

Abstract
Targeting multiple components of the MAPK pathway can prolong the survival of patients with BRAFV600E melanoma. This approach is not curative, as some BRAF-mutated melanoma cells are intrinsically resistant to MAPK inhibitors (MAPKi). At the systemic level, our knowledge of how signaling pathways underlie drug resistance needs to be further expanded. Here, we have shown that intrinsically resistant BRAF-mutated melanoma cells with a low basal level of mitochondrial biogenesis depend on this process to survive MAPKi. Intrinsically resistant cells exploited an integrated stress response, exhibited an increase in mitochondrial DNA content, and required oxidative phosphorylation to meet their bioenergetic needs. We determined that intrinsically resistant cells rely on the genes encoding TFAM, which controls mitochondrial genome replication and transcription, and TRAP1, which regulates mitochondrial protein folding. Therefore, we targeted mitochondrial biogenesis with a mitochondrium-targeted, small-molecule HSP90 inhibitor (Gamitrinib), which eradicated intrinsically resistant cells and augmented the efficacy of MAPKi by inducing mitochondrial dysfunction and inhibiting tumor bioenergetics. A subset of tumor biopsies from patients with disease progression despite MAPKi treatment showed increased mitochondrial biogenesis and tumor bioenergetics. A subset of acquired drug-resistant melanoma cell lines was sensitive to Gamitrinib. Our study establishes mitochondrial biogenesis, coupled with aberrant tumor bioenergetics, as a potential therapy escape mechanism and paves the way for a rationale-based combinatorial strategy to improve the efficacy of MAPKi.
Introduction
Cutaneous melanoma is a devastating disease, with a 10-year survival rate of less than 10% in patients diagnosed with stage IV melanoma (1). Approximately 50% of melanoma patients’ tumors harbor a BRAFV600 mutation, resulting in constitutively activated MAPK signaling (2). Selective MAPK inhibitors (MAPKi) directly target the MAPK pathway and significantly improve the overall and progression-free survival of patients with BRAF-mutant melanomas (3–8). Despite the clinical efficacy of targeted therapies, the initial tumor regression often precedes a rapid tumor relapse caused by the survival of residual tumor cells and the subsequent acquisition of drug resistance. The core mechanisms of acquired resistance encompass reactivation of the MAPK and PI3K/AKT signaling pathways (9).
Notably, approximately 10% to 15% of patients with BRAF-mutated melanomas do not respond to initial treatment with targeted therapies, and approximately 40% to 50% of patients experience stable or partial responses at best, suggesting that intrinsic resistance is a major hurdle to effectively eradicate all tumor cells. Genetically, accumulating evidence has suggested that FOXD3, ERBB3, BCL2A1, PDK1, PGC1A, MITF, and NF1 underlie intrinsic resistance to targeted therapies (10–14).
Abnormal metabolic reprogramming is a unique mechanism by which cancer cells not only adapt to the microenvironment but also generate energy (15). The Warburg effect illustrates that aerobic glycolysis is the predominant metabolic pathway for cancer cells to produce energy. However, slow-cycling melanoma cells that are characterized by high expression levels of the histone demethylase JARID1B predominantly utilize oxidative phosphorylation (OxPhos) to generate ATP and are intrinsically resistant to multiple signaling inhibitors (16, 17). A subset of human melanoma cell lines with high expression levels of PGC1A are less glycolytic and rely more heavily on mitochondrial OxPhos to generate ATP (18). When BRAF-mutated melanoma cells were treated with vemurafenib, the MITF/PGC1A signaling axis was upregulated, resulting in metabolic reprogramming toward OxPhos and conferring intrinsic resistance to BRAF inhibitors (13). Similarly, the treatment of leukemia cells with tyrosine kinase inhibitors also altered the metabolic state of surviving cells that remained sensitive to oligomycin-A, which targeted mitochondrial respiration (19).
Understanding which signaling pathway(s) are premier therapeutic targets for overcoming drug resistance remains largely elusive. Mitochondrial biogenesis is a biological process involving the formation of new mitochondria due to the regulation of mitochondrial fusion and fission. Numerous nuclear genome– and mitochondrial genome–encoding factors are controlling mitochondrial biogenesis in response to stress stimuli (20). Here, we focus on the mitochondrial biogenesis signature and investigate its role in mediating resistance to MAPKi. We address the molecular basis of this novel resistance mechanism, which is tightly coupled to aberrant tumor bioenergetics. Furthermore, we show that the small-molecule inhibitor Gamitrinib, which targets mitochondrial HSP90–directed (or TRAP1-directed) protein folding, is effective in circumventing mitochondrial biogenesis. By overcoming both intrinsic and acquired resistance, we can progress toward the long-term goal of eliminating all malignant cells as a precondition for achieving cures.
Results
BRAF-mutated melanoma cell lines with lower expression of mitochondrial biogenesis and mitochondrial mass at the basal level were more resistant to MAPKi.
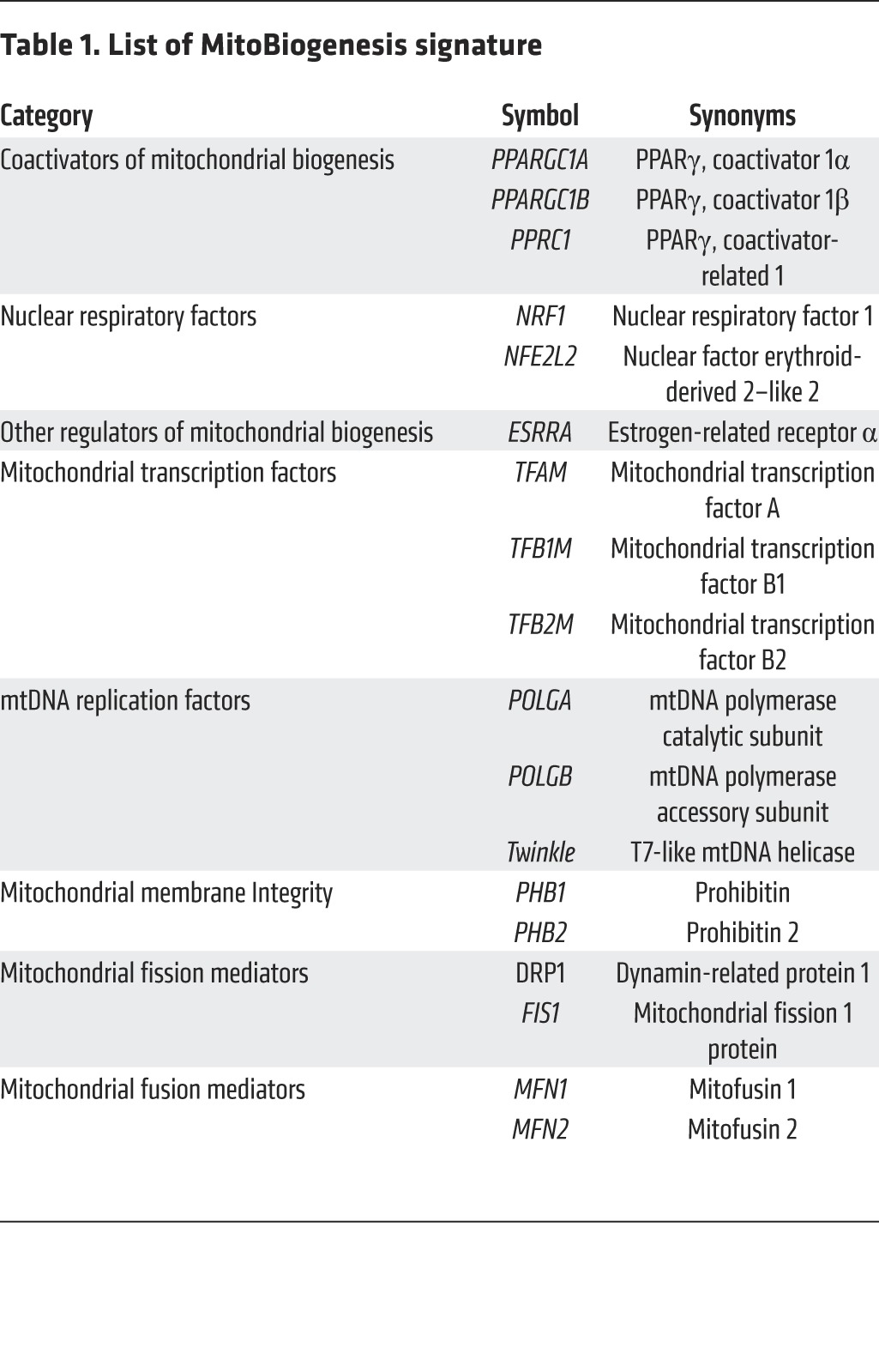
We first consulted the current literature and curated a list of 18 genes that are essential for controlling mitochondrial biogenesis to compose the gene signature “MitoBiogenesis.” Specifically, these genes are: (a) nuclear respiratory factors (NRF1 and NFE2L2 or NRF2) and PGC-1 family coactivators (PPARGC1A or PGC1A, PPARGC1B or PGC1B, PPRC1, and ESRRA), which coordinately control mitochondrial transcription specificity factors (TFAM, TFB1M, and TFB2M) (21); (b) POLGA, POLGB, and Twinkle, which collectively regulate mitochondrial transcription, mitochondrial DNA (mtDNA) replication, and packaging in the mitochondrial genome (22); (c) prohibitin 1 (PHB1) and prohibitin 2 (PHB2), which reside in the inner mitochondrial membrane to safeguard mitochondrial quality control (23); and (d) MFN1, MFN2, DRP1, and FIS1, which regulate mitochondrial fusion and fission (ref. 24 and Table 1).
Table 1. List of MitoBiogenesis signature.
We then sought to determine whether MitoBiogenesis is one of the defining features that distinguish melanoma from other cancer types. Toward this goal, we analyzed genome-wide gene expression data for 947 human cancer cell lines collected for the Cancer Cell Line Encyclopedia (CCLE) project (25). In order to identify gene sets that are only enriched in melanoma, we compared melanoma cell lines with nonmelanoma cancer cell lines and performed gene set enrichment analysis (GSEA). Not surprisingly, the analysis showed that “BRAF Targets,” “MEK Targets,” “Melanogenesis,” and “Lysosome” were among the top 10 ranked gene sets that were enriched in melanoma (Supplemental Figure 1; supplemental material available online with this article; doi:10.1172/JCI82661DS1). “MitoBiogenesis,” however, was not identified among those top-10-ranked gene sets. This led us to test our alternative hypothesis that MitoBiogenesis is enriched only in a subset of melanoma cell lines. We were able to separate 61 CCLE human melanoma cell lines into 2 subgroups: higher and lower expression of MitoBiogenesis (Figure 1A). Next, we obtained 16 additional mitochondrial gene sets from the Molecular Signatures Database (MSigDB) and investigated their correlations with MitoBiogenesis. For each cell line, we performed single-sample GSEA (ssGSEA) in order to calculate the enrichment score for each gene set. “KEGG Glycolysis and Gluconeogenesis” and “KEGG Oxidative Phosphorylation” were also included as controls. The analysis revealed that MitoBiogenesis was clustered with 16 MSigDB mitochondrial gene sets (Figure 1B).
Figure 1. BRAF-mutated melanoma cells with lower mitochondrial biogenesis and mass at the basal level are resistant to MAPKi.

(A) Heatmap of microarray data for MitoBiogenesis in 61 CCLE melanoma cell lines. (B and C) Heatmaps of enrichment scores for 16 mitochondrial gene sets, 2 metabolic gene sets, and 2 MAPK pathway gene sets in 61 CCLE (B) and 10 Wistar Institute (C) melanoma cell lines. (D–G) Drug IC50 of CCLE BRAF-mutated melanoma cell lines RAF265 (D), PLX4720 (E), PD0325901 (F), and AZD6244 (G). A 2-sample t test was used to determine the P values in D–G. (H) Percentage of PSVue 643+ cells in each BRAF-mutated melanoma cell line treated with DMSO or MAPKi for 72 hours. (I and J) Mean fluorescence intensity (MFI) of 2-NBDG (I) and MitoTracker Red (J) in melanoma cell lines. Data were normalized to the MFI derived from the unstained sample of each cell line. (H–J) n = 3; data represent 2 independent experiments. A 2-tailed, unpaired t test was used to determine the P values in H and I. MitoB, MitoTracker B. Horizontal bars in panels D–J denote the mean of each group.
Furthermore, we performed the Illumina genome-wide gene expression experiment to profile 10 of our own BRAF-mutated melanoma cell lines. ssGSEA revealed that 4 of 10 cell lines, including WM1158, WM1799, 1205Lu, and WM9, had lower expression of MitoBiogenesis, in which all mitochondrial gene sets were expressed at a lower level (Figure 1C).
Drug-sensitivity data on 4 MAPKi were available for CCLE BRAF-mutated melanoma cell lines, including 2 BRAF inhibitors, RAF265 and PLX4720, as well as 2 MEK inhibitors, PD0325901 and AZD6244. Therefore, we tested whether there is any correlation between MitoBiogenesis expression and drug sensitivities. Interestingly, our analysis showed that BRAF-mutated melanoma cell lines with lower expression of MitoBiogenesis had a significantly higher drug IC50 for all 4 inhibitors, suggesting that these cell lines were more resistant to MAPKi (Figure 1, D–G).
Next, we treated 20 of our own BRAF-mutated melanoma cell lines with PLX4720 plus PD0325901 for 72 hours. We classified these cell lines into “sensitive” (n = 8) and “resistant” (n = 9) subgroups on the basis of the degree of apoptosis and cell death induced by PLX4720 plus PD0325901 treatment (Figure 1H). This result demonstrated the heterogeneous responses of the melanoma cell lines to MAPKi. For 9 cell lines in the “resistant” group, 6 were profiled by genome-wide gene expression array experiments, and the analysis showed that 5 of 6 cell lines, including WM1158, 1205Lu, WM9, WM115, and A2058, had lower expression levels of MitoBiogenesis (Figure 1, B and C). For 8 cell lines in the “sensitive” group, 7 were profiled by genome-wide gene expression array experiments, and the analysis showed that 6 of 7 cell lines, including WM88, UACC-903, UACC-62, WM983B, A375, and WM266-4, had higher expression levels of MitoBiogenesis (Figure 1, B and C).
Subsequently, we tried to correlate drug sensitivities with the glycolytic capacities and the levels of mitochondrial mass of these cell lines, respectively. The glucose uptake assay did not reveal a statistically significant difference between 2 subgroups, suggesting that the glycolytic capacity was not correlated with drug sensitivity (Figure 1I). Intriguingly, “resistant” cell lines had a significantly lower mitochondrial membrane potential than that of “sensitive” cell lines and hence a lower level of mitochondrial mass (Figure 1J).
Taken together, our data demonstrated that a subgroup of BRAF-mutated melanoma cell lines with lower expression of MitoBiogenesis and mitochondrial mass at the basal level was more resistant to MAPKi.
Patients with higher expression of MitoBiogenesis and tumor metabolism had worse overall survival.
To interrogate the expression of MitoBiogenesis in tumor biopsies derived from patients with malignant melanoma, we performed ssGSEA in 2 independent cohorts of patients, including 470 patients listed in The Cancer Genome Atlas (TCGA) with skin cutaneous melanoma (SKCM) and 104 patients with melanoma (Gene Expression Omnibus [GEO] database accession number GSE46517). The analysis of MitoBiogenesis expression showed that there were 2 subgroups of patients’ tumors in each data set (Figure 2, A and B, and refs. 26, 27). This extended analysis from cell lines to patients’ tumors confirmed our hypothesis that MitoBiogenesis is highly expressed in a subset of, but not in all, melanoma cell lines or patients’ tumors.
Figure 2. TCGA melanoma patients have a worse overall survival outcome if their tumors express high levels of MitoBiogenesis.

(A and B) Heatmaps of enrichment scores for 16 mitochondrial gene sets and 2 metabolic genes in 470 TCGA melanoma patients (A) and 104 melanoma patients (GSE46517) (B). (C–E) Kaplan-Meier survival curves for TCGA melanoma patients who were divided into 2 subgroups of high and low expression of MitoTracker B (C), 2 subgroups of high and low expression of PGC1A (D), and 4 subgroups on the basis of their expression of 62 glycolytic and 135 OxPhos genes (E). P values in C–E were determined by log-rank test.
We next explored the clinical association of MitoBiogenesis expression with patients’ overall survival rates using TCGA SKCM data set. The Kaplan-Meier survival analysis showed that patients with higher expression of MitoBiogenesis in their tumors had a worse overall survival than did patients with lower expression of MitoBiogenesis (Figure 2C).
Because PPARGC1A (PGC1A) is a master regulator of MitoBiogenesis, we also separated TCGA melanoma patients into 2 subgroups on the basis of their PGC1A expression levels. Consistent with the previous finding, patients with higher expression levels of PGC1A had a worse overall survival outcome (Figure 2D and ref. 18).
Similarly, we subdivided TCGA melanoma patients into 4 subgroups on the basis of expression of glycolysis and OxPhos gene sets in their tumors. Intriguingly, we found that a subgroup of patients (n = 25) whose tumor biopsies highly expressed both glycolysis and OxPhos had the worst prognosis (Figure 2E).
MAPKi regulated expression of MitoBiogenesis in BRAF-mutated melanomas.
The finding that lower expression of MitoBiogenesis and mitochondrial mass was associated with drug resistance prompted us to investigate whether and how short-term treatment with MAPKi alters MitoBiogenesis. We conducted an Illumina genome-wide gene expression array experiment to profile 4 BRAF-mutated melanoma cell lines: UACC-62, WM989, and WM164, with higher expression of MitoBiogenesis, and WM9, with lower expression of MitoBiogenesis — all of which were treated with PLX4720. We also obtained publicly available genome-wide gene expression data for 4 more BRAF-mutated melanoma cell lines that had higher expression of MitoBiogenesis: SK-MEL-28, A375, Malme-3M, and COLO829, which were treated with vemurafenib or PD0325901.
The analysis of genome-wide gene expression data showed that MitoBiogenesis expression was inhibited by MAPKi in cell lines with higher expression of MitoBiogenesis, including UACC-62, WM989, WM164, SK-MEL-28, and A375, which were treated with PLX4720 or vemurafenib (Figure 3, A–C and Figure 4, A–C), and Malme-3M and COLO829, which were treated with PD0325901 (Supplemental Figure 2, A and B, and ref. 28). The analysis of time-course genome-wide gene expression data also revealed that PLX4720 increased MitoBiogenesis expression, as exhibited by WM9 cells that had lower expression of MitoBiogenesis (Figure 4C).
Figure 3. MAPKi regulate the expression of MitoBiogenesis in BRAF-mutated melanoma cell lines at the RNA level.

Heatmaps of enrichment scores for 16 mitochondrial gene sets, 2 metabolic gene sets, and 2 MAPK pathway gene sets in UACC-62 (A), WM989 (B), and WM164 cells (C) treated with 10 μM DMSO or PLX4720 for 96 hours.
Figure 4. MAPKi regulate the expression of MitoBiogenesis in BRAF-mutated melanoma cell lines at the RNA level.

Heatmaps of enrichment scores for 16 mitochondrial gene sets, 2 metabolic gene sets, and 2 MAPK pathway gene sets in SK-MEL-28 cells (A) treated with vemurafenib for 48 hours, in A375 cells (B) treated with the control and vemurafenib for 48 hours, and in WM9 cells (C) treated with 10 μM PLX4720 for 96 hours. C, control; V, vemurafenib.
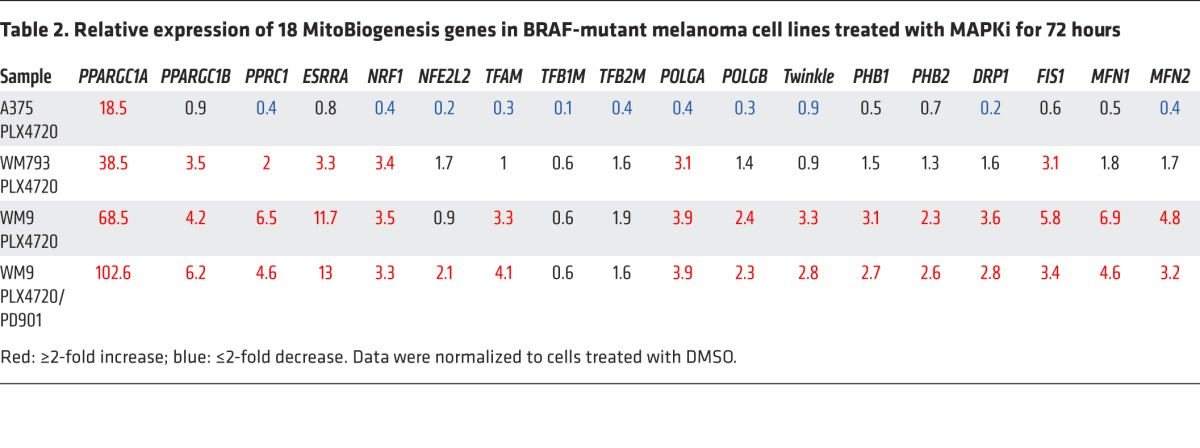
A quantitative real-time PCR (qRT-PCR) experiment confirmed that MAPKi blocked expression of MitoBiogenesis in A375 cells with higher expression of MitoBiogenesis, but upregulated MitoBiogenesis in WM793 and WM9 cells with lower expression of MitoBiogenesis (Table 2).
Table 2. Relative expression of 18 MitoBiogenesis genes in BRAF-mutant melanoma cell lines treated with MAPKi for 72 hours.
In summary, our data suggested that MAPKi suppressed MitoBiogenesis in melanoma cell lines with higher expression of MitoBiogenesis, resulting in a lower drug IC50, and upregulated MitoBiogenesis in melanoma cell lines with lower expression of MitoBiogenesis, leading to a higher drug IC50.
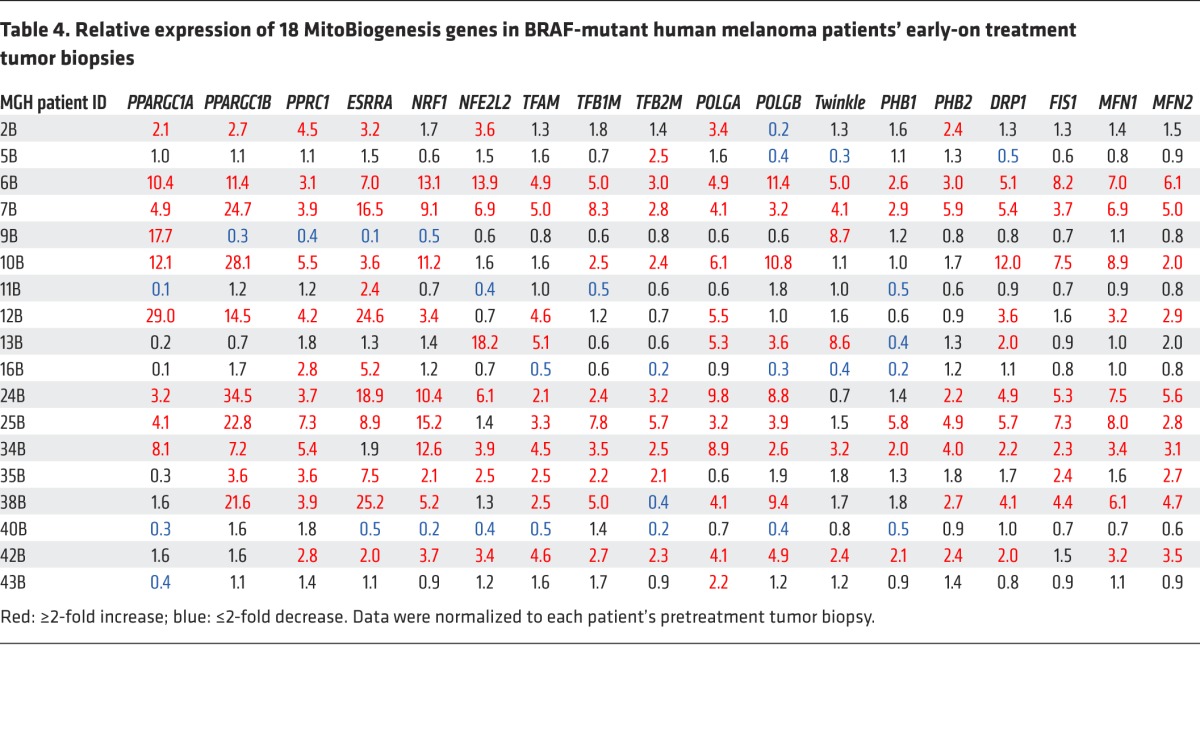
To address the clinical implication of this phenomenon, we determined the expression levels of MitoBiogenesis in 18 BRAF-mutated melanoma patients’ paired pre- and early-on treatment tumor biopsies that were procured between days 14 and 16 following the first dose of MAPKi (Table 3). Eight patients’ early-on treatment tumor biopsies (Massachusetts General Hospital [MGH] patients 6, 7, 10, 12, 24, 25, 34, and 42) showed a more than 55% increase in expression of 18 MitoBiogenesis genes (Table 4).
Table 3. Clinical information for 24 BRAF-mutated melanoma patients treated at Massachusetts General Hospital.
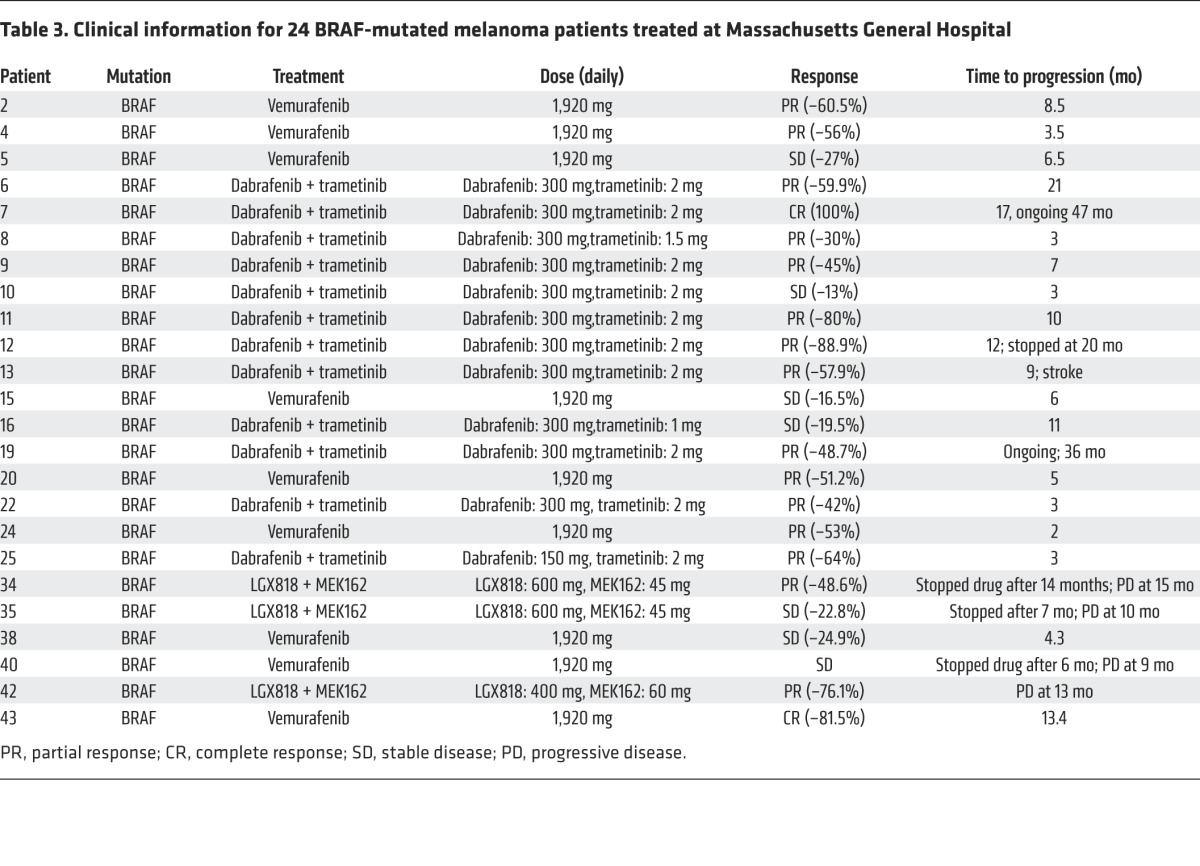
Table 4. Relative expression of 18 MitoBiogenesis genes in BRAF-mutant human melanoma patients’ early-on treatment tumor biopsies.
We also corroborated our genome-wide gene expression array and qRT-PCR data on SK-MEL-28, A375, and WM9 cells with immunoblotting data. We included 4 more mitochondrial proteins, including HSP60, which is implicated in mitochondrial protein import and macromolecular assembly; VDAC1, which is implicated as a gatekeeper for the transport of mitochondrial metabolites; TUFM, which is implicated in mitochondrial protein translation; and SOD2, which regulates the detoxification of ROS to protect against oxidative stress (Supplemental Table 1 and refs. 29–32). For SK-MEL-28 and A375 melanoma cells that were sensitive to MAPKi and had higher expression of MitoBiogenesis at the basal level, MAPKi downregulated the expression of ESRRA, NRF2, TUFM, TFAM, and PGC1α/β (Figure 5, A–C). For WM9 cells that were resistant to MAPKi and had lower expression of MitoBiogenesis, MAPKi activated MitoBiogenesis, as evidenced by upregulation of PGC1α, TFAM, NRF2, PHB1, PHB2, SOD2, and ESRRA (Figure 5D). We further showed that expression of ESRRA, TFAM, PHB1, PHB2, VDAC1, TUFM, HSP60, and SOD2 was upregulated in WM9 cells by PLX4720 or PLX4720 plus PD0325901 in a time-dependent manner (Figure 5, E and F).
Figure 5. MAPKi regulate the expression of MitoBiogenesis in BRAF-mutated melanoma cell lines at the protein level.

(A–F) Immunoblotting of proteins related to MitoBiogenesis and the MAPK pathway in SK-MEL-28 (A), A375 (B and C), and WM9 (D–F) cells treated with DMSO or the indicated MAPKi for 72 hours.
MAPKi led to an increase in mtDNA content and mitochondrial mass in BRAF-mutated melanoma cells.
Next, we focused on 3 key biological aspects of MitoBiogenesis, including mtDNA content, mitochondrial mass, and ROS.
For 3 intrinsically resistant melanoma cell lines (WM9, 1205Lu, and MEL624) treated with MAPKi, we found that the mtDNA content was significantly increased in surviving cells (Figure 6, A and B, and Supplemental Figure 3A). We also demonstrated that mitochondrial mass was significantly enhanced (Figure 6C and Supplemental Figure 3B). MAPKi significantly suppressed mitochondrial mass in 4 sensitive cell lines (A375, WM266-4, UACC-62, and 451Lu) (Supplemental Figure 3, C–F).
Figure 6. MAPKi increase mtDNA copy numbers, mitochondrial mass, ROS, and expression of SOD2 in a subset of BRAF-mutated melanoma cell lines and patients’ tumor biopsies.

(A and B) Relative mtDNA copy numbers in WM9 (A) and 1205Lu (B) cells treated with DMSO or the indicated MAPKi for 72 hours. (C) MFI for MitoTracker Red in WM9 cells treated with DMSO or the indicated MAPKi for 72 hours. (D) Relative mtDNA copy numbers in patients’ tumor biopsies. Each patient’s pretreatment tumor biopsy was used as an internal control. (E) Relative gene expression of TFAM determined by qRT-PCR in patients’ tumor biopsies. (F and G) MFI of CellROX Deep Red in WM9 (F) and 1205Lu (G) cells treated with DMSO or the indicated MAPKi for 72 hours. (H and I) Relative gene expression of SOD2 determined by qRT-PCR in samples included in F and in patients’ tumor biopsies (I). (A–C and F–H) n = 3 biological replicates; data are representative of 2 independent experiments. (E and I) The pretreatment tumor biopsy from MGH patient 5 was used as a control for all other samples. n = 3 technical replicates; data represent the average. (C, F, and G) Data were normalized to the MFI derived from the unstained sample in each experimental condition. n = 3 (biological replicates); data are representative of 2 independent experiments. (D, E, and I) n = 3 technical replicates. (A–D and F–H) *P <0.05, **P <0.005, and ***P <0.0005, by 2-tailed, unpaired t test.
In 3 of 10 early-on treatment tumor biopsies derived from patients (MGH patients 7, 24, and 35), mtDNA content was significantly increased compared with that of the respective pretreatment tumor biopsies (Figure 6D). Furthermore, transcriptional factor A, mitochondrial (TFAM), a transcriptional regulator of mtDNA content, was significantly upregulated in these early-on treatment tumor biopsies (Table 4, Figure 6E, and ref. 33).
Finally, we showed that MAPKi resulted in a significant increase in ROS production in both WM9 and 1205Lu cell lines (Figure 6, F and G). The increase in ROS production was accompanied by an upregulation of the antioxidant gene superoxide dismutase 2 (SOD2) at both mRNA and protein levels (Figure 5, D–F, Figure 6H, and Supplemental Figure 3G). Using paired pre- and early-on treatment tumor biopsies, we demonstrated that the expression of SOD2 was increased in early-on treatment tumor biopsies (Figure 6I).
Taken together, our data showed that MAPKi substantially enhanced the expression of MitoBiogenesis in surviving cells in a subset of BRAF-mutated melanoma cell lines or patients’ tumor biopsies that escaped short-term inhibition of the MAPK pathway.
MAPKi significantly induced OxPhos, lysosomes, and ATP-binding cassette transporters in melanoma cells with high expression of MitoBiogenesis that were slowly cycling.
In each BRAF-mutated melanoma cell line that was tested, short-term treatment with MAPKi always led to the enrichment of surviving cells (Figure 1H). By testing WM9 and 1205Lu cell lines, we confirmed that surviving cells were arrested in the G0/1 phase of the cell cycle (Supplemental Figure 4, A and B).
In order to investigate the kinetics of cell division shown by surviving cells, prior to MAPKi treatment, we labeled an intrinsically resistant cell line, WM9, and 2 sensitive cell lines, A375 and WM266-4, with a cell proliferation fluorescent dye, CellTrace Violet. We tracked the fluorescence intensity of CellTrace Violet at multiple time points during the treatment and observed that, regardless of their expression of MitoBiogenesis and drug sensitivities, surviving cells in all 3 cell lines were dividing at a much slower rate than were control cells (Figure 7, A–C). Treatment with PLX4720 plus PD0325901 resulted in a much more pronounced slow-cycling phenotype compared with that observed with PLX4720 treatment, as demonstrated by surviving cells in WM9 and A375, but not WM266-4, cells (Figure 7, A–C).
Figure 7. Surviving cells adopt a slow-growing phenotype and activate OxPhos, lysosomes, and ABC transporters in response to MAPKi.

(A–C) Log2 transformation of the MFI of CellTrace Violet in WM9 (A), A375 (B), and WM266-4 (C) cells treated with DMSO or the indicated MAPKi for 15 days. n = 3 biological replicates; data are representative of 2 independent experiments. ***P <0.0005, by 2-way ANOVA. (D) WM9 cells were treated with 10 μM DMSO or PLX4720, and cells were harvested at 12, 24, 48, 60, 72, and 96 hours for a time-course gene expression microarray study. Upper panel: heatmaps of 10 significantly altered genes chosen from each of 5 gene sets. Lower panel: GSEA plots of the 5 top-ranked gene sets shown in the upper panel. WM9 cells treated with DMSO for 96 hours were included as a control. NES, normalized enrichment score.
Taken together, our data revealed that, although surviving cells were initially arrested in the G0/1 phase, they were capable of slow proliferation in the presence of MAPKi.
By focusing on WM9 surviving cells, we decided to elucidate the molecular basis underlying the increase in MitoBiogenesis. We analyzed time-course Illumina genome-wide gene expression microarray data. GSEA revealed a striking temporal expression profile change. Forty-eight hours was a critical transition time point, at and beyond which 6 upregulated pathways, including “Oxidative Phosphorylation (OxPhos),” “Lysosome,” “Parkinson’s Disease,” “Alzheimer’s Disease,” “Huntington’s Disease,” and “ABC Transporters,” were identified as top pathways that were enriched in surviving cells (Figure 7D and Supplemental Table 1). “MEK Targets,” “BRAF Targets,” and “Cell Cycle” emerged as the top-3-ranked downregulated pathways, confirming that PLX4720 inhibited the MAPK pathway and cell-cycle progression (Figure 7D and Supplemental Table 2).
To validate the results based on genome-wide gene expression data, we first performed a qRT-PCR experiment and showed that expression of DUSP6 and FOXM1 was suppressed by PLX4720 in surviving cells (Supplemental Figure 4, C and D). We also observed that in surviving cells, a continuous treatment with PLX4720 for 120 hours resulted in the upregulation of 5 representative mitochondrial respiratory chain subunits including NDUFA8, SDHB, UQCRB, MT-CO1, and ATP5G1, which was accompanied by increased expression of PDP2 and TXNIP, both of which were negative regulators of glycolysis (Figure 8A). Additionally, we demonstrated that the expression of 30 mitochondria respiratory chain complex subunits was substantially increased in surviving cells that escaped the 72-hour treatment with MAPKi (Figure 8B and Supplemental Figure 4E). Moreover, we showed that the expression of 3 representative respiratory chain subunits, NDUFA8, MT-CO1, and COX7B, was significantly upregulated in patients’ early-on treatment tumor biopsies compared with that seen in paired pretreatment tumor biopsies (Figure 8C). MAPKi upregulated 6 mitochondrial respiratory chain complex subunits, NDUFB8, SDHA, SDHB, UQCRC2, COX II, and ATP5A, at the protein level in surviving cells, which was accompanied by suppression of phospho-ERK (p-ERK), p-Rb, and p-S6 as well as upregulation of p27 and the DNA damage marker γ-H2AX (Figure 8D). The increase in OxPhos in WM9 surviving cells was demonstrated by an increase in oxygen consumption, particularly maximal respiration (Figure 8, E and F).
Figure 8. Surviving cells and patients’ early-on treatment tumor biopsies highly express mitochondrial respiratory chain complex subunits and increase maximal respiration.

(A–C) Relative gene expression of the indicated genes was determined by qRT-PCR in WM9 cells treated with 10 μM PLX4720 for 120 hours and harvested at the indicated time points (A); in WM9 cells treated for 72 hours with DMSO or the indicated MAPKi (B); and in patients’ tumor biopsies (C). n = 3 technical replicates; data are representative of 2 independent experiments. (C) The pretreatment tumor biopsy from MGH patient 2 was used as the baseline for all other samples. (D) Immunoblot analysis of proteins in WM9 cells treated with DMSO or the indicated MAPKi for 72 hours. (E) Mitochondrial respiration, indicated as the OCR of WM9 cells treated with the indicated MAPKi for 72 hours, was measured by a Seahorse XF24 Analyzer. Coupled and maximal respirations were determined by the sequential addition of oligomycin, DNP, and antimycin and rotenone, respectively. Data are representative of 4 biological replicates. (F) Maximal respiration measured in the samples in E. *P <0.05, **P <0.005, and ***P <0.0005, by 2-tailed, unpaired t test.
In the late stage of autophagy, autophagosomes fuse with lysosomes to degrade captured cellular substrates or damaged organelles. Expression of LC3B-II was induced in WM9 surviving cells by MAPKi (Figure 5H), which was in line with the increase in autophagic flux as assessed by the ratio of mCherry over eGFP in WM9 cells expressing a fluorescent autophagy reporter construct, mCherry-EGFP-LC3B (Supplemental Figure 4F). Furthermore, expression of the ER stress genes CHOP, GRP78, GRP94, ATF4, GADD34, and ERDJ4 was markedly induced in surviving cells (Supplemental Figure 4G). Together, our data showed that MAPKi induced a profound stress response program and upregulated the expression of MitoBiogenesis in surviving cells.
Finally, we confirmed that expression of the representative ATP-binding cassette (ABC) transporters ABCC1, ABCG2, ABCC2, and ABCB5 was substantially upregulated by MAPKi in surviving cells (Supplemental Figure 4, H and I).
Altogether, our experiments identified and confirmed that OxPhos, ER stress, autophagy/lysosomes, and ABC transporters were strongly induced in surviving cells in which MitoBiogenesis was highly expressed. Our data also underscored the potential roles of these processes as a survival mechanism by which BRAF-mutated melanoma cells escape MAPKi.
Depletion of TFAM or TRAP1, but not PGC1A, inhibited MitoBiogenesis.
Our next goal was to pinpoint which genes were essential for surviving cells. We performed 2 selective siRNA screens in WM9 cells by depleting 6 genes related to autophagy and ER stress response and 18 genes related to MitoBiogenesis and tumor bioenergetics and subsequently treating cells with PLX4720 plus PD0325901 (Supplemental Figure 5, A and B).
In the first siRNA screen, we tested the combination of MAPKi and siRNAs that inhibited the autophagy and ER stress response pathways. The autophagy inhibitor spautin-1 was included as a control. We observed that, although the combination of siRNAs targeting the autophagy–ER stress response and MAPKi indeed effectively led to a decrease in the autophagic flux that was enhanced by MAPKi alone (Supplemental Figure 5, C and E), MAPKi, combined with either siRNAs (with the exception of siRNA targeting VPS34) or the autophagy inhibitor spautin-1, was not able to trigger a significant induction of apoptosis and cell death (Supplemental Figure 5, D and F). These data suggested that, upon the dual inhibition of the autophagy–ER stress response and the MAPK pathway, surviving cells may activate other compensatory pathways. Another possibility was that the autophagy–ER stress response pathways did not necessarily affect the efficacy of MAPKi.
In the second siRNA screen, we focused on the combination of MAPKi and siRNAs targeting 8 MitoBiogenesis genes, TRAP1, 5 OxPhos genes, and 4 glycolytic genes. The mitochondrial HSP90 inhibitor Gamitrinib was included as a control. TRAP1 is critical in directing mitochondrial protein folding to control central metabolic networks (34–36). Targeting TRAP1-directed protein folding in mitochondria with Gamitrinib inhibits both glycolysis and OxPhos in a diverse panel of cancer cell lines including melanomas (35). Gamitrinib is a metabolic poison that is cytotoxic to cancer cells in vitro and retards tumor growth in xenografts. Unlike the cytosolic HSP90 inhibitor 17-AAG, Gamitrinib specifically inhibited the expression of proteins related to OxPhos and MitoBiogenesis, but not the clients of cytosolic HSP90 (Supplemental Figure 5G).
Gamitrinib administered at 1 or 2.5 μM, in combination with MAPKi, resulted in a more remarkable induction of apoptosis and cell death compared with that observed with either Gamitrinib or MAPKi alone (Figure 9A). Intriguingly, specific siRNAs targeting TFAM, TRAP1, PPRC1, and ESRRA, but not PGC1A, substantially augmented the efficacy of MAPKi and emerged as top hits. The effects of these siRNAs were comparable to those of Gamitrinib when administered at 1 or 2.5 μM (Figure 9A).
Figure 9. Knockdown of TFAM or TRAP1 improves the efficacy of MAPKi.

(A) Percentage of PSVue 643+ WM9 cells that were transfected with the indicated siRNA clone and treated with MAPKi for 72 hours. Cells transfected with siNS (nontargeting sequence) were included as a negative control. Gamitrinib, used at 0.5, 1, and 2.5 μM, is indicated by a red star in the graph. The average of 2 biological replicates was plotted, and data are representative of 2 independent experiments. Gami, Gamitrinib. (B) Immunoblot analysis of proteins in WM9 cells transfected with siTFAM or siTRAP1 and treated with DMSO or the indicated MAPKi for 72 hours.
Next, we proved that siRNAs targeting TRAP1 or TFAM suppressed the expression of MitoBiogenesis in surviving cells that was induced by MAPKi (Table 5 and Figure 9B). We also demonstrated that, although 2 siRNA clones targeting PGC1A were able to inhibit the expression of MitoBiogenesis in WM9 control cells, unexpectedly, these clones failed to further augment MAPKi-induced apoptosis and cell death (Figure 9A) and to inhibit the expression of MitoBiogenesis in WM9 surviving cells (Table 6).
Table 5. Relative expression of 18 MitoBiogenesis genes in WM9 cells that were transfected with siTFAM or siTRAP1 and treated with MAPKi for 72 hours.
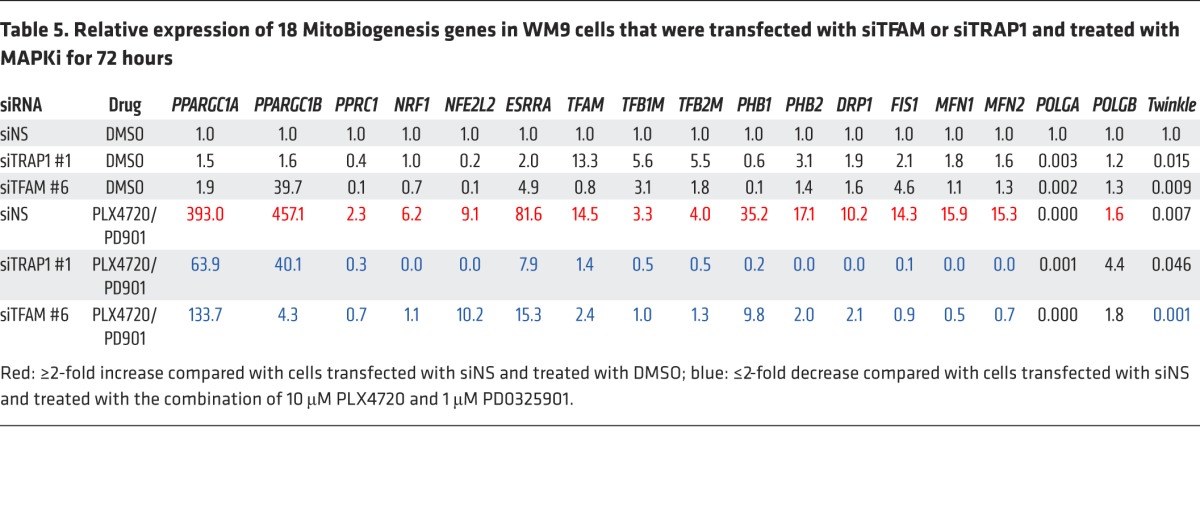
Table 6. Relative expression of 18 MitoBiogenesis genes in WM9 cells that were transfected with siPPARGC1α and treated with MAPKi for 72 hours.
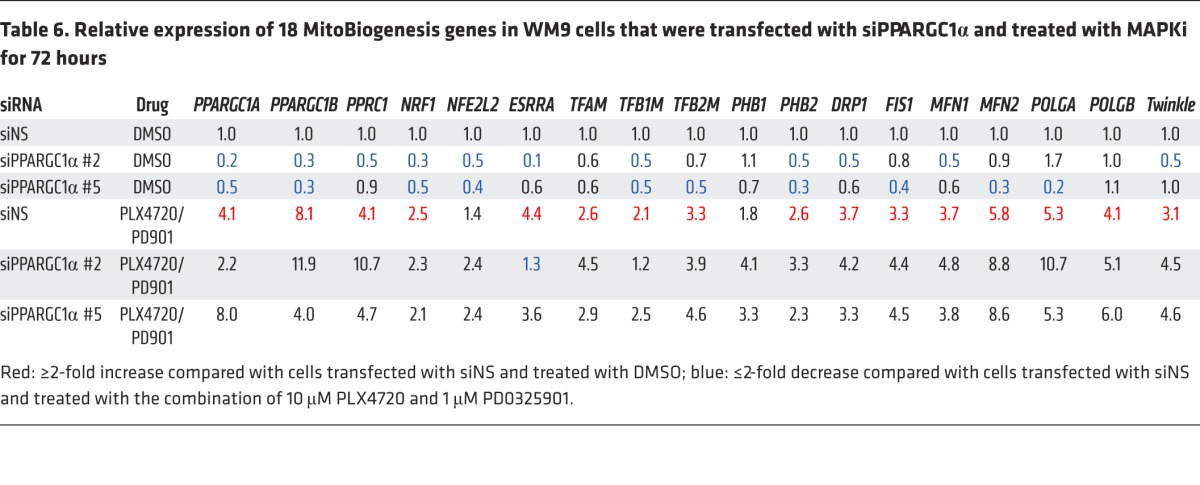
It is known that the PGC1A/PGC1B/PPRC1/NRF1/ESRRA signaling axis is essential for the regulation of TFAM in order to drive mitochondrial biogenesis. Taken together, our data now pointed to a core network consisting of PPRC1/ESRRA/TFAM and demonstrated that the loss of each component of this network augmented the increase in apoptosis and cell death when combined with MAPKi. Our data based on 2 siRNA clones targeting TRAP1 and the mitochondrial HSP90 inhibitor Gamitrinib were consistent and of particular interest, because TRAP1 was proven to be the target of Gamitrinib. Furthermore, our data suggested that there might be a switch in the regulation of mitochondrial biogenesis from PGC1A to TFAM and/or TRAP1, particularly in surviving cells.
Gamitrinib inhibited MitoBiogenesis and tumor bioenergetics.
To identify the optimal combination therapy that would trigger a greater induction of apoptosis and cell death, we conducted an additional selective drug screen. In addition to Gamitrinib, we included (a) 2 mTOR inhibitors, rapamycin and BEZ235, because another mTOR inhibitor, AZD8055, inhibits OxPhos by suppressing PGC1α (37); (b) Phenformin, which has been implicated in inhibiting the mitochondrial respiratory complex I (17); (c) 2,4-dinitrophenol (2,4-DNP), which has been implicated as a mitochondrial uncoupler (13); and (d) the autophagy inhibitor spautin-1.
Our data showed that the combination of Gamitrinib or Phenformin and MAPKi led to a greater increase in apoptosis and cell death, outperforming other inhibitors (Figure 10A). We focused on Gamitrinib in subsequent studies, because our previous studies had explored the combination of Phenformin and MAPKi. However, the combination of Gamitrinib and MAPKi has not been investigated yet. This novel combination therapy was subsequently tested in 22 of our own BRAF-mutated melanoma cell lines, and we showed that it led to a greater increase in apoptosis and cell death than did MAPKi alone in many cell lines (Figure 10B).
Figure 10. Gamitrinib suppresses MitoBiogenesis and tumor bioenergetics.

(A) Percentage of PSVue 643+ WM9 cells treated with DMSO or the indicated MAPKi in combination with Gamitrinib (Gami), rapamycin (Rapa), Phenformin (Phen), BEZ235, 2,4-DNP, or Spautin-1 for 72 hours and harvested on days 1, 2, and 3 for FACS analysis. (B) Percentage of PSVue 643+ cells in 22 BRAF-mutated melanoma cell lines treated with a combination of the indicated MAPKi and Gamitrinib for 72 hours (PLX4720, 10 μM; PD901, 1 μM; Gamitrinib, 2.5 M). (A and B) Cells treated with DMSO were used as a control. The average of 2 biological replicates was plotted, and data are representative of 2 independent experiments. (C) Long-term cell growth assay of WM9 cells treated with 10 μM DMSO or PLX4720 in combination with Gamitrinib for 15 days. Cells were fixed and stained with crystal violet on day 15. Data are representative of 2 independent experiments. (D) Two heatmaps of RPPA data on WM9 cells treated with DMSO or the indicated MAPKi in combination with 1 μM Gamitrinib for 72 hours. Upper panel: PLX4720, 10 μM; lower panel: PLX4720, 10 μM and PD0325901, 1 μM. n = 3 biological replicates. (E) Immunoblot analysis of proteins in WM9 cells treated with 10 μM DMSO or PLX4720, with or without 1 μM Gamitrinib, for 72 hours.
The prolonged combination of Gamitrinib and PLX4720 substantially inhibited the viability of WM9 cells compared with that of cells treated with PLX4720 alone, suggesting that the upfront combination therapy was able to overcome the acquisition of drug resistance to PLX4720 (Figure 10C).
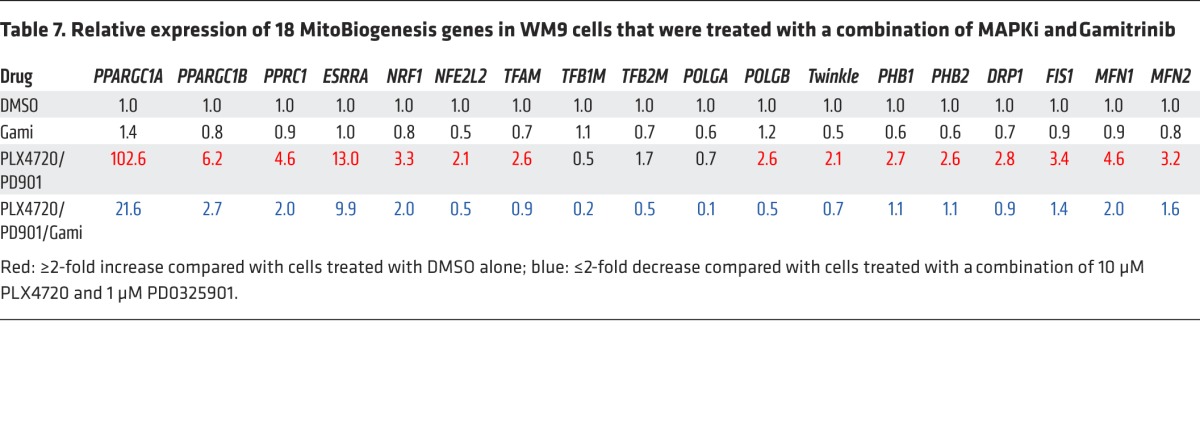
To gain mechanistic insights into this combination therapy, we conducted a reverse-phase protein array (RPPA) experiment. The analysis of RPPA data showed that Gamitrinib caused a significant reduction in expression of the mitochondrial respiratory chain complex subunits SDHA, SDHB, UQCRC2, ATP5H, and cyclophilin D, which were induced by MAPKi (Figure 10D and Supplemental Figure 6, A and B). The RPPA data were corroborated by immunoblotting data showing that all 5 mitochondrial respiratory chain complex subunits were downregulated by Gamitrinib (Figure 10E). Importantly, we demonstrated that Gamitrinib inhibited the expression of MitoBiogenesis that was enhanced by MAPKi (Table 7).
Table 7. Relative expression of 18 MitoBiogenesis genes in WM9 cells that were treated with a combination of MAPKi and Gamitrinib.
To elucidate the biological consequences of this combination therapy, we treated WM9 cells with a combination of Gamitrinib and MAPKi (PLX4720 plus PD0325901 or LGX818 plus MEK162) and found that the combination led to decreases in mtDNA content (Figure 11, A and B) and mitochondrial mass (Figure 11C). We also profiled the metabolic activities of WM9 cells that were treated with the combination of MAPKi (PLX4720, PLX4720 plus PD0325901, or LGX818 plus MEK162) and Gamitrinib by testing for real-time oxygen consumption rates (OCRs). Gamitrinib inhibited all key OCR parameters, including basal respiration, proton leak, ATP production, maximal respiration, and spare respiratory capacity, compared with what was seen in the control cells that were not treated with Gamitrinib (Figure 11, D and E, and Supplemental Figure 6C).
Figure 11. Gamitrinib leads to decreased mtDNA copy numbers, mass, and respiration and inhibits glycolysis and ATP production.

(A and B) Relative mtDNA copy numbers in WM9 cells treated with DMSO or the indicated MAPKi, with or without 1 μM Gamitrinib, for 72 hours. (C) MFI of MitoTracker Red for WM9 cells treated for 72 hours with DMSO or the indicated MAPKi, with or without 1 or 2.5 μM Gamitrinib. n = 2 biological replicates, and the average was plotted. Data are representative of 2 independent experiments. (D and E) Real-time oxygen consumption of WM9 cells subjected to a metabolic stress test with a Seahorse XF 24 Analyzer. Cells were treated for 72 hours with DMSO or the indicated MAPK inhibitor, with or without 1 or 2.5 μM Gamitrinib. n = 4 biological replicates. (F) Relative gene expression was assessed by qRT-PCR in WM9 cells treated for 48 hours with DMSO or the indicated MAPKi, with or without 2.5 μM Gamitrinib. (G and H) MFI of 2-NBDG (G) and relative ATP production (H) in WM9 cells treated for 72 hours with DMSO or the indicated MAPKi, with or without 1 or 2.5 μM Gamitrinib. (A, B, and F) n = 3 technical replicates, and data are representative of 2 independent experiments. (G and H) n = 3 biological replicates, which were included in each sample, and the data are representative of 2 independent experiments. *P <0.05, **P <0.005, and ***P <0.0005, by 2-tailed, unpaired t test.
The combination of Gamitrinib and PLX4720 substantially downregulated the expression of HKI, HKII, PDHE1, PDK1, PDK2, LDHA, PKM1, GLUT1, and GLUT3, all of which are key regulators of aerobic glycolysis (Figure 10E and Figure 11F). This was in line with the decrease in the uptake of an analog of glucose, 2-NBDG, when WM9 cells were treated with the combination of Gamitrinib and PLX4720 (Figure 11G).
Collectively, RPPA and immunoblotting data also showed that Gamitrinib downregulated LC3B-II and p-S6 and upregulated p-AMPKα. The upregulation of p-AMPKα was indicative of metabolic stress, which was probably due to a decrease in the cellular ratio of ATP over ADP (Figure 10, D and E). In fact, the combination of Gamitrinib and PLX4720 led to a significant decrease in ATP production (Figure 11H).
The M-MITF/PGC1A signaling axis is important in regulating mitochondrial biogenesis and OxPhos in BRAF-mutated melanoma cells (13, 37). Although our data did not support a direct role of PGC1A in the regulation of MitoBiogenesis, we showed that in surviving cells, Gamitrinib downregulated both PGC1A and M-MITF, which were upregulated by MAPKi (Table 7 and Supplemental Figure 5D)
Taken together, our data not only showed that the combination of Gamitrinib and MAPKi phenocopied the combination of siRNAs targeting TFAM or TRAP1 and MAPKi, but also demonstrated that targeting MitoBiogenesis was effective in improving the efficacy of MAPKi and preventing the acquisition of drug resistance to MAPKi. The finding that Gamitrinib was able to convert the survival into apoptosis and cell death prompted us to investigate the molecular mechanisms underlying the efficacy of the combination of Gamitrinib and MAPKi.
The combination of Gamitrinib and MAPKi resulted in mitochondrial dysfunction and inhibited tumor growth.
The decrease in oxygen consumption demonstrated by melanoma cells that were treated with the combination of MAPKi and Gamitrinib suggested that mitochondrial respiration was inhibited and that the electron transport chain did not function properly. We hypothesized that the ROS production increased because electrons would leak from the electron transport chain and prematurely react with oxygen. Indeed, the combination of MAPKi and Gamitrinib led to a marked increase in ROS production compared with that of cells treated with MAPKi alone (Figure 12A). This indicated that the combination of Gamitrinib and MAPKi resulted in mitochondrial dysfunction and oxidative stress elevation.
Figure 12. The combination of Gamitrinib and MAPKi results in mitochondrial dysfunction and delays tumor growth in vivo.

(A) MFI of CellROX Deep Red in WM9 cells treated with DMSO or the indicated MAPKi, with or without 1 μM Gamitrinib, for 72 hours. (B) Relative gene expression of SOD2 was assessed by qRT-PCR in WM9 cells treated with DMSO or the indicated MAPKi, with or without 1 μM Gamitrinib, for 72 hours. n = 3 technical replicates, and data are representative of 2 independent experiments. (C) Immunoblot analysis of p-ERK and SOD2 in WM9 cells treated with DMSO or the indicated MAPKi, with or without 1 μM Gamitrinib, for 72 hours. (D) Percentage of PSVue 643+ WM9 cells treated with DMSO, the indicated MAPKi, 1 μM Gamitrinib, and 5 mM NAC, used either alone or in combination, for 72 hours. (E–G) Tumor volume of 1205Lu xenografts treated for 15 days with the vehicle control or PLX4720, either alone or in combination with 2,4-DNP (E), 1205Lu xenografts treated for 17 days with the vehicle control or PLX4720, either alone or in combination with Gamitrinib (F), and WM9 xenografts treated for 22 days with the vehicle control or PLX4720, either alone or in combination with Gamitrinib (G). (E and F) n = 10 mice per group; (G) n = 5 mice per group. (A and D) n = 3 biological replicates, which were included in each sample. Data are representative of 2 independent experiments. **P <0.005 and ***P <0.0005, by 2-tailed, unpaired t test (A and D). A 2-tailed, unpaired t test was also used to determine the P values in E–G.
For cells to survive under oxidative stress, SOD2 is normally induced to regulate the detoxification of mitochondrial ROS. Interestingly, the combination of Gamitrinib and MAPKi led to the upregulation of SOD2 compared with that observed in cells treated with MAPKi alone (Figure 12, B and C). By blocking oxidative stress with an ROS scavenger, N-acetyl-L-cysteine (NAC), we demonstrated that NAC markedly suppressed the apoptosis and cell death that were induced by the combination of Gamitrinib and MAPKi (Figure 12D). This suggested that enhancing oxidative stress beyond a tolerable threshold would determine how the combination of Gamitrinib and MAPKi causes the mitochondrial dysfunction that leads to apoptosis and cell death.
Next, we tested the in vivo efficacy of the combination of PLX4720 and Gamitrinib using 2 xenograft models bearing 1205Lu and WM9 melanoma cells. Because of its antimelanoma activity as a single agent (13), we first tested the antitumor activity of the combination of the mitochondrial uncoupler 2,4-DNP and PLX4720. Our data showed that 2,4-DNP failed to inhibit tumor growth and that the combination of 2,4-DNP and PLX4720 did not result in a greater effect when compared with PLX4720 in 1205Lu xenografts (Figure 12E and Supplemental Table 3). Next, we tested the antitumor activity of the combination of Gamitrinib and PLX4720 in both 1205Lu and WM9 xenografts. Gamitrinib administered at 15 mg/kg alone or combination with PLX4720 significantly impaired the tumor growth of 1205Lu and WM9 xenografts (Figure 12, F and G, and Supplemental Tables 4 and 5).
Targeting MitoBiogenesis and tumor bioenergetics was effective in a subset of acquired-resistance, BRAF-mutated tumor cells.
Next, to investigate the clinical relevance of MitoBiogenesis and tumor bioenergetics in the context of acquired drug resistance, we analyzed genome-wide gene expression data available from 5 publicly available data sets (GEO GSE50535, GSE61992, GSE50509, GSE65185, and European Genome-Phenome Archive [EGA] EGAS00001000992) by performing ssGSEA, which collectively profiled paired pre- and post-treatment tumor biopsies from 153 melanoma patients whose BRAF-mutated tumors relapsed with MAPKi treatment.
In GSE50535, the expression of MitoBiogenesis, glycolysis and gluconeogenesis, and OxPhos was increased in the post-treatment tumor biopsy from patient 3 (Figure 13A). In GSE50509, the expression of MitoBiogenesis was increased in post-treatment tumor biopsies derived from patients 3, 4, 9, 10, 17, 18, 23, 28, and 30; the expression of glycolysis and gluconeogenesis was increased in post-treatment tumor biopsies derived from patients 2, 3, 4, 7, 10, 14, 25, and 28; and the expression of OxPhos was increased in post-treatment tumor biopsies derived from patients 3, 7, 9, 10, 12, 18, 21, 25, 28, and 30 (Figure 13B). In GSE61992, the expression of MitoBiogenesis was increased in post-treatment tumor biopsies derived from patients 3, 4, 5, 6, 7, and 9; the expression of glycolysis and gluconeogenesis was increased in post-treatment tumor biopsies derived from patients 1, 2, 4, 6, 7, 9, and 10; and the expression of OxPhos was increased in post-treatment tumor biopsies derived from patients 1, 4, and 9 (Figure 13C). In GSE65185, the expression of MitoBiogenesis was increased in post-treatment tumor biopsies derived from patients 1, 2, 3, 5, 8, 9, 10, 17, and 21; the expression of glycolysis and gluconeogenesis was increased in post-treatment tumor biopsies derived from patients 1, 2, 5, 6, 9, 10, 15, 17, 19, 20, 21, and 24; and the expression of OxPhos was increased in post-treatment tumor biopsies derived from patients 1, 2, 8, 9, 10, 15, 16, 17, 18, 19, 23, and 24 (Figure 13D). In EGAS00001000992, the expression of MitoBiogenesis was not increased in any of the post-treatment tumor biopsies from patients; the expression of glycolysis and gluconeogenesis was increased in the post-treatment tumor biopsy from patient 16; and the expression of OxPhos was increased in the post-treatment tumor biopsy from patient 25 (Supplemental Figure 7).
Figure 13. Clinical relevance of mitochondrial biogenesis and tumor bioenergetics in the context of acquired drug resistance.

(A–D) Heatmaps of enrichment scores for 16 mitochondrial gene sets and 2 metabolic gene sets in patients’ paired pre- and post-treatment tumor biopsies that are included in GSE50535 (A), GSE50509 (B), GSE61992 (C), and GSE65185 (D). Pre, before treatment; Post, after treatment; Prog, progression.
Additionally, we performed immunohistochemical staining of TFAM, TRAP1, and MT-CO1 in paired pre- and post-treatment tumor biopsies. Compared with the paired pretreatment tumor biopsies, TFAM expression was substantially increased in 3 (University of Pennsylvania patient [Penn] 11-47, Penn 12-148, and Penn 503) of 9 post-treatment tumor biopsies (Figure 14A and Supplemental Table 6); TRAP1 expression was substantially increased in 3 (Penn 11-35, Penn 12-148, and Penn 503) of 9 post-treatment tumor biopsies (Figure 14B and Supplemental Table 6); and MT-CO1 expression was substantially increased in 1 early-on treatment tumor biopsy (MGH patient 24) as well as in 5 (MGH patients 9, 20, and 26 and Penn patients 11-3 and 578) of 18 post-treatment tumor biopsies (Figure 14C, Table 2, and Supplemental Table 6).
Figure 14. Increases of mitochondrial biogenesis proteins exhibited in patients’ progressive tumor biopsies and targeting acquired drug resistant cells with Gamitrinib.

(A–C) Immunohistochemical staining of TFAM (A), TRAP1 (B), and MT-CO1 (C) in patients’ paired pre-, early-on, and post-treatment tumor biopsies. Scale bars represent 40 μm. (D) Percentage of PSVue 643+ cells in 23 BRAF-mutated resistant melanoma cell lines that were treated with 2.5 μM Gamitrinib for 72 hours. n =2 biological replicates, which were included for each sample. Data are representative of 2 independent experiments. *P <0.05, **P <0.005, and ***P <0.0005, by 2-tailed, unpaired t test was used. PDXs, patient-derived xenografts.(E) Schematic model showing cotargeting the MAPK pathway and mitochondrial biogenesis in BRAF-mutated melanoma cells as a viable therapeutic strategy.
To investigate the prognostic values of mitochondrial biogenesis genes, we performed a Cox regression analysis and found that (a) patients with higher expression of DNM1L, HSPD1, or VDAC1 in pretreatment tumor biopsies would likely experience faster disease progression; (b) patients with increased expression of DNM1L or MFN1 in post-treatment tumor biopsies would likely experience slower disease progression; (c) patients with higher expression of VDAC1 in pretreatment tumor biopsies would likely have a worse overall survival; and (d) patients with higher expression of TUFM in post-treatment tumor biopsies would likely have a worse overall survival (Supplemental Tables 7–10).
Finally, we tested whether cell lines with acquired resistance to BRAF-targeted therapies or immune checkpoint inhibitors are susceptible to Gamitrinib. All cell lines except WM4265-1 and WM4265-2 acquired resistance to MAPKi. WM4265-1 and WM4265-2 cell lines were established from 2 PDX models that were derived from 2 different brain metastatic lesions that were surgically removed from a patient after this patient continued to progress on multiple therapies including cisplatin, vinblastine, temozolomide, interleukin-2, interferon alfa-2b, ipilimumab, and pembrolizumab. In fact, Gamitrinib significantly induced apoptosis and cell death in 18 of 23 resistant cell lines (Figure 14D).
In conclusion, our data show that MitoBiogenesis and tumor bioenergetics are highly upregulated in a subset of resistant tumors. Our data suggest that patients who are refractory to MAPKi treatment may benefit from the addition of Gamitrinib to their second-line therapies to overcome acquired drug resistance (Figure 14E).
Discussion
In this study, we elucidated the role of MitoBiogenesis in mediating both intrinsic and acquired resistance to MAPKi. Our results demonstrate the existence of intrinsically resistant BRAFV600E melanoma cells that activate mitochondrial biogenesis and OxPhos to meet their bioenergetic needs and to survive MAPKi. Our study establishes the molecular basis for how mitochondrial biogenesis underlies and couples both intrinsic and acquired drug resistance. Furthermore, our data pave the way for combining Gamitrinib with MAPKi to trigger synthetic lethality.
We found that the MitoBiogenesis gene signature is primarily expressed in a subset of melanoma cell lines or tumors. MAPKi downregulate MitoBiogenesis in sensitive melanoma cell lines with high basal levels of MitoBiogenesis and upregulate MitoBiogenesis in resistant melanoma cell lines with low basal levels of MitoBiogenesis. Intrinsically resistant cells that highly express mitochondrial biogenesis have high mtDNA copy numbers, mitochondrial mass, and OCRs and produce more ROS. Clinically, the expression of MitoBiogenesis and mtDNA content is highly upregulated in early-on treatment tumor biopsies from a subset of patients.
We uncovered a dynamic expression profile change in intrinsically resistant cells responding to MAPKi and identified 3 major pathways, OxPhos, autophagy/lysosomes, and ABC transporters, that may confer the intrinsic resistance phenotype. Interestingly, KRAS-mutant pancreatic cancer cells also depend on OxPhos for survival when the mutant KRAS is blunted (30).
To pinpoint the causal and passive roles of MitoBiogenesis, tumor bioenergetics, and autophagy in underlying intrinsic resistance, we performed selected siRNA- and drug-based screens. The autophagy inhibitor and siRNAs against representative mitochondrial respiratory chain subunits did not enhance the apoptosis and cell death that were induced by MAPKi. We reason that it is possible that redundant or alternative pathways are activated to compensate for the genetic and pharmacologic inhibition. Importantly, we demonstrate that the depletion of TFAM or TRAP1, but not PGC1A, synergizes with MAPK. Our data underscore the idea that a core signaling network involving PPRC1, ESRRA, and TFAM confers resistance to MAPKi. TRAP1 is exclusively located in mitochondria and controls proper mitochondrial protein folding and integrity. The potent effect of the combination of siTRAP1 and MAPKi encouraged us to test the efficacy of Gamitrinib, which is designed to target TRAP1 (mitochondrial HSP90) in inhibiting MitoBiogenesis.
The combination of Gamitrinib and MAPKi results in an unparalleled synthetic lethality, outperforming many other metabolic inhibitors, except Phenformin. We further demonstrate that the combination of Gamitrinib and MAPKi downregulates TFAM, PGC1A, and M-MITF and impairs many key parameters of mitochondrial biogenesis as well as glucose uptake and ATP production. The combination of Gamitrinib and MAPKi leads to an increase in mitochondrial oxidative stress that is beyond the tolerable threshold for cell detoxification and survival. Our data support the use of Gamitrinib as an effective therapeutic option to inhibit aberrant tumor metabolism. A recent study showed the efficacy of BCL2 inhibition by targeting OxPhos to eradicate leukemia stem cells with low ROS and warrants the investigation of its efficacy in targeting resistant melanoma cells (38).
We show that Gamitrinib not only circumvents the acquisition of resistance to MAPKi but also impairs the viability of cells with acquired resistance. Furthermore, our data show that, in progressive tumor biopsies from a subset of patients, the expression of MitoBiogenesis and tumor bioenergetics is upregulated.
The clinical relevance of mitochondrial biogenesis, glycolysis, and OxPhos in patients with melanoma is very intriguing. TCGA melanoma patients with high expression of MitoBiogenesis or coexpression of glycolysis and OxPhos have worse overall survival rates. Thus, mitochondrial metabolism might be a tractable therapeutic target for that subset of patients. Further investigation of the expression of mitochondrial biogenesis, glycolysis, and OxPhos genes and the association with clinical status in other types of cancers is clearly warranted.
Methods
Cell lines established from human melanomas and patient-derived xenografts.
See the Supplemental Methods for details.
Illumina gene expression microarray data on WM9 cells.
Time-course Illumina gene expression microarray data on WM9 cells were deposited in the NCBI’s GEO database (GEO GSE58721).
Melanoma xenotransplantation and in vivo studies.
Ten thousand 1205Lu or WM9 cells were harvested from cell culture and resuspended in culture medium and Matrigel at a 1:1 ratio. Cells were s.c. injected into mice, which were treated with the indicated inhibitors when the tumor volume reached 100 mm3. Mice were sacrificed at the end time point and solid tumors collected.
Statistics.
Unless otherwise indicated, data in the figures are presented as the mean ± SEM for 3 biological or technical replicates. Significant differences between experimental conditions were determined using a 2-tailed, unpaired t test. For survival data, Kaplan–Meier survival curves were generated, and their differences were examined using a log-rank test. For tumor growth data, mixed-effects models were used to determine the differences between treatment groups in tumor volume change at the end of the experiment. A 2-sided P value of less than 0.05 was considered statistically significant. See the Supplemental Methods for detailed statistical information.
Study approval.
All clinical data and patients’ samples were collected following IRB approval of the Massachusetts General Hospital and the Hospital of the University of Pennsylvania, and informed consent was obtained from all study participants. All animal studies were conducted in accordance with NIH animal care and use guidelines, and mice were maintained according to the guidelines of the IACUC of The Wistar Institute. All animal experiments were performed in accordance with IACUC protocol 112330 of The Wistar Institute, and the study designs were approved by the IACUC of The Wistar Institute.
Author contributions
GZ and MH designed the study. DTF, LW, CK, SS, YCC, XX, HC, ED, OO, BS, DZ, MG, MX, SR, KS, CX, AG, PB, and YL conducted experiments. ZW, SS, MG, Weiting Xu, TT, JZ, SZ, and QL analyzed data. DTF, Wei Xu, JL, GCK, LMS, TCG, and KTF provided patients’ samples and clinical information. GZ and MH wrote the manuscript, with input and scientific advice from LW, BS, DB, RKA, AW, MAD, JAW, NGA, GBM, DCA, and KTF.
Supplementary Material
Acknowledgments
We thank all former and current laboratory members for their comments and helpful discussions; J. Hayden (Wistar Microscopy Facility), C. Chang, S. Billouin, and T. Nguyen (Wistar Genomics Facility), J.S. Faust, and D. Ambrose (Wistar Flow Cytometry Facility) for their technical support; M.B. Powell for providing human melanoma cells (Stanford University); and M.S. Soengas and E. Riveiro-Falkenbach for providing plasmids (Centro Nacional de Investigaciones Oncológicas [CNIO]). We apologize to those whose work was not cited or mentioned here due to space constraints. The research was funded by NIH grants P01 CA114046, P01 CA025874, P30 CA010815, and R01 CA047159 and by the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation and the Melanoma Research Foundation. The support for Shared Resources used in this study was provided by Cancer Center Support Grant (CCSG) CA010815 (to the Wistar Institute).
Footnotes
Conflict of interest: K.T. Flaherty has received consulting fees from GlaxoSmithKline (GSK), Roche, and Novartis. G.B. Mills serves as a consultant for AstraZeneca, Blend Therapeutics, Critical Outcome Technologies Inc., HanAl Bio Korea, Illumina, Nuevolution, Pfizer, Provista Diagnostics, Roche, SignalChem Lifesciences, Symphogen, and Tau Therapeutics; owns stock in Catena Pharmaceuticals, PTV Healthcare Capital, Spindle Top Capital; and has received research funding from Adelson Medical Research Foundation, AstraZeneca, Critical Outcome Technologies Inc., GSK, and Illumina.
Reference information:J Clin Invest. 2016;126(5):1834–1856. doi:10.1172/JCI82661.
Contributor Information
Gao Zhang, Email: gzhang@Wistar.org.
Lawrence Wu, Email: lawrenwu@sas.upenn.edu.
Zhi Wei, Email: zhiwei04@gmail.com.
Clemens Krepler, Email: ckrepler@Wistar.org.
Satish Srinivasan, Email: satishs@vet.upenn.edu.
Young Chan Chae, Email: ychae@wistar.org.
Xiaowei Xu, Email: xug@mail.med.upenn.edu.
Harry Choi, Email: hehrhee@gmail.com.
Elaida Dimwamwa, Email: elaidad@seas.upenn.edu.
Omotayo Ope, Email: jeromees2000@gmail.com.
Batool Shannan, Email: Batool.Shannan@uk-essen.de.
Devraj Basu, Email: devbasu@mail.med.upenn.edu.
Dongmei Zhang, Email: dongmeizhangkx@gmail.com.
Manti Guha, Email: manti@vet.upenn.edu.
Min Xiao, Email: mxiao@wistar.org.
Sergio Randell, Email: serrja@gmail.com.
Katrin Sproesser, Email: sproesser@Wistar.org.
Wei Xu, Email: Wei.Xu@uphs.upenn.edu.
Jephrey Liu, Email: jephyliu@gmail.com.
Tara C. Gangadhar, Email: Tara.Gangadhar@uphs.upenn.edu.
Mengnan Gu, Email: mg339@njit.edu.
Caiyue Xu, Email: caiyuexu@mail.med.upenn.edu.
Abheek Ghosh, Email: abheekg@gmail.com.
Weiting Xu, Email: wx37@njit.edu.
Tian Tian, Email: tt72@njit.edu.
Jie Zhang, Email: jiezhang535161432@gmail.com.
Shijie Zha, Email: sz279@njit.edu.
Qin Liu, Email: qliu@Wistar.org.
Patricia Brafford, Email: brafford@Wistar.org.
Ashani Weeraratna, Email: aweeraratna@wistar.org.
Yiling Lu, Email: yilinglu@mdanderson.org.
Dario C. Altieri, Email: daltieri@wistar.org.
Meenhard Herlyn, Email: herlynm@wistar.org.
References
- 1.Bhatia S, Tykodi SS, Thompson JA. Treatment of metastatic melanoma: an overview. Oncology (Williston Park) 2009;23(6):488–496. [PMC free article] [PubMed] [Google Scholar]
- 2.Davies H, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 3.McArthur GA, et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014;15(3):323–332. doi: 10.1016/S1470-2045(14)70012-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Flaherty KT, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363(9):809–819. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bollag G, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010;467(7315):596–599. doi: 10.1038/nature09454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chapman PB, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Flaherty KT, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367(2):107–114. doi: 10.1056/NEJMoa1203421. [DOI] [PubMed] [Google Scholar]
- 8.Flaherty KT, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367(18):1694–1703. doi: 10.1056/NEJMoa1210093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shi H, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014;4(1):80–93. doi: 10.1158/2159-8290.CD-13-0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Whittaker SR, et al. A genome-scale RNA interference screen implicates NF1 loss in resistance to RAF inhibition. Cancer Discov. 2013;3(3):350–362. doi: 10.1158/2159-8290.CD-12-0470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaplon J, et al. A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature. 2013;498(7452):109–112. doi: 10.1038/nature12154. [DOI] [PubMed] [Google Scholar]
- 12.Haq R, et al. BCL2A1 is a lineage-specific antiapoptotic melanoma oncogene that confers resistance to BRAF inhibition. Proc Natl Acad Sci U S A. 2013;110(11):4321–4326. doi: 10.1073/pnas.1205575110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haq R, et al. Oncogenic BRAF regulates oxidative metabolism via PGC1α and MITF. Cancer Cell. 2013;23(3):302–315. doi: 10.1016/j.ccr.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abel EV, et al. Melanoma adapts to RAF/MEK inhibitors through FOXD3-mediated upregulation of ERBB3. J Clin Invest. 2013;123(5):2155–2168. doi: 10.1172/JCI65780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 16.Roesch A, et al. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell. 2010;141(4):583–594. doi: 10.1016/j.cell.2010.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roesch A, et al. Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1B(high) cells. Cancer Cell. 2013;23(6):811–825. doi: 10.1016/j.ccr.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vazquez F, et al. PGC1α expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell. 2013;23(3):287–301. doi: 10.1016/j.ccr.2012.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alvarez-Calderon F, et al. Tyrosine kinase inhibition in leukemia induces an altered metabolic state sensitive to mitochondrial perturbations. Clin Cancer Res. 2015;21(6):1360–1372. doi: 10.1158/1078-0432.CCR-14-2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kelly DP, Scarpulla RC. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev. 2004;18(4):357–368. doi: 10.1101/gad.1177604. [DOI] [PubMed] [Google Scholar]
- 21.Gleyzer N, Vercauteren K, Scarpulla RC. Control of mitochondrial transcription specificity factors (TFB1M and TFB2M) by nuclear respiratory factors (NRF-1 and NRF-2) and PGC-1 family coactivators. Mol Cell Biol. 2005;25(4):1354–1366. doi: 10.1128/MCB.25.4.1354-1366.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Di Re M, et al. The accessory subunit of mitochondrial DNA polymerase gamma determines the DNA content of mitochondrial nucleoids in human cultured cells. Nucleic Acids Res. 2009;37(17):5701–5713. doi: 10.1093/nar/gkp614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Supale S, et al. Loss of prohibitin induces mitochondrial damages altering β-cell function and survival and is responsible for gradual diabetes development. Diabetes. 2013;62(10):3488–3499. doi: 10.2337/db13-0152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shirendeb U, et al. Abnormal mitochondrial dynamics, mitochondrial loss and mutant huntingtin oligomers in Huntington’s disease: implications for selective neuronal damage. Hum Mol Genet. 2011;20(7):1438–1455. doi: 10.1093/hmg/ddr024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barretina J, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483(7391):603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kabbarah O, et al. Integrative genome comparison of primary and metastatic melanomas. PLoS One. 2010;5(5): doi: 10.1371/journal.pone.0010770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cancer Genome Atlas Network Genomic classification of cutaneous melanoma. Cell. 2015;161(7):1681–1696. doi: 10.1016/j.cell.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Litvin O, et al. Interferon α/β enhances the cytotoxic response of MEK inhibition in melanoma. Mol Cell. 2015;57(5):784–796. doi: 10.1016/j.molcel.2014.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kokoszka JE, Coskun P, Esposito LA, Wallace DC. Increased mitochondrial oxidative stress in the Sod2 (+/–) mouse results in the age-related decline of mitochondrial function culminating in increased apoptosis. Proc Natl Acad Sci U S A. 2001;98(5):2278–2283. doi: 10.1073/pnas.051627098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Viale A, et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature. 2014;514(7524):628–632. doi: 10.1038/nature13611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cheng MY, Hartl FU, Horwich AL. The mitochondrial chaperonin hsp60 is required for its own assembly. Nature. 1990;348(6300):455–458. doi: 10.1038/348455a0. [DOI] [PubMed] [Google Scholar]
- 32.Skrtic M, et al. Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell. 2011;20(5):674–688. doi: 10.1016/j.ccr.2011.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ekstrand MI, et al. Mitochondrial transcription factor A regulates mtDNA copy number in mammals. Hum Mol Genet. 2004;13(9):935–944. doi: 10.1093/hmg/ddh109. [DOI] [PubMed] [Google Scholar]
- 34.Kang BH, et al. Combinatorial drug design targeting multiple cancer signaling networks controlled by mitochondrial Hsp90. J Clin Invest. 2009;119(3):454–464. doi: 10.1172/JCI37613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chae YC, et al. Landscape of the mitochondrial Hsp90 metabolome in tumours. Nat Commun. 2013;4: doi: 10.1038/ncomms3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chae YC, et al. Control of tumor bioenergetics and survival stress signaling by mitochondrial HSP90s. Cancer Cell. 2012;22(3):331–344. doi: 10.1016/j.ccr.2012.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gopal YN, et al. Inhibition of mTORC1/2 overcomes resistance to MAPK pathway inhibitors mediated by PGC1alpha and oxidative phosphorylation in melanoma. Cancer Res. 2014;74(23):7037–7047. doi: 10.1158/0008-5472.CAN-14-1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lagadinou ED, et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell. 2013;12(3):329–341. doi: 10.1016/j.stem.2012.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.