI 24 hours after splenocyte transfer, and plasma creatinine was evaluated 24 hours after IRI. (A) In pilot studies, the protective effect of donor splenocytes was proportional to the number of cells transferred, and the greatest difference between splenocytes from sham- and VNS-treated donors was observed with 1 million cells. (B) Using transfer of 1 million cells, significant protection was seen in mice that received splenocytes from VNS-treated donor mice compared with administration of PBS (i.v.) or splenocytes from sham-treated donor mice. n = 3 each in A and n = 6 each in B. Data in B were analyzed using 1-way ANOVA. Means were compared by post hoc multiple-comparison test (Tukey’s). ***P < 0.001.
Figure 8. Protection against IRI by VNS is absent in α7 KO mice.

WT (progeny control), Chrna7+/– (α7HE), and α7KO mice underwent VNS 24 hours prior to IRI. The protective effect by VNS was lost in Chrna7+/– and α7KO mice, as demonstrated by plasma creatinine (A) and ATN (B, representative H&E staining of kidney sections; C, scored from H&E samples). n = 5. Data were analyzed with 1-way ANOVA. Means were compared by post hoc multiple-comparison test (Tukey’s). *P < 0.05; **P < 0.01; ***P < 0.001. Scale bars: 100 μm; 50 μm (inset).
Figure 9. Protective effect of adoptively transferred splenocytes from VNS-treated mice requires α7nAChRs.

α7KO mice and WT (progeny controls) were used as donor mice. Donor mice underwent VNS or sham VNS treatment 1 day before splenocyte transfer. Twenty-four hours later, 1 × 106 splenocytes from donor mice were injected i.v. into the recipient mice (WT). The recipient mice were subjected to IRI 24 hours after the transfer, and plasma creatinine was evaluated 24 hours later. The recipient mice that received splenocytes from VNS-treated WT mice were protected against IRI, but this protection was abolished when the mice received splenocytes from VNS-treated α7KO mice. n = 5 each (n = 4 for PBS-treated group). Data were analyzed using 1-way ANOVA. Means were compared by post hoc multiple-comparison test (Tukey’s). ***P < 0.001.
M1 and M2 macrophages play an important role in early initiation and late resolution of AKI, respectively (27). In the CAP, macrophages are believed to play an important role in the protective effect of CAP activation (26). Therefore, we evaluated by flow cytometry the number and phenotype of macrophages infiltrating the kidney after IRI (gating strategy is shown in Supplemental Figure 2). The number of macrophages and granulocytes in kidney increased with time after IRI in WT (Figure 10, A and C) and α7KO mice (Figure 10, B and D), but there were no differences between the groups that received sham treatment or VNS prior to IRI, with the exception that prior treatment with VNS blunted the increase in granulocytes 24 hours after IRI in WT mice (Figure 10, A and C). We further investigated the phenotype of flow cytometry–sorted macrophages using qPCR for M1 (Nos2, Tnf, Il12a, and Il1b) and M2 markers (Mrc1, Msr1, Chil3, and Arg1). Relative gene expression compared with the control group (untreated mice) was calculated, and based on the data, a heat map was created (Figure 10, E and F; raw data in Supplemental Figure 3). The combination of IRI and VNS increased expression of most of the genes, but IRI alone suppressed Arg1 expression and prior VNS rescued its expression in WT mice (Supplemental Figure 3). In α7KO mice, Arg1 expression was not rescued by prior VNS.
Figure 10. Prior VNS does not change the number of macrophages infiltrating the kidney, but changes their phenotype.

α7KO mice and WT (progeny controls) mice underwent IRI 24 hours after VNS or sham VNS treatment. Mice were euthanized after 0, 4, or 24 hours of reperfusion, and the number of macrophages/monocytes and granulocytes in the kidney was evaluated by flow cytometry (gating strategy in Supplemental Figure 2). (A and B) The number of macrophages/monocytes infiltrating the kidney increased with time after IRI in WT (A) and α7KO mice (B), but prior VNS did not change the number. (C and D) The number of granulocytes infiltrating the kidney increased with time after IRI in WT (C) and α7KO mice (D), and this increase was suppressed 24 hours after IRI in VNS-treated WT mice (C), but not in α7KO mice (D). (E and F) qPCR was performed using FACS-sorted macrophages/monocytes from the kidney of WT (E) and α7KO mice (F) (raw data in Supplemental Figure 3). Relative gene expressions compared with control group were calculated, and clustering was performed. Data were analyzed using 2-way ANOVA. Means were compared by post hoc multiple-comparison test (Tukey’s). *P < 0.05. n = 3 in A–D. n = 3 for control (untreated) and n = 6 for sham-IRI and VNS-IRI (E and F).
Afferent VNS may protect kidneys through a different mechanism than efferent VNS.
Vagus nerve is known to affect many organs through a variety of mechanisms. We investigated whether renal sympathetic nerve is involved in VNS-mediated protection from IRI. To eliminate the effect of renal sympathetic nerve, renal denervation (RND) was conducted using locally applied phenol. RND caused a significant reduction of NE in the kidney (10 kidneys/group; sham 681.7 ± 12.6 and RND 68.8 ± 17.8 ng/gram of kidney; P < 0.001) and completely protected the kidney from IRI based on plasma creatinine (5 mice/group; sham-IRI 1.89 ± 0.05 and RND-IRI 0.46 ± 0.20 mg/dl; P < 0.001), as shown previously by others (28). This observation precluded additional studies to examine the effects of RND on the protective effects of VNS. Alternatively, renal sympathetic activity was recorded directly during VNS. As shown in Supplemental Figure 4, when the intact vagus nerve was stimulated, an evoked potential was recorded in the renal sympathetic nerve. In contrast, no evoked potential was seen in renal sympathetic nerve during efferent vagal stimulation alone. These results indicate that afferent but not efferent VNS activates renal sympathetic nerve in mice. This connection has also been observed in rats and operates via presympathetic neurons located in the rostral ventrolateral medulla (29).
We showed that either vagus afferent, vagus efferent, or intact VNS protects kidney almost equally (Figure 3) and left afferent VNS elicits a robust evoked response in right efferent vagal nerve (Figure 1D). In addition, the importance of spleen in the CAP and in mediating the protective effect of efferent VNS was revealed by splenectomy and splenocyte adoptive transfer studies (Figures 6 and 7). However, there is no direct evidence to show that efferent vagal stimulation is necessary for the protective effect. Therefore, we performed experiments using vagus afferent stimulation with contralateral vagal nerve blocked using local anesthesia and found, somewhat surprisingly, that left vagus stimulation in this paradigm still protected kidney from IRI (Supplemental Figure 5). This result indicates that the protection elicited by vagus afferent nerve stimulation does not require activation of the contralateral efferent vagus nerve (Figure 11). In addition, efferent nerve stimulation is sufficient but not necessary to exert a protective effect.
Figure 11. CAP in protection from kidney IRI.

VNS applied 24 to 48 hours before an ischemic episode protects the kidney from IRI. Stimulation of vagal afferents or efferents protects the kidneys through α7nAChR-positive (α7-positive)splenocytes. Vagal afferent stimulation also protects kidney, but through a different and unidentified pathway.
Discussion
The essential new findings are as follows (Figure 11). VNS applied 24 to 48 hours before an ischemic episode protects the kidney from ischemic injury. This protection is equivalent to the previously reported beneficial effect of US. Renal protection by VNS and US operates via activation of the CAP. Stimulation of vagal afferents or efferents is equally effective, possibly because both modes of stimulation activate vagal preganglionic neurons. In addition, vagal afferent stimulation appears to protect the kidneys via a mechanism that does not require intact contralateral vagal efferents.
US and VNS protect the kidneys from ischemic injury by activating the splenic cholinergic antiinflammatory reflex.
This study revealed numerous similarities between the effects of US and VNS on AKI (7, 8). Judging by the creatinine level, renal histology, and other criteria 24 hours after renal ischemia, the protective effect of US and VNS is comparable in mice. In clinical trials to assess the benefit of VNS in RA, stimulation parameters such as current, pulse width, and frequency and duration of stimulation can be adjusted based on the effects and symptoms (30). We have not tried to optimize these parameters in the present study and may not have produced the maximum renal protection possible via VNS.
In both cases (US and VNS), the renal protection required the spleen and α7nAChRs. CD4+ T cells are required for renal protection by US, and this was explained by the dependence on an intact spleen for the restoration of US-induced protection by CD4+ T cells in Rag1–/– mice that lack B and T cells (7). VNS was reported to induce or increase the production of ACh, the endogenous α7nAChR agonist, by a splenic CD4+ T cell subset (15). Thus, the roles of spleen, α7nAChRs, inflammatory cytokines, and immune cells are shared by US and VNS (7, 10, 31). Considering these similarities, we conclude that the splenic CAP, which includes ChAT-positive T cells that synthesize ACh (15, 32), α7nAChR-positive macrophages (26), and subsequent downstream effects, is activated both by US and by VNS.
Nicotinic stimulation of functional α7nAChR on macrophages can dampen LPS-induced TNF release from macrophages, α7nAChRs are necessary for the antiinflammatory response to VNS (ref. 26 and data in our current study), and α7nAChRs are necessary for the protective effect in our splenocyte transfer studies; however, the requirement of α7nAChR+ macrophages per se for the antiinflammatory effects of CAP stimulation has not been demonstrated. Nevertheless, macrophages are key players in injury and repair. M1 macrophages produce proinflammatory mediators, while M2 macrophages have antiinflammatory functions and are involved in wound healing and fibrosis (33–35). Accumulating evidence suggests that M2 macrophages play a reparative role during the recovery phase of kidney disease, especially in the kidney IRI model (27, 36). The mechanisms underlying the phenotypic switch of kidney macrophages from proinflammatory to antiinflammatory are not well defined, although the process is believed to require at least a few days after injury, nor is the role of macrophages in VNS-mediated protection from IRI. Our data showing increased expression of M1 markers are typical of an early inflammatory response within 24 hours of IRI. Although VNS suppressed only the IRI-induced increase in Il1b expression, blunting this proinflammatory cytokine is consistent with the protective effect of VNS. However, we found that IRI suppressed Arg1 (a, M2 marker) expression of macrophages/monocytes in the kidney just 1 day after IRI and prior VNS abolished the suppression in WT mice, but not in α7KO mice. These data suggest that VNS enhances or maintains a macrophage switch to M2 phenotype and that this might be one mechanism of the protection by VNS. Other M2 markers were not altered by IRI within the acute phase of injury, but polarization would be expected to play a more prominent role in the later recovery phase from injury (37), which is beyond the end point of the current study.
The way in which US activates the splenic CAP is unknown. The pathways responsible for the activation of the splenic CAP by vagal stimulation are discussed below.
Protection induced by VNS is long lasting, but not immediate.
Protection from injury was observed when VNS was applied 24 hours, but not 10 minutes, before IRI. Considering the speed of nerve conduction, the lack of immediate protective effect suggests a more complex interaction among neuronal input, neurochemical signaling, inflammation, and tissue injury. It is possible that dynamic changes in specific populations of immune cells occur during the 24-hour period between VNS and IRI. The spleen, which is necessary for both US- (7) and VNS-induced protection, is a critical site in an inflammatory reflex for the neural control of distant organ inflammation and provides a potential therapeutic target for immune-mediated diseases (9). Thus, the spleen plays an important role as the “coordinating center” to integrate these 2 apparently disparate signaling pathways. We also showed that the protective effect of VNS can last at least 48 hours. Future studies will be needed to elucidate the mechanisms involved in modulating immune cells by VNS, how the protective effects endure, and the mechanisms mediating immune cell effects on kidney protection.
Neural pathways responsible for the protective effect of VNS on AKI.
Given the critical importance of the spleen to the CAP, the original hypothesis espoused that vagal efferents innervate the spleen via a synaptic relay in the celiac superior mesenteric plexus (24). More specifically, vagal preganglionic neurons were thought to contact and activate noradrenergic postganglionic neurons coursing in the splenic nerve. However, no anatomical or electrophysiological evidence of such synapses has been found (38), and we have confirmed that VNS does not elicit a detectable evoked response in the splenic nerve of anesthetized rats (C. Abe, unpublished observations). Interestingly, the spleen seems to be an exception among visceral structures because it does not have a vagal sensory innervation (39). Because beta blockers and spleen denervation reduce the antiinflammatory effect elicited by vagal stimulation, an alternative hypothesis posits that the CAP is elicited via the sympathetic system and that VNS may activate the splenic nerve via a vagosympathetic reflex (10). Clearly, under anesthesia, vagal afferent stimulation evokes a response in sympathetic nerves and this response does have an excitatory component (29). In addition, the sympathetic system facilitates the rapid inflammatory response (increased TNF-α) elicited by an acute injection of LPS in anesthetized rats (38). However, the vagosympathetic reflex hypothesis does not explain satisfactorily why the antiinflammatory effect is elicited by stimulating the peripheral end of the cut vagus nerve (24), an observation replicated in the present renal ischemia model; this hypothesis also does not explain why the protective effect of VNS is delayed (VNS done 10 minutes before ischemia offers no protection) and long lasting (at least 2 days). The present results provide 2 new elements to this ongoing debate: VNS is effective against IRI regardless of whether vagal afferent or efferent fibers are being stimulated, and stimulating vagal afferents on the left side activates vagal efferents on the opposite side. These observations suggest that activation of vagal efferents, directly or reflexly, could be a common trigger of the CAP and underlie at least some of the beneficial effects of VNS in the present ischemic kidney injury model.
The fact that CAP activation can be produced by activating the central end of the vagus nerve in anesthetized animals further suggests that a lower brain stem reflex could be at play in eliciting this response because long-loop reflexes involving more rostral structures are unlikely to operate under anesthesia. Vagovagal reflexes are well described in gastrointestinal physiology (40). Vagal afferents activated by inflammation are believed to trigger a vagovagal reflex that elicits the CAP (41, 42). The present results are compatible with the possibility that VNS attenuates ischemic kidney injury by eliciting such a reflex. However, the connection between vagal efferents and the spleen is not clarified. Vagal efferents innervate the thymus and other myeloid organs (43, 44). These organs/tissues could conceivably be the missing link in the chain of events initiated by VNS and culminating in the spleen.
However, the present study also suggests that the protective effect elicited by electrical stimulation of the central end of a cut vagus nerve may not be caused by a vagovagal reflex. Mice cannot survive bilateral vagotomy; therefore, this surgical procedure could not be implemented to test whether stimulation of the central end of the cut left vagus nerve exerts its protective effect via a contralateral vagovagal reflex. As a substitute, we blocked the right vagal nerve with a local anesthetic while the central end of the left vagus nerve was being stimulated. The loss of the response evoked in the right vagal nerve by stimulating the left central end was taken as evidence of the efficacy of the local anesthetic (data not shown). Surprisingly, left afferent vagal stimulation performed while the right vagal nerve was blocked with local anesthetic still protected the kidneys from ischemic injury. Therefore, the renal protection elicited by activating vagal afferents selectively can occur independently of the activation of contralateral vagal efferents. This observation does not rule out a later contribution of the vagus nerve, since local anesthesia is reversible, but it suggests that other mechanisms may be at play, such as a vagosympathetic reflex or the activation of the hypothalamic-pituitary-adrenal axis (45).
Feasibility of VNS as a means of reducing renal injury.
Vagus afferent stimulation at high frequency (20–30 Hz) is used clinically for the treatment of drug-resistant epilepsy and depression, and low-frequency (5 Hz) is used for efferent stimulation to activate the CAP to produce antiinflammatory effects (31). As of August 2014, over 100,000 VNS devices were implanted in more than 75,000 patients worldwide (46). VNS side effects are usually related to stimulation and often improve with time. They are usually mild to moderate and seldom require that the device be explanted (47). Recently, 2 types of noninvasive transcutaneous VNS (t-VNS) devices, safe and tolerable alternative treatments, were developed, and they are in clinical trials for epilepsy, depression, and migraine (48–51). These noninvasive devices might be effective in disorders classically treated with VNS and could potentially be applied to inflammation-related disorders. We believe that VNS could have therapeutic potential for the prevention of AKI dependent on α7nAChR-positive splenocytes. In addition, future VNS studies will likely uncover mechanisms that will inform the use of therapeutic US, which is even less invasive than VNS, in protecting kidneys and other organs from acute injury.
Methods
Mice.
Male mice (8–12 weeks of age, 20–25 g) were used for all experiments. WT C57BL/6 mice were purchased from the National Cancer Institute, Chrna7–/– (referred to as α7KO) mice (B6.129S7-Chrna7tm1Bay/J) were obtained from Jackson Laboratories, and WT (Chrna7+/+) progeny were used as controls in experiments depicted in Figures 8 and 10.
VNS and miscellaneous recordings.
All mice used to test the effect of VNS on IRI were anesthetized with an i.p. injection of ketamine (120 mg/kg) and xylazine (12 mg/kg). We stimulated the left vagus nerve because this nerve is usually selected for stimulation in animal and human experiments (15, 18, 52). The left cervical vagus nerve was isolated via a midline cervical incision and placed on a bipolar silver wire electrode for stimulation (AS633; Cooner Wire). In a subgroup of mice, the nerve was left intact. In other mice, the nerve was cut and the central end was stimulated to activate vagal afferents selectively. In other mice, the peripheral end of the cut nerve was stimulated in order to activate vagal efferents. In another group of mice, we applied bupivacaine (10–30 μl of 2.5 mg/ml) directly to the targeted nerve in order to block nerve conduction. In all cases, electrical stimulation (square wave; 50 μA intensity; frequency, 5 Hz; duration, 1 ms) was applied for 10 minutes using a Grass model S88 stimulator and stimulus isolation unit (Astro-Med Inc.). In sham-operated animals, the vagus nerve was exposed but not stimulated.
The stimulation parameters were chosen after observing their effects on BP and HR in 2 series of preliminary experiments. In C57BL/6J male mice anesthetized with ketamine/xylazine, BP was recorded conventionally by threading a polyethylene catheter (PE-10; BD) into the left common carotid artery, and HR was derived from the pressure pulse or recorded using ECG electrodes (lead II) inserted s.c. VNS at 5 Hz (50 μA, 1 ms pulses) reliably produced a small reduction in HR without effect on BP (see Results). In a second series of mice, both vagus nerves were isolated, the central end of the left nerve was stimulated as described above (1 ms, 50 μA), and the central end of the right vagus nerve was placed on a bipolar electrode in order to record spontaneous and stimulation-evoked efferent activity. The goal of this experiment was to test for the presence of a vagovagal evoked response, which would assess whether nerve activation of the right central end of the vagus occurs during contralateral left vagal afferent stimulation. For this purpose, 2 mice were anesthetized with a mixture of urethane (500 mg/ml) and α-chloralose (50 mg/ml, 2 ml/kg i.p.) and another 2 mice were anesthetized with ketamine/xylazine. These mice were intubated and mechanically ventilated with 100% O2 through a tracheal tube (MiniVent Model 845 ventilator; Hugo Sachs Elektronik–Harvard Apparatus GmbH). Vagal efferent multiunit activity was recorded with a differential amplifier (BMA-400 AC/DC Bioamplifier, CWE Inc.; band-pass 100-1,000 Hz) (53). Signals were acquired using an analog-to-digital converter (Spike 2; Cambridge Electronics) at a rate of 1,000 Hz. They were rectified and further processed using Spike version 7.03 software (Cambridge Electronics).
Recording of renal sympathetic nerve activity.
Recordings of renal sympathetic nerve activity were performed under urethane anesthesia (1.6 g/kg, i.p). Depth of anesthesia was assessed by absence of the corneal and hind-paw withdrawal reflex. Additional anesthetic was administered as necessary (10% of the original dose). Body temperature was maintained at 37.2 ± 0.5°C with a servo-controlled temperature pad (TC-1000; CWE Inc.). The left cervical vagus nerve was isolated and prepared for stimulation as described above. To record renal sympathetic nerve activity, the postganglionic sympathetic nerve was isolated through a left-flank incision. Two stainless-steel electrodes (AS633; Cooner Wire) were placed around the postganglionic sympathetic nerve. The nerve and electrodes were covered and fixed with silicone gel (Kwik-Sil; World Precision Instruments). Physiological signals were filtered and amplified (AP, 10–1000 Hz, ×1,000; vagus, 30–3,000 Hz, ×10,000) (BMA-400; CWE Inc.) and acquired in Spike 2 software (v7.06; CED). The stimulation-induced (rectangle wave; amplitude, 50 μA; frequency, 5 Hz; duration, 1 ms) evoked potential of renal sympathetic nerve was recorded. At the end of the experiment, we confirmed the disappearance of renal sympathetic nerve activity in response to an i.m. bolus injection of a ganglionic blocker, hexamethonium bromide (30 mg/kg), and recorded the noise level, which was treated as the zero level of renal sympathetic nerve activity.
IRI and splenectomy.
Twenty-four hours after VNS or sham treatments, mice were anesthetized with an i.p. injection of ketamine (120 mg/kg) and xylazine (12 mg/kg) and underwent renal IRI, as described (7). Bilateral renal IRI was performed through flank incisions by clamping the renal pedicles for 26 minutes. The clamps were then removed and the wound sutured after restoration of blood flow was visually observed. Sham-operated mice underwent the same procedure except that the renal pedicles were not clamped. For splenectomy, mice were anesthetized with an i.p. injection of ketamine (120 mg/kg), xylazine (12 mg/kg), and atropine (0.324 mg/kg). The splenic vasculature was then ligated and the spleen removed through a small flank incision. Sham-operated mice underwent the same procedure, with the exception of splenic artery ligation and removal of the spleen. Sham-operated and splenectomized mice were allowed to recover for 7 days prior to IRI studies. Mice received buprenorphine (0.15 mg/kg) as a postoperative analgesic for both IRI and splenectomy.
LPS studies.
A series of preliminary experiments was conducted to ascertain that our VNS parameters triggered the antiinflammatory reflex as expected by determining whether VNS reduced LPS-induced TNF release in vivo (26). Four days before the experiment, C57BL/6J male mice (n = 12) were anesthetized with a mixture of ketamine (120 mg/kg) and xylazine (12 mg/kg) given i.p., and we inserted an implantable and programmable microinfusion pump system (iPRECIO Micro Infusion Pump System, SMP-300, a gift from Primetech Corp.). Just before the implantation of the microinfusion pump, we programmed the infusion start time, infusion end time, and infusion rate using the provided software (iPRECIO IMS-300 management software, Primetech Corp.). The catheter part of the microinfusion pump was inserted into the left external jugular vein via a midcervical incision. The main body of the microinfusion pump was implanted s.c. in the lumbar region. Ampicillin (125 mg/kg) and ketoprofen (4 mg/kg) were injected i.m. after closing the incision. During the recovery period, normal saline was continuously infused at the rate of 1 μl/h for 4 days.
One day before LPS infusion, mice were anesthetized (ketamine [120 mg/kg] and xylazine [12 mg/kg]) and the left cervical vagus nerve (uncut) was electrically stimulated (50 μA, 5 Hz, 1 ms) for 10 minutes as described above. At the infusion start time, normal saline in the microinfusion pump was changed to the LPS solution (10 μg/ml) under isoflurane anesthesia. LPS was infused at the rate of 10 μl/h for 3 hours. At the end of infusion, mice were anesthetized (ketamine [120 mg/kg] and xylazine [12 mg/kg]) and blood was collected from the periorbital sinus. Plasma TNF-α was measured with a commercially available ELISA kit (Affymetrix).
Adoptive transfer studies.
Spleens were harvested from VNS or sham-treated animals 24 hours after treatment. Single-cell suspensions were generated by passing whole spleen through 40-μm filters into PBS. The cell pellet was collected by centrifugation (500 g for 5 minutes) and then treated with red blood cell lysis buffer (BioLegend) for 3 minutes according to the manufacturer’s protocol, with some modifications, as described here. After cell lysis, the samples were centrifuged, the resulting cell pellet was diluted, and 1 × 105 or 1 × 106 cells were injected via tail vein 24 hours prior to IRI.
Plasma creatinine and stereological analysis of tissue morphology.
Plasma was prepared by centrifuging heparinized blood at 4,800 g for 5 minutes. Plasma creatinine (mg/dl) was determined by using an enzymatic method, with minor modifications from the manufacturer’s protocol (using twice the recommended volume of sample and standard and a 2-fold serial dilution of the calibrator [standard] provided in the kit; Diazyme Laboratories). We validated the enzymatic kit by comparing with analysis of creatinine by liquid chromatography–mass spectrometry (LC-MS) performed at the George F. O’Brien Center (University of Alabama, Birmingham, Alabama, USA). As depicted in Supplemental Figure 6, a very strong correlation between HPLC and the enzymatic method was confirmed, as shown previously by others (54).
Kidneys were dissected and the capsule removed. A center transverse section was cut and placed in 4% PLP (4% paraformaldehyde, 1.4% DL-lysine, 0.2% sodium periodate in 0.1 M sodium phosphate buffer, pH 7.4) for 24 hours and then stored in 70% EtOH until paraffin embedding (UVA Research Histology Core). Paraffin sections (5 μm) were cut and stained with H&E. The sections were viewed by light microscopy (Zeiss AxioImager Z1/Apotome microscope, Carl Zeiss Microscopy). Photographs were taken with an AxioCam MRc camera (Zeiss), and brightness/contrast and white balance adjustments were made using StereoInvestigator software (MBF Bioscience).
The extent of ATN was assessed in an unbiased, systematic manner using design-based stereology to achieve statistically accurate random sampling of kidney sections, yielding the percentage of total area of the section occupied by injured tubules. The investigator was blinded to the experimental identity of the sections. Sections were imaged by using a Zeiss Axio Imager Z1/Apotome Microscope fitted with motorized focus drives and motorized XYZ microscope stage and integrated to a work station running Stereo Investigator software (MBF Bioscience). The area fraction fractionator probe was used for stereological analysis of the fractional area of the section occupied by tubular necrosis or interstitial fibrosis. The following parameters were defined: counting frame, 400 × 400 μm; sample grid, 800 × 800 μm; grid spacing, 85 μm. These values were determined empirically such that adequate numbers of sample sites were visited and adequate numbers of markers (indicating injured tubules) were acquired, in keeping with accepted counting rules for stereology. Injured tubules were identified based on the presence of cast formation, tubule dilation, and/or tubular epithelial denucleation. A total of 258 ± 6.4 (mean ± SEM) grid sites were evaluated per section.
Real-time PCR and cytokine analysis.
Renal mRNA was isolated by following the ethanol-precipitation method, and RNA concentration was determined based on spectrophometric determination of a 260/280 ratio. For RNA isolation from 5,000 flow-sorted macrophages/monocytes, RNeasy Mini Kit (QIAGEN) was used. cDNA was generated from the resultant tissue RNA using the iScript cDNA Synthesis Kit (Bio-Rad) as described by the manufacturer. Resultant cDNA was then used to determine relative mRNA expression of various genes using the iTAC Universal SYBR Green Supermix (Bio-Rad). Clustering was performed using Cluster 3.0 (55), and a heat map was created with Java TreeView 1.1 (56). Primer sequences are shown in Supplemental Table 1.
A panel of serum cytokines and chemokines was assessed using Mouse Cytokine/Chemokine Magnetic Bead Multiplex Assay (Millipore) as described by the manufacturer. The panel included a total of 32 cytokines: CSF3, CCL11, CSF2, IFN-γ, IL-1a, IL-1b, IL-2, IL-4, IL-3, IL-5, IL-6, IL-7, IL-9, IL-10, IL-12b, IL-12p70, LIF, IL-13, CXCL5, IL-15, IL-17a, CXCL10, CXCL1, CCL2, CCL3, CCL4, CSF1, CXCL2, CXCL9, CCL5, VEGFa, and TNF. Plasma samples were analyzed as recommended by the manufacturer using a Luminex IS 100 system (UVA Flow Cytometry Core Facility). Plasma TNF concentration was measured using the mouse TNF-α ELISA Ready-SET-Go kit (Affymetrix) as described by the manufacturer. ELx 405 (BioTek Instruments) was used as an ELISA plate washer. Synergy HTX (BioTek Instruments) was used as an ELISA plate reader.
Kidney tissue digestion and FACS analysis of leukocytes.
Kidney suspensions were prepared from mice subjected to IRI or sham operation with or without prior VNS. Kidneys were weighed, minced, and incubated with collagenase (Liberase TM, 10 μg/ml; Sigma-Aldrich) and DNAseI in cold RPMI buffer with penicillin/streptomycin, l-glutamine, 10% FCS, and HEPES (pH7.4, 25 mM) for 40 minutes at 37°C. The digested kidney tissue suspension was teased through a 53-μm and 35-μm cell strainer (Endecotts Ltd.) via the rubber end of a 5-ml syringe plunger and then centrifuged at 500 g for 8 minutes at 4°C. Red blood cell lysis buffer (5 ml) was added to the cell pellet; after disrupting the pellet, the cells were incubated for 3 minutes at room temperature, and 10 ml cold PBS was added to stop the reaction. The cells were centrifuged again, the supernatant was discarded, and the cells were resuspended with 0.5% BSA in PBS containing 0.1% sodium azide (Sigma-Aldrich). After blocking nonspecific Fc binding with anti-mouse CD16/32 (2.4G2), fresh kidney suspensions were incubated with the following antibodies: anti-mouse CD45-APC–Alexa Fluor 780 (30-F11, Affymetrix), CD11b-FITC (M1/70, Affymetrix), Ly6C–Brilliant Violet 421 (HK1.4, BioLegend), Ly6G–Brilliant Violet 510 (1A8, BioLegend), MHC class II [IA]–Alexa Fluor 647 (M5/114.15.2, BioLegend), CD11c-PE/Cy7 (N418, BioLegend) and F4/80-PE (BM8, BioLegend). 7-AAD (Thermo Fisher Scientific) was used to exclude dead cells.
Counting Beads (CountBright Absolute Counting Beads, Thermo Fisher Scientific) were used to calculate the cell number (g−1 kidney) as follows: CD45 cell absolute count (g−1 kidney) = (events of CD45 cells counted/total number of beads counted × input bead number)/g kidney. The leukocyte subset cell number (g−1 kidney) was multiplied by the CD45 cell number and by the percentage of the subset. For compensation, compensation beads (UltraComp eBeads, Affymetrix) were used. Flow cytometry data were acquired on a BD FACSCalibur (BD Biosciences) with Cytek 8 Color Flow Cytometry Upgrade (Cytek Development Inc.) and analyzed by FlowJo software 10.0 (Tree Star Inc.). Flow cytometry sorting with BD Influx (BD Biosciences) was performed to isolate macrophages/monocytes from the kidney, which yielded a population with 98.5% purity (post-sort purity). The purity shows the percentage of macrophage/monocyte population among live cells (7AAD negative) after eliminating debris based on FSC.
Renal nerve denervation.
Renal nerve denervation on both sides was performed under anesthesia (ketamine [120 mg/kg] and xylazine [12 mg/kg]). Depth of anesthesia was assessed by absence of the corneal and hind-paw withdrawal reflex. Additional anesthetic was administered as necessary (10% of the original dose, i.p.). Body temperature was maintained at 37.2 ± 0.5°C with a servo-controlled temperature pad (TC-1000; CWE Inc.). To perform renal nerve denervation, the postganglionic sympathetic nerve was identified through a left-flank incision. Renal nerve denervation was conducted by application of 10% phenol in ethanol to the renal sympathetic nerve. After application of phenol, the denervation site was washed by 0.9% NaCl solution. Mice received ketoprofen (4 mg/kg) as a postoperative analgesic and ampicillin (125 mg/kg) immediately after surgery. Denervation was confirmed by measuring kidney content of NE. Kidneys were removed 7 days after denervation and were snap-frozen in liquid nitrogen. Tissue was spiked with internal standard and extracted, and NE was measured by HPLC with electrochemical detection (in the laboratory of John Elsworth, Department of Psychiatry, Yale University School of Medicine, New Haven, Connecticut, USA).
Statistics.
Data were analyzed using 1-way or 2-way ANOVA and Student’s t test (2-tailed) for experiments with only 2 subgroups, with a significant difference defined as P < 0.05. Repeated experiments were analyzed as a randomized complete block design. Means were compared by post hoc multiple-comparison test (Tukey’s), and all values are presented as mean ± SEM and as individual values in dot plots. Sham treatment data are included only for reference in Figures 6, 7, and 9 and Table 1. All the analyses were performed with SPSS version 22 (IBM) or GraphPad Prism version 6 (GraphPad Software).
Study approval.
All animals were handled and procedures were performed in adherence to the NIH Guide for the Care and Use of Laboratory Animals (8th ed. National Academies Press. 2011.). All protocols were approved by the UVA Institutional Animal Care and Use Committee.
Author contributions
TI, CA, SSJS, DLR, PGG, and MDO designed research studies. TI, CA, SM, JJ, LH, and HY conducted experiments and acquired and analyzed data. TI, CA, DLR, PGG, and MDO wrote the manuscript.
Supplementary Material
Acknowledgments
Research reported in this publication was supported by the National Institute of Diabetes and Digestive and Kidney Diseases of the NIH (R01DK085259 and R01DK062324 to M.D. Okusa), the National Heart, Lung, and Blood Institute of the NIH (RO1HL028785 to P.G. Guyenet), and by 2 Japan Society for the Promotion of Science Postdoctoral Fellowships for Overseas Researchers (awarded separately to T. Inoue and C. Abe). The stereology data described here were gathered on an MBF Bioscience and Zeiss microscope system for stereology and tissue morphology funded by NIH grant 1S10RR026799-01 (to M.D. Okusa). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. We thank John D. Elsworth (Department of Psychiatry, Yale University School of Medicine, New Haven, Connecticut, USA) for measuring NE, the George F. O’Brien Center, University of Alabama (P30DK079337; principal investigator, Anupam Agarwal) for measuring plasma creatinine by LC-MS, the UVA Research Histology Core for their assistance in preparation of histology slides, and the UVA Flow Cytometry Facility for sample analysis using the Luminex 100 IS system and FACS sorting using BD Influx. We also thank Primetech Corp. (Tokyo, Japan) for supplying the iPRECIO microinfusion pump system.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2016;126(5):1939–1952. doi:10.1172/JCI83658.
See the related Commentary beginning on page 1640.
Contributor Information
Tsuyoshi Inoue, Email: TI5S@hscmail.mcc.virginia.edu.
Chikara Abe, Email: ca8sc@virginia.edu.
Stefan Moscalu, Email: sm8du@virginia.edu.
Jakub Jankowski, Email: jj3fu@Virginia.EDU.
Liping Huang, Email: lh7c@virginia.edu.
Hong Ye, Email: yehonghong@gmail.com.
Diane L. Rosin, Email: dr5e@virginia.edu.
Mark D. Okusa, Email: mdo7y@virginia.edu.
References
- 1.Hsu RK, McCulloch CE, Dudley RA, Lo LJ, Hsu CY. Temporal changes in incidence of dialysis-requiring AKI. J Am Soc Nephrol. 2013;24(1):37–42. doi: 10.1681/ASN.2012080800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Molitoris BA, et al. Design of clinical trials in AKI: a report from an NIDDK workshop. Trials of patients with sepsis and in selected hospital settings. Clin J Am Soc Nephrol. 2012;7(5):856–860. doi: 10.2215/CJN.12821211. [DOI] [PubMed] [Google Scholar]
- 3.Okusa MD, et al. Design of clinical trials in acute kidney injury: a report from an NIDDK workshop — prevention trials. Clin J Am Soc Nephrol. 2012;7(5):851–855. doi: 10.2215/CJN.12811211. [DOI] [PubMed] [Google Scholar]
- 4.Lameire NH, et al. Acute kidney injury: an increasing global concern. Lancet. 2013;382(9887):170–179. doi: 10.1016/S0140-6736(13)60647-9. [DOI] [PubMed] [Google Scholar]
- 5.Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest. 2011;121(11):4210–4221. doi: 10.1172/JCI45161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ratliff BB, Rabadi MM, Vasko R, Yasuda K, Goligorsky MS. Messengers without borders: mediators of systemic inflammatory response in AKI. J Am Soc Nephrol. 2013;24(4):529–536. doi: 10.1681/ASN.2012060633. [DOI] [PubMed] [Google Scholar]
- 7.Gigliotti JC, et al. Ultrasound prevents renal ischemia-reperfusion injury by stimulating the splenic cholinergic anti-inflammatory pathway. J Am Soc Nephrol. 2013;24(9):1451–1460. doi: 10.1681/ASN.2013010084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gigliotti JC, et al. Ultrasound modulates the splenic neuroimmune axis in attenuating AKI. J Am Soc Nephrol. 2015;26(10):2470–2481. doi: 10.1681/ASN.2014080769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Andersson U, Tracey KJ. Neural reflexes in inflammation and immunity. J Exp Med. 2012;209(6):1057–1068. doi: 10.1084/jem.20120571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martelli D, McKinley MJ, McAllen RM. The cholinergic anti-inflammatory pathway: a critical review. Auton Neurosci. 2014;182:65–69. doi: 10.1016/j.autneu.2013.12.007. [DOI] [PubMed] [Google Scholar]
- 11.Andersson U, Tracey KJ. Reflex principles of immunological homeostasis. Annu Rev Immunol. 2012;30:313–335. doi: 10.1146/annurev-immunol-020711-075015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tracey KJ. Physiology and immunology of the cholinergic antiinflammatory pathway. J Clin Invest. 2007;117(2):289–296. doi: 10.1172/JCI30555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chavan SS, Tracey KJ. Regulating innate immunity with dopamine and electroacupuncture. Nat Med. 2014;20(3):239–241. doi: 10.1038/nm.3501. [DOI] [PubMed] [Google Scholar]
- 14.Olofsson PS, Rosas-Ballina M, Levine YA, Tracey KJ. Rethinking inflammation: neural circuits in the regulation of immunity. Immunol Rev. 2012;248(1):188–204. doi: 10.1111/j.1600-065X.2012.01138.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rosas-Ballina M, et al. Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science. 2011;334(6052):98–101. doi: 10.1126/science.1209985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cailotto C, et al. Neuro-anatomical evidence indicating indirect modulation of macrophages by vagal efferents in the intestine but not in the spleen. PLoS One. 2014;9(1): doi: 10.1371/journal.pone.0087785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reardon C, et al. Lymphocyte-derived ACh regulates local innate but not adaptive immunity. Proc Natl Acad Sci U S A. 2013;110(4):1410–1415. doi: 10.1073/pnas.1221655110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stacey WC, Litt B. Technology insight: neuroengineering and epilepsy-designing devices for seizure control. Nat Clin Pract Neurol. 2008;4(4):190–201. doi: 10.1038/ncpneuro0750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cai PY, et al. Vagus nerve stimulation in ischemic stroke: old wine in a new bottle. Front Neurol. 2014;5: doi: 10.3389/fneur.2014.00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun P, et al. Involvement of MAPK/NF-κB signaling in the activation of the cholinergic anti-inflammatory pathway in experimental colitis by chronic vagus nerve stimulation. PLoS One. 2013;8(8): doi: 10.1371/journal.pone.0069424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yeboah MM, et al. Cholinergic agonists attenuate renal ischemia-reperfusion injury in rats. Kidney Int. 2008;74(1):62–69. doi: 10.1038/ki.2008.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Borovikova LV, et al. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature. 2000;405(6785):458–462. doi: 10.1038/35013070. [DOI] [PubMed] [Google Scholar]
- 23.Huston JM, et al. Splenectomy inactivates the cholinergic antiinflammatory pathway during lethal endotoxemia and polymicrobial sepsis. J Exp Med. 2006;203(7):1623–1628. doi: 10.1084/jem.20052362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rosas-Ballina M, et al. Splenic nerve is required for cholinergic antiinflammatory pathway control of TNF in endotoxemia. Proc Natl Acad Sci U S A. 2008;105(31):11008–11013. doi: 10.1073/pnas.0803237105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sadis C, et al. Nicotine protects kidney from renal ischemia/reperfusion injury through the cholinergic anti-inflammatory pathway. PLoS One. 2007;2(5): doi: 10.1371/journal.pone.0000469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang H, et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature. 2003;421(6921):384–388. doi: 10.1038/nature01339. [DOI] [PubMed] [Google Scholar]
- 27.Lee S, et al. Distinct macrophage phenotypes contribute to kidney injury and repair. J Am Soc Nephrol. 2011;22(2):317–326. doi: 10.1681/ASN.2009060615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fujii T, et al. The role of renal sympathetic nervous system in the pathogenesis of ischemic acute renal failure. Eur J Pharmacol. 2003;481(2–3):241–248. doi: 10.1016/j.ejphar.2003.09.036. [DOI] [PubMed] [Google Scholar]
- 29.Sun MK, Guyenet PG. Arterial baroreceptor and vagal inputs to sympathoexcitatory neurons in rat medulla. Am J Physiol. 1987;252(4 pt 2):R699–R709. doi: 10.1152/ajpregu.1987.252.4.R699. [DOI] [PubMed] [Google Scholar]
- 30.Koopman FA, Schuurman PR, Vervoordeldonk MJ, Tak PP. Vagus nerve stimulation: A new bioelectronics approach to treat rheumatoid arthritis? Best Pract Res Clin Rheumatol. 2014;28(4):625–635. doi: 10.1016/j.berh.2014.10.015. [DOI] [PubMed] [Google Scholar]
- 31.Bonaz B, Picq C, Sinniger V, Mayol JF, Clarencon D. Vagus nerve stimulation: from epilepsy to the cholinergic anti-inflammatory pathway. Neurogastroenterol Motil. 2013;25(3):208–221. doi: 10.1111/nmo.12076. [DOI] [PubMed] [Google Scholar]
- 32.Gautron L, Rutkowski JM, Burton MD, Wei W, Wan Y, Elmquist JK. Neuronal and nonneuronal cholinergic structures in the mouse gastrointestinal tract and spleen. J Comp Neurol. 2013;521(16):3741–3767. doi: 10.1002/cne.23376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11(11):723–737. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122(3):787–795. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cao Q, Harris DC, Wang Y. Macrophages in kidney injury, inflammation, and fibrosis. Physiology (Bethesda) 2015;30(3):183–194. doi: 10.1152/physiol.00046.2014. [DOI] [PubMed] [Google Scholar]
- 36.Huen SC, Cantley LG. Macrophage-mediated injury and repair after ischemic kidney injury. Pediatr Nephrol. 2015;30(2):199–209. doi: 10.1007/s00467-013-2726-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tian S, Chen SY. Macrophage polarization in kidney diseases. Macrophage (Houst) 2015;2(1): doi: 10.14800/macrophage.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martelli D, Yao ST, McKinley MJ, McAllen RM. Reflex control of inflammation by sympathetic nerves, not the vagus. J Physiol. 2014;592(pt 7):1677–1686. doi: 10.1113/jphysiol.2013.268573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Berthoud HR, Neuhuber WL. Functional and chemical anatomy of the afferent vagal system. Auton Neurosci. 2000;85(1–3):1–17. doi: 10.1016/S1566-0702(00)00215-0. [DOI] [PubMed] [Google Scholar]
- 40.Browning KN, Travagli RA. Central nervous system control of gastrointestinal motility and secretion and modulation of gastrointestinal functions. Compr Physiol. 2014;4(4):1339–1368. doi: 10.1002/cphy.c130055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dovas A, et al. Collaterals of recurrent laryngeal nerve fibres innervate the thymus: a fluorescent tracer and HRP investigation of efferent vagal neurons in the rat brainstem. Brain Res. 1998;809(2):141–148. doi: 10.1016/S0006-8993(98)00705-7. [DOI] [PubMed] [Google Scholar]
- 42.Antonica A, Ayroldi E, Magni F, Paolocci N. Lymphocyte traffic changes induced by monolateral vagal denervation in mouse thymus and peripheral lymphoid organs. J Neuroimmunol. 1996;64(2):115–122. doi: 10.1016/0165-5728(95)00157-3. [DOI] [PubMed] [Google Scholar]
- 43.Niijima A. An electrophysiological study on the vagal innervation of the thymus in the rat. Brain Res Bull. 1995;38(4):319–323. doi: 10.1016/0361-9230(95)00103-L. [DOI] [PubMed] [Google Scholar]
- 44.Antonica A, Magni F, Mearini L, Paolocci N. Vagal control of lymphocyte release from rat thymus. J Auton Nerv Syst. 1994;48(3):187–197. doi: 10.1016/0165-1838(94)90047-7. [DOI] [PubMed] [Google Scholar]
- 45.Hosoi T, Okuma Y, Nomura Y. Electrical stimulation of afferent vagus nerve induces IL-1beta expression in the brain and activates HPA axis. Am J Physiol Regul Integr Comp Physiol. 2000;279(1):R141–R147. doi: 10.1152/ajpregu.2000.279.1.R141. [DOI] [PubMed] [Google Scholar]
- 46.Ben-Menachem E, Revesz D, Simon BJ, Silberstein S. Surgically implanted and non-invasive vagus nerve stimulation: a review of efficacy, safety and tolerability. Eur J Neurol. 2015;22(9):1260–1268. doi: 10.1111/ene.12629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ben-Menachem E. Vagus nerve stimulation, side effects, and long-term safety. J Clin Neurophysiol. 2001;18(5):415–418. doi: 10.1097/00004691-200109000-00005. [DOI] [PubMed] [Google Scholar]
- 48.Stefan H, et al. Transcutaneous vagus nerve stimulation (t-VNS) in pharmacoresistant epilepsies: a proof of concept trial. Epilepsia. 2012;53(7):e115–e118. doi: 10.1111/j.1528-1167.2012.03492.x. [DOI] [PubMed] [Google Scholar]
- 49.He W, et al. Transcutaneous auricular vagus nerve stimulation as a complementary therapy for pediatric epilepsy: a pilot trial. Epilepsy Behav. 2013;28(3):343–346. doi: 10.1016/j.yebeh.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 50.Hein E, et al. Auricular transcutaneous electrical nerve stimulation in depressed patients: a randomized controlled pilot study. J Neural Transm. 2013;120(5):821–827. doi: 10.1007/s00702-012-0908-6. [DOI] [PubMed] [Google Scholar]
- 51.Goadsby PJ, Grosberg BM, Mauskop A, Cady R, Simmons KA. Effect of noninvasive vagus nerve stimulation on acute migraine: an open-label pilot study. Cephalalgia. 2014;34(12):986–993. doi: 10.1177/0333102414524494. [DOI] [PubMed] [Google Scholar]
- 52.Levine YA, et al. Neurostimulation of the cholinergic anti-inflammatory pathway ameliorates disease in rat collagen-induced arthritis. PLoS One. 2014;9(8): doi: 10.1371/journal.pone.0104530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Abbott SB, Holloway BB, Viar KE, Guyenet PG. Vesicular glutamate transporter 2 is required for the respiratory and parasympathetic activation produced by optogenetic stimulation of catecholaminergic neurons in the rostral ventrolateral medulla of mice in vivo. Eur J Neurosci. 2014;39(1):98–106. doi: 10.1111/ejn.12421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Keppler A, et al. Plasma creatinine determination in mice and rats: an enzymatic method compares favorably with a high-performance liquid chromatography assay. Kidney Int. 2007;71(1):74–78. doi: 10.1038/sj.ki.5001988. [DOI] [PubMed] [Google Scholar]
- 55.de Hoon MJ, Imoto S, Nolan J, Miyano S. Open source clustering software. Bioinformatics. 2004;20(9):1453–1454. doi: 10.1093/bioinformatics/bth078. [DOI] [PubMed] [Google Scholar]
- 56.Saldanha AJ. Java Treeview — extensible visualization of microarray data. Bioinformatics. 2004;20(17):3246–3248. doi: 10.1093/bioinformatics/bth349. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.