

Abstract
Immune aging results in progressive loss of both protective immunity and T cell–mediated suppression, thereby conferring susceptibility to a combination of immunodeficiency and chronic inflammatory disease. Here, we determined that older individuals fail to generate immunosuppressive CD8+CCR7+ Tregs, a defect that is even more pronounced in the age-related vasculitic syndrome giant cell arteritis. In young, healthy individuals, CD8+CCR7+ Tregs are localized in T cell zones of secondary lymphoid organs, suppress activation and expansion of CD4 T cells by inhibiting the phosphorylation of membrane-proximal signaling molecules, and effectively inhibit proliferative expansion of CD4 T cells in vitro and in vivo. We identified deficiency of NADPH oxidase 2 (NOX2) as the molecular underpinning of CD8 Treg failure in the older individuals and in patients with giant cell arteritis. CD8 Tregs suppress by releasing exosomes that carry preassembled NOX2 membrane clusters and are taken up by CD4 T cells. Overexpression of NOX2 in aged CD8 Tregs promptly restored suppressive function. Together, our data support NOX2 as a critical component of the suppressive machinery of CD8 Tregs and suggest that repairing NOX2 deficiency in these cells may protect older individuals from tissue-destructive inflammatory disease, such as large-vessel vasculitis.
Introduction
The immune system has evolved to protect the host against pathogens and cancerous cells, while keeping tissue-damaging inflammation at a minimum (1). Tregs serve the imperative role of dampening and halting immune responses to prevent autoimmunity and chronic inflammation (1–3). Conversely, excess Treg activity weakens host protection against microorganisms and tumors (2–5). Understanding the details of Treg-mediated suppression may enable elegant immune-regulatory therapy, both in enhancing immunity and suppressing unwanted immune activity.
With progressive age, infections and autoimmune diseases prominently contribute to morbidity, particularly in vulnerable older humans, as both protective and regulatory immunity undergo aging-induced decline (6–8). Aging may affect the repertoire, frequency, subset distribution, and functional activity of Tregs (6, 9, 10). Tregs are developmentally and functionally heterogeneous and suppress through a variety of mechanisms (1, 5, 11). Enhancing antiinflammatory Treg activity with age could enable therapeutic management of inflammaging, a condition tightly linked to all-cause morbidity and mortality.
While CD4+FoxP3+ T cells are considered quintessential suppressor cells, their CD8 counterparts offer several advantages for therapeutic exploitation (12). CD4+FoxP3+ cells rapidly invade but insufficiently suppress peripheral inflammatory lesions (13). Conversely, CD8 Tregs interfere with immune responses in secondary lymphoid tissues (14, 15). CD8 Treg-mediated suppression affects T follicular helper cell expansion, enhances antitumor immunity, and curbs antiviral immune responses (16, 17). Adoptively transferred CD8 Tregs suppress collagen-induced arthritis and attenuate graft-versus-host disease (18, 19). The therapeutic exploitation of phenotypically and functionally diverse human CD8 Tregs has been hampered by insufficient knowledge of their mechanism of action (6, 20, 21).
Here, we have defined molecular mechanisms through which human CD8 Tregs contribute to immune homeostasis and have identified molecular defects underlying aging-related failure of CD8 Tregs. Human CD8 Tregs expressed CCR7, sought out the T cell zones of secondary lymphoid organs, and inhibited expansion of the CD4 T cell compartment. Their suppressive function relies on NADPH oxidase 2–derived (NOX2-derived) ROS. Upon activation, CD8 Tregs assembled bulky NOX2 membrane clusters and released NOX2-containing microvesicles. Once absorbed by contacting CD4 T cells, NOX2-derived ROS abrogates phosphorylation of the upstream signaling molecules ZAP70 and linker of activated T cells (LAT). CD8 Tregs from older donors, particularly those with the inflammatory vasculopathy giant cell arteritis (GCA), failed to upregulate NOX2, and NOX2 overexpression was sufficient to rescue their suppressive function. Implicating NOX2 in controlling T cell homeostasis and inflammaging defines the oxidase as a critical immune regulator and identifies a druggable target to correct uncontrolled inflammation in older individuals.
Results
CD8 Tregs localize to secondary lymphoid organs and suppress CD4 T cell activation.
To understand how CD8 Tregs affect immunity in humans, we traced CD8+FoxP3+ T cells in the blood, in tonsils, in lymph nodes, and in inflammatory infiltrates. Circulating CD8+FoxP3+ T cells accounted for 0.9% ± 0.6% of peripheral mononuclear cells (PBMCs) (n = 8 healthy donors) and expressed the lymphoid homing receptor CCR7 (Figure 1A). In human tonsils and lymph nodes, CD8+FoxP3+ localized to the T cell–rich zones surrounding germinal centers (Figure 1B and Supplemental Figure 1; supplemental material available online with this article; doi:10.1172/JCI84181DS1) but were explicitly infrequent in inflammatory synovitis and essentially undetectable in the tissue lesions of GCA (data not shown).
Figure 1. CD8 Tregs express CCR7, home to secondary lymphoid tissues, and suppress CD4 T cell activation.

(A) Expression of FoxP3 and CCR7 was analyzed within gated peripheral blood CD8 T cells. One representative dot blot from 7 healthy donors. (B) Frozen sections from human tonsils were stained with mouse anti-human FoxP3 and rabbit anti-human CD8 antibodies. CD8 T cells were visualized with Alexa Fluor 488 fluorescence-labeled goat anti-rabbit antibodies. FoxP3 was detected with peroxidase-labeled goat anti-mouse antibodies. Merged images demonstrate CD8+FoxP3+ T cells in T cell–rich zones. Images are representative for 4 different tissues. Scale bar: 20 μm. (C–G) CD8 Tregs were isolated directly from PBMCs or generated ex vivo. Noncultured, fresh CD8 T cells served as controls. Naive CD4 T cells were mixed with CD8 T cells (1:1 ratio) and activated with anti-CD3/CD28 beads. (C) pZAP70 in CD4 T cells was measured by flow cytometry. A representative example of pZAP70 expression after 5 minutes of stimulation in the presence and absence of CD8 Tregs. (D) Frequencies of pZAP70+ CD4 T cells were monitored over a time span of 30 minutes following activation. Results are shown as mean ± SD from 4 independent experiments. (E) Frequencies of pZAP70+ CD4 T cells were measured after 5 minutes of stimulation in 19 independent experiments (mean ± SD). (F) CD4 T cells were mixed with CD8 Tregs at different ratios, and pZAP70 in CD4 T cells was measured by flow cytometry. Date are mean ± SD from 4 independent experiments. (G) CD8+CD39+CD26– Tregs were sorted from peripheral blood and immediately tested for suppressive function by mixing them with CD4 T cells. Percentages of pZAP70+ CD4 T cells (mean ± SD) were measured by flow cytometry in 3 independent experiments. Unpaired 2-tailed Student’s t test was used for comparisons.
To characterize their mechanism of action, suppressive kinetics of ex vivo–generated CD8+CCR7+ Tregs (22) were analyzed. CD8 Tregs effectively prevented ZAP70 phosphorylation (pZAP70) in neighboring CD4 T cells that were undergoing T cell receptor–mediated (TCR-mediated) activation (Figure 1, C and D). Suppression was fast, reaching maximal effect within 5 minutes. About 60% of TCR-dependent pZAP70 was sensitive to suppression (Figure 1E). In contrast, freshly purified CD8 T cells could not inhibit activation of CD4 T cells (Figure 1, D and E). Suppressive activity was maintained even when one CD8 Treg was mixed with 8 CD4 target cells (Figure 1F), suggesting that one Treg may interact with several CD4 T cells.
To test whether naturally occurring CD8+CCR7+FoxP3+ cells share suppressive function with ex vivo–induced CD8+CCR7+ Tregs, we devised a strategy to sort CD8+CCR7+FoxP3+ cells directly from the blood. Treg function has been associated with expression of the ectonucleoside triphosphate diphosphohydrolase 1 (known as CD39) (23), which hydrolyzes ATP/ADP to AMP. Since CD39 is also upregulated on activated cells, we searched for additional markers expressed on CD8+FoxP3+ cells. We confirmed that CD26, the adenosine deaminase complexing protein 2, was absent on CD8+FoxP3+ cells (24). We therefore sorted CD8+CD39+CD26– cells and tested them immediately for their suppressive capacity (Figure 1G). Freshly isolated CD8+CD39+CD26– cells were effective suppressor cells, comparable to ex vivo–induced CD8+CCR7+ Tregs.
CD8 Tregs suppress the proliferative turnover and expansion of CD4 T cells.
T-T cell suppression involved close membrane contact between both partners. CD8 Tregs reorganized CD3 molecules into an immune synapse at the CD8-CD4 interface (Figure 2A). Of interest, the CD4-CD8 Treg interaction had lasting consequences for CD4 T cells. After CD8 Tregs were removed, CD4 T cells did not recover for several days, and proliferative expansion remained impaired (Figure 2, B and C), reaching only 50% to 60% of that in control cultures. Thus, the suppressive mechanism of CD8 Tregs relies on an extremely early, membrane-proximal event, targeting the TCR activation cascade upstream of ZAP70 with long-lasting effects.
Figure 2. CD8 Tregs regulate CD4 T cell expansion in vitro and in vivo.

(A) CD8 Tregs form an immune synapse when contacting CD4 T cells. CD8 Tregs were labeled with CellTracker Red, mixed with naive CD4 T cells (1:1 ratio), and stimulated with anti-CD3/CD28 beads. Cells were fixed, permeabilized, stained with Alexa Fluor 488–labeled anti-CD3 antibody, and analyzed by microscopy. Two representative samples are shown. Scale bar: 5 μm. (B and C) The suppressive effect of CD8 Tregs is long lasting. Naive CD4 T cells were CFSE labeled and incubated with or without CD8 Tregs (1:1 ratio) for 30 minutes. CD8 Tregs were removed with magnetic beads, CD4 T cells were stimulated with anti-CD3/CD28 beads for 4 days, and proliferation was analyzed by assessing CFSE dilution with flow cytometry. Data are mean ± SD from 3 independent experiments. (D–F) CD8 Tregs were adoptively transferred into NSG mice that were reconstituted with naive CD4 T cells and monocytes. Proliferative expansion of CD4 T cells was quantified after 9 days by enumerating the frequency and number of human CD4 T cells in the murine spleen by flow cytometry. CD4 and CD8 T cells were derived from the same human donor. Results from 5 independent experiments examining 5 different donors are shown as mean ± SD. (G–I) NSG mice were reconstituted with CD4 T cells, monocytes, and CD8 Tregs, as above. CD4 T cells were CFSE labeled prior to the transfer. Proliferation of CD4 T cells was assessed by flow cytometric analysis of CFSE dilution in splenocytes harvested after 9 days. A representative CFSE dilution curve is shown, and results from 5 independent experiments are summarized as mean ± SD of division index and proliferation index. Unpaired 2-tailed Student’s t test was used for comparisons.
To test in vivo functions of CD8 Tregs, NSG mice were reconstituted with naive human CD4 T cells and autologous CD8 Tregs at a 1:1 ratio. In such NSG chimeras, human T cells form organized lymphoid structures in the murine spleen. On day 9 after reconstitution, human CD4 T cells in the spleen amounted to 100 × 103 to 200 × 103 cells, but expansion of these cells was suppressed in mice that had also received CD8 Tregs. Cotransferred CD8 T cells freshly isolated from PBMCs had no effect (Figure 2, D–F). Proliferative behavior of CD4 T cells was directly assessed by CFSE dilution assays (Figure 2, G–I). The division and proliferation indices of CD4 T cells were strongly influenced by CD8 Tregs, which considerably slowed down the turnover of CD4 T cells in the host.
Defective suppressive activity of CD8 Tregs in aging individuals.
Older individuals are at risk for inflammaging, a process involving systemic inflammation and tissue-destructive inflammatory disease. Inflammatory biomarkers steadily increase with progressing age and, in the oldest inflammatory cytokines, are excellent predictors of death (25). Young (<30 years) and older healthy individuals (>60 years) have similar frequencies of circulating CD8+CCR7+FoxP3+ Tregs (Supplemental Figure 2). However, ex vivo–generated CD8 Tregs from older donors are much less efficient at suppressing CD4 T cell activation (Figure 3A), having lost almost all of their suppressive ability. At a ratio of CD8 Tregs to CD4 target cells of 1:4, old CD8 Tregs were about half as efficient as young counterparts (Figure 3B). At a 1:8 ratio, old CD8 Tregs were essentially unable to suppress. CD8+CD39+CD26– cells sorted out of the blood of young and older donors yielded similar results (Figure 3C). Only high numbers of old CD8+CD39+CD26– cells were able to inhibit the activation process in CD4 T cells.
Figure 3. The suppressive function of CD8 Tregs is deficient in older individuals.

(A) CD8 Tregs induced from young (age < 30 yrs) and older (age > 60 yrs) healthy individuals were tested for their ability to suppress pZAP70 in naive CD4 T cells 10 minutes after stimulation. Results are shown as mean ± SD MFI from 9 independent experiments. (B) CD8 Tregs generated from young and older subjects were mixed with CD4 T cells at increasing ratios. pZAP70 in CD4 T cells was analyzed by flow cytometry. Results are shown as mean ± SD from 4 independent experiments. (C) CD8+CD39+CD26– Tregs were sorted from peripheral blood and immediately assessed for their suppressive activity by mixing them at increasing ratios with CD4 T cells. pZAP70 in CD4 T cells was measured after 10 minutes stimulation. Results are shown as mean ± SD from 6 independent experiments. (D and E) NSG mice were reconstituted with naive CD4 T cells, monocytes, and CD8 Tregs generated from young or older donors. Nine days later, expansion of CD4 T cells was quantified by enumerating the (D) frequency and (E) number of human CD4 T cells in the murine spleen. Results from 7 independent experiments are shown as box-and-whisker plots. Boxes represent the 25th and 75th percentile, and lines within the boxes are medians. The 10th and 90th percentiles are shown as whiskers. (F–H) CD4 T cells were labeled with CFSE prior to the transfer. Proliferation of CD4 T cells was assessed by flow cytometric analysis of CFSE dilution in splenocytes harvested after 9 days. (F) A representative image is shown, and results from 6 independent experiments are summarized as box-and-whisker plots of (G) division index and (H) proliferation index. Unpaired 2-tailed Student’s t test was used for comparisons.
To compare CD8 Tregs from young and older donors in vivo, NSG mice were reconstituted with naive CD4 T cells plus CD8 Tregs from young or older donors. Numbers and proliferation of human CD4 T cells within the murine spleen were measured on day 9 after reconstitution. CD4 T cells cotransferred with young CD8 Tregs were minimally expanded but grew to a 4-fold larger size if the accompanying CD8 Tregs derived from healthy older individuals (Figure 3, D and E). Monitoring in vivo CD4 proliferation through CFSE dilution studies confirmed the difference in suppressive capacity of young and old CD8 Tregs (Figure 3, F–H). CD8 Tregs originating from older subjects had essentially lost the ability to suppress CD4 T cell proliferation in vivo.
CD8 Tregs from older individuals are NOX2 deficient.
Guided by pilot experiments that demonstrated sensitivity of the suppressive mechanism to ROS scavenging, we proceeded with targeted gene expression profiling for ROS-generating enzymes in young and old CD8 Tregs. These experiments identified NOX2 as a gene of interest. CD8 Tregs expressed low levels of the NOX isoforms 1, 3, 4, and 5 and DUOX1 and moderate levels of DUOX2, but they expressed abundant concentrations of NOX2 transcripts (Figure 4A). Ex vivo CD8 Treg induction was accompanied by robust NOX2 upregulation (Figure 4B), which was markedly higher in CD8 Tregs from young individuals. To seek confirmation in vivo, NOX2 expression on CD8+CCR7+ Tregs in the peripheral blood of young and older healthy subjects was compared. Frequencies of NOX2-expressing CD8+CCR7+ Tregs were notably low in the older individuals (Figure 4, C and D), and expression intensity of NOX2 was reduced by more than 50% (Figure 4E). These data suggested that NOX2 expression is a biomarker for CD8 Tregs.
Figure 4. NOX2 insufficiency in CD8 Tregs from older individuals.

(A) CD8+ CCR7+ Tregs were induced ex vivo and profiled for the expression of the NADPH oxidases NOX1, NOX2, NOX3, NOX4, NOX5, DUOX1, and DUOX2 by RT-PCR. Results from 6 donors are shown as a heat map. (B) Expression of NOX2-specific transcripts in CD8 Tregs derived from either young or older individuals was compared by RT-PCR. Results (mean ± SD) are from 4 young-old donor pairs. (C) Cellular expression of NOX2 was analyzed in gated CD8+FoxP3+CCR7+ T cells in the peripheral blood of healthy young or older donors, patients with GCA with or without treatment, middle-aged and older patients with PsA, and middle-aged patients with SVV. One representative data set is shown. (D and E) Cellular expression of NOX2 in gated CD8+CCR7+ Tregs was determined in the peripheral blood of young healthy individuals (n = 7), older healthy individuals (n = 10), patients with GCA without treatment (n = 7), patients with GCA with treatment (n = 6), middle-aged patients with PsA (n = 5), older patients with PsA (n = 5), and middle-aged patients with SVV (n = 9). Results are shown as mean ± SD of (D) frequencies and (E) MFI of cell surface expression. Unpaired 2-tailed Student’s t test was used for comparisons.
To evaluate whether systemic inflammatory disease in older individuals is reflected in CD8 Treg availability, we examined a cohort of patients with GCA. GCA occurs exclusively in the 6th to 9th decade of life and manifests with an inflammatory attack on medium and large arteries (26, 27). NOX2+CD8+CCR7+ Tregs were distinctly infrequent in patients with biopsy-proven GCA (Figure 4, C–E). Loss of NOX2+CD8 Tregs in patients with GCA was independent of treatment; untreated and steroid-treated patients both had the defect, and treatment-induced reversal of the systemic inflammatory syndrome was insufficient to reverse the deficiency for CD8+CCR7+NOX2+ cells. To test whether CD8 Treg deficiency is a feature of all inflammatory diseases, we recruited middle-aged and older patients with psoriatic arthritis (PsA), an aggressive inflammatory syndrome of the skin and the joints (28). Middle-aged patients with PsA were able to generate NOX2-expressing CD8 Tregs, and patients with PsA over 60 years of age were indistinguishable from healthy older individuals (Figure 4, C–E). To explore whether the inability to generate NOX2+CD8 Tregs is shared among all types of vasculitic syndromes, we examined patients with small-vessel vasculitis (SVV). Frequencies of NOX2+CD8 Tregs were age appropriate in patients with SVV. Overall, young healthy individuals expressed NOX2 on 40% to 50% of their CD8+CCR7+ Tregs, healthy older donors expressed NOX2 on 23% of their CD8+CCR7+ Tregs, middle-aged patients with PsA expressed NOX2 on 45% of their CD8+CCR7+ Tregs, older patients with PsA expressed NOX2 on 26% of their CD8+CCR7+ Tregs, middle-aged patients with SVV expressed NOX2 on 46% of their CD8+CCR7+ Tregs, and patients with GCA expressed NOX2 on only 6% of their CD8+CCR7+ Tregs (Figure 4, C–E).
Thus, CD8 Treg functionality is closely correlated with the induction of membrane NOX2. Poorly suppressive CD8 Tregs derived from the older individuals are typically NOX2 deficient, and patients with GCA essentially lack NOX2+ CD8 Tregs. This loss of NOX2+CD8 Tregs appears to be disease specific and not solely a result of systemic inflammation.
CD8 Treg-mediated suppression requires ROS production and NOX function.
The superoxide-generating enzyme NOX2 is a critical component of the microbicidal defense machinery in phagocytes (29, 30), but it also mediates tissue damage by ROS production. When loaded with oxidation-sensitive fluorogenic probes, CD8 Tregs and nonsuppressive, freshly isolated CD8 T cells could be clearly distinguished in that only CD8 Tregs were able to produce high levels of intracellular ROS (Figure 5, A and B). To examine whether ROS production has a direct impact on their suppressive activity, CD8 Tregs were pretreated with the SOD mimetic tempol. ROS scavenging effectively rescued CD4 responsiveness (Figure 5, C and D), directly implicating ROS generation in T-T cell suppression. ROS originate from different cellular sources, with mitochondria and dedicated NADPH oxidases being the major suppliers (31). Pretreatment with neither rotenone nor mitoTEMPO rescued pZAP70 in the CD4 T cells (Figure 5, E and F), eliminating mitochondrial ROS as a functional element in the CD8-mediated suppressive machinery.
Figure 5. CD8 Treg-mediated suppression requires ROS production and NOX2 function.

(A and B) CD8 Tregs were loaded with oxidation-sensitive CellROX and analyzed by flow cytometry to detect ROS levels. Fresh CD8 T cells from the same donor were used as controls. (A) Representative histograms and (B) a summary of 5 independent experiments (mean ± SD) are shown. (C and D) CD8 Tregs were pretreated with or without the ROS scavenger tempol, mixed with naive CD4 T cells, and stimulated with anti-CD3/CD28 beads. pZAP70 in CD4 T cells was analyzed. (C) One representative dot blot and (D) results (mean ± SD) from 3 independent experiments are shown. (E) CD8 Tregs were pretreated with the mitochondrial complex I inhibitor rotenone (ROT) or (F) the mitochondrial ROS scavenger mitoTEMPO (mito-Temp) and tested for their suppressive activity by mixing them with naive CD4 T cells and quantifying pZAP70 after anti-CD3/CD28 stimulation. Frequencies of pZAP70+ CD4 T cells (mean ± SD) in 5 independent experiments are shown. (G) CD8 Tregs were pretreated with the NADPH oxidase inhibitor DPI or (H) gp91ds-tat, an inhibitor of the NADPH oxidase complex assembly, or (I) were transfected with NOX2 shRNA or control shRNA. CD8 Tregs were tested for their suppressive activity in a CD4-CD8 coculture assay by quantifying pZAP70 in CD4 T cells. Results are from (G and H) 4 and (I) 5 independent experiments and are presented as mean ± SD. (J and K) CD8 Tregs were pretreated with DPI, gp91ds-tat, or NOX2 knockdown, as above, and their suppressive activity was assessed in CD4-CD8 coculture assays. CD4 activation was quantified by measuring the phosphorylation of the signaling molecule LAT. Results are from 3 independent experiments (mean ± SD). Unpaired 2-tailed Student’s t test was used for comparisons.
To test whether NADPH oxidase is critically important for enabling CD8 Tregs to function as suppressor cells, CD8 Tregs were pretreated with the NADPH oxidase inhibitor diphenyleneiodonium (DPI). Disabling NOX-dependent ROS production essentially depleted all suppressive activity (Figure 5G). Functional NOX2 requires assembly of a hetero-oligomeric membrane complex, which can be inhibited by gp91ds-tat peptide (32). Pretreatment of CD8 Tregs with gp91ds-tat dampened suppressor function (Figure 5H). Finally, reducing NOX2 expression via interfering RNA was sufficient to significantly impair suppressive capability (Figure 5I), confirming NOX2 as a crucial effector in CD8 Treg-mediated suppression.
To understand how NOX2 contributes to the overall ROS production in CD8 Tregs and whether other oxidases could possibly be part of the suppressive mechanism, we compared ROS levels in CD8 Tregs following three interventions: shRNA-mediated NOX2 knockdown, treatment with DPI, or treatment with gp91ds-tat. To mimic conditions in young versus old CD8 Tregs (Figure 4), we aimed at reducing NOX2 transcript levels to about 50%. This reduction of NOX2 transcripts translated into a decline of intracellular ROS levels by 60% (Supplemental Figure 3). DPI pretreatment was the most effective in lowering ROS levels (Supplemental Figure 4), eliminating 80% to 90% of ROS and abrogating the suppressive activity to a similar degree. Almost all ROS generation in CD8 Tregs that was sensitive to DPI was also sensitive to gp91ds-tat treatment, identifying NOX2 as the major source of ROS production in these cells (Supplemental Figure 4). We used a second readout parameter to confirm that CD8 Treg-mediated suppression of CD4 T cell activation is ROS and NOX2 dependent (Figure 5, J and K). TCR triggering initiates LAT phosphorylation by the ZAP70 protein kinase, and phosphorylated LAT functions as a docking site for the assembly of multimolecular complexes in the TCR signaling cascade. Untreated CD8 Tregs efficiently reduced LAT phosphorylation accumulation in CD4 T cells. Suppression was impaired after pretreatment of CD8 Tregs with either gp91ds or NOX2 knockdown. DPI pretreatment essentially abrogated the inhibitory capacity of CD8 Tregs.
To examine whether interfering with NOX function was effective in altering downstream effects of the CD8 Treg–CD4 T cell interaction, we quantified proliferative expansion of CD4 T cells 4 days after contact with CD8 Tregs in a CFSE dilution assay (Supplemental Figure 5). DPI-pretreated CD8 Tregs lost almost all of their ability to stop CD4 T cells from clonal expansion. Proliferative potential was restored in CD4 T cells cocultured with CD8 Tregs pretreated with gp91ds-tat or NOX2 shRNA.
CD8 Tregs function by transferring NOX2 onto CD4 T cells.
Immunostaining for NOX2/gp91 revealed that resting and stimulated CD4 T cells are essentially negative for NOX2 expression (Figure 6A). In contrast, the oxidase is abundantly expressed in the membranes of resting and activated CD8 Tregs (Figure 6A). Stimulation resulted in a striking redistribution of membrane NOX2. About 60% of resting CD8 Tregs had a circumferential NOX2 expression pattern. Upon activation, CD8 Tregs organized NOX2 into membrane clusters (Figure 6, A and B). Activation-induced clustering of membrane-integrated NOX2 caused a marked increase of staining intensity (Figure 6C). Membrane NOX2 expression was an exclusive feature of CD8 Tregs and was not shared by unselected resting and activated CD8 T cells (Figure 6A). To understand whether NOX is universally encountered on Treg populations, we analyzed CD4+FoxP3+ cells in healthy individuals (Supplemental Figure 6). Only a small proportion (10%–15%) of CD4 Tregs had a low signal for NOX2 expression, identifying NOX2 as a CD8 Treg marker.
Figure 6. CD8 Tregs function by transferring NOX2 onto CD4 T cells.

(A–C) Resting and activated CD8 Tregs, naive CD4 T cells, and freshly isolated total CD8 T cells were stained with anti-NOX2/gp91phox. Antibody binding was visualized with Alexa Fluor 488–labeled anti-rabbit antibody. (A) Representative images are shown. Scale bar: 5 μm. (B) Activation of CD8 Tregs induces clustering of membrane NOX2. A minimum of 45 cells from each of 3 independent experiments was analyzed for the frequency of CD8 Tregs with circumferential versus clustered NOX2 expression patterns. (C) Activation of CD8 Tregs increases the intensity of membrane NOX2 expression. CD8 Tregs were stained with anti-NOX2/gp91phox antibodies before and after stimulation. Staining intensity is expressed as pixel numbers. Each dot represents the data from a single cell. Unpaired 2-tailed Student’s t test was used for comparisons. (D) CD8 Tregs were stained with NOX2/gp91phox and subsequently cocultured with naive CD4 T cells (marked with CellTracker Red) for 60 minutes with (anti-CD3/CD28 beads) or without stimulation. Live-cell imaging demonstrates NOX2/gp91phox on the CD8 Tregs distributed in membrane-integrated clusters. Small clusters of NOX2/gp91phox were transferred to CD4 T cells, where they appeared as yellow dots (CellTracker Red overlaying green NOX2/gp91phox). Transfer of NOX2/gp91phox requires activation of the CD8 Tregs. Representative images are from 3 independent experiments. Scale bar: 5 μm.
Live-cell imaging demonstrated the transport of NOX2 clusters from CD8 Tregs to contacting CD4 T cells (Figure 6D). Membrane-residing NOX2 in CD8 Tregs was stained with anti-NOX2/gp91 antibody, and CD4 T cells were labeled with CellTracker Red. TCR stimulation promptly induced NOX2 clustering and transfer of NOX2-containing clusters from the CD8 Tregs to the CD4 T cells. Such transfer occurred only if the CD8 Tregs were attached to stimulatory anti-CD3/CD28 beads (Figure 6D).
CD8 Tregs produce NOX2-containing exosomes.
To elucidate mechanisms underlying the shuffling of membrane NOX2 in CD4 T cell–CD8 Treg communication, we monitored the evolution of immune synapse formation (Figure 2A) between the two partners. These experiments revealed a transfer of labeled cell membrane originating from CD8 Tregs to contacting CD4 T cells. We consistently observed small fragments of green PKH67 (used to label CD8 Tregs) on CellTracker Red–labeled CD4 T cells, creating a yellow signal (Figure 7A). In some cases, larger green membrane fragments were moved from the CD8 Tregs to the CD4 T cells (Figure 7A). The inverse event of finding yellow membrane particles on the green CD8 Tregs did not occur. Capture of membrane particles by CD4 T cells was confirmed by flow cytometry (Figure 7B). CD4 T cells incubated in the absence of CD8 Tregs were negative for membrane-integrated PKH67, while coincubation of CD4 T cells and PKH67-labeled CD8 Tregs consistently resulted in “chimeric” CD4 T cells, carrying PKH67 stain (Figure 7B). Within 1 hour, almost 30% of the CD4 T cells had absorbed detectable PKH67 label from CD8 Tregs (Figure 7C).
Figure 7. CD4 T cells uptake NOX2-containing exosomes released by CD8 Tregs.

(A) PKH67-labeled CD8 Tregs (green) and CellTracker Red–labeled naive CD4 T cells (red) were mixed for 60 minutes. Transfer of PKH67-labeled membranes from CD8 Tregs into CD4 T cells (yellow) was observed by live-cell microscopy. Scale bar: 5 μm. (B) Uptake of PKH67-labeled membrane fragments by CD4 T cells was analyzed by flow cytometry. A representative histogram of CD4 T cells that had or had not been in contact with PKH67-labeled CD8 Tregs cells is shown. (C) Frequencies of CD4 T cells that had taken up PKH67-labeled membrane fragments from CD8 Tregs. Results are from 3 independent experiments (mean ± SD). (D and E) Exosomes were isolated from the supernatant of PKH67-labeled CD8 Tregs and added into the culture of naive CD4 T cells at the indicated ratios for 60 minutes. The frequency of CD4 T cells that had taken up PKH67-containing exosomes was determined by flow cytometry. Results are presented as mean ± SD and are from 5 independent experiments. (F and G) Exosomes isolated from the supernatant of CD8 Tregs were stained with anti-human NOX2/gp91phox antibody and analyzed by flow cytometry. One representative image and results from 4 independent experiments (mean ± SD) are shown. (H and I) CD8 Tregs and CD8 Treg-derived exosomes were loaded with oxidation-sensitive fluorogens and analyzed by flow cytometry to detect ROS levels. Fresh CD8 T cells from the same donor were used as controls. Representative histograms and a summary of 7 independent experiments (mean ± SD) are shown. Unpaired 2-tailed Student’s t test was used for comparisons.
To explore whether only intact CD8 Tregs could transfer membrane to CD4 T cells, exosomes were produced and purified from the supernatant of PKH67-labeled CD8 Tregs. Incubation of CD4 T cells with such exosomes at ratios of 1:250 and 1:2,500 was sufficient to stably integrate exosomes into the recipient CD4 T cells. With the lower dose of exosomes, 8%–9% of CD4 T cells took up PKH67-containing membrane. Higher exosome doses rendered 15%–17% of CD4 T cells positive for a PKH67 signal (Figure 7, D and E). Flow cytometry demonstrated that exosomes generated from CD8 Tregs stained strongly positive for NOX2/gp91 and contained high levels of ROS (Figure 7, F–I). In contrast, CD8 Treg-derived exosomes lacked mitochondrial 12S rRNA (Supplemental Figure 7).
CD8 Treg-derived exosomes function through ROS production.
Functional activity of transferred exosomes was tested in suppression assays using pZAP70 as a readout, as above (Figure 8, A and B). Preincubation of CD4 T cells with CD8-derived exosomes effectively inhibited pZAP70. CD8 Treg-derived exosomes suppressed CD4 T cell activation in a dose-dependent manner (Figure 8B). Pretreatment of the CD8 Treg-derived exosomes with the NOX inhibitor DPI abrogated CD4 T cell suppression (Figure 8C), whereas gp91ds-pretreated exosomes retained their inhibitory function (Figure 8D). Incubation of CD4 T cells with CD8 Treg-derived exosomes lead to a measurable increase of ROS levels within the CD4 T cells (Supplemental Figure 8)
Figure 8. The suppressive function of CD8 Treg-derived exosomes depends on ROS production.

(A) Naive CD4 T cells were cultured with or without CD8 Treg-derived exosomes (Exo, 1:2,500 ratio) and stimulated with anti-CD3/CD28 beads. pZAP70 in CD4 T cells was analyzed by flow cytometry. A representative dot blot is shown. (B) Naive CD4 T cells were incubated with either CD8 Treg-derived exosomes or intact CD8 Tregs and stimulated with anti-CD3/CD28 beads. pZAP70 in CD4 T cells was analyzed. Data are presented as mean ± SD from 5 independent experiments. (C and D) Exosomes were generated from CD8 Tregs, as above; pretreated with (C) DPI or (D) gp91ds-tat; and incubated with naive CD4 T cells to test for suppressive activity. Frequencies of pZAP70+ CD4 T cells were measured by flow cytometry. Results (mean ± SD) are from (C) 5 and (D) 3 independent experiments. (E) CD8 Tregs were cultured with or without the membrane transport inhibitor brefeldin A (BFA), mixed with naive CD4 T cells, and then stimulated with anti-CD3/CD28 beads. TCR signaling within CD4 cells was quantified by flow cytometry analysis of pZAP70. Results from 5 independent experiments are presented as mean ± SD. For comparisons, trend test was used in B (P < 0.001) and unpaired 2-tailed Student’s t test was used in C–E.
To examine whether CD8 Tregs actively released exosomes, the membrane transport inhibitor brefeldin A was added to the cells (refs. 33, 34, and Figure 8E). Brefeldin A–treated CD8 Tregs could no longer inhibit the TCR signaling cascade in CD4 T cells.
In essence, CD8 Tregs function by transferring exosomes to neighboring T cells, which effectively disrupts the TCR-dependent signaling cascade.
Forced overexpression of NOX2 rescues the suppressive activity of CD8 Tregs.
If NOX2 deficiency is the primary defect of CD8 Tregs from older individuals, then modulating enzyme expression could be developed into a druggable approach for inflammaging. Transfection of old CD8 Tregs with a NOX2 expression vector corrected the low surface expression (Figure 9, A and B) and forced overexpression. Restoring NOX2 surface expression was sufficient to rescue the suppressive activity of age-impaired CD8 Tregs (Figure 9C). In 10 independent experiments, forced NOX2 overexpression repaired the suppressive capacity of old CD8 Tregs, rendering them equally effective as CD8 Tregs generated from the young (Figure 1E and Figure 9C).
Figure 9. Overexpression of NOX2 rescues the suppressive activity of old CD8 Tregs.

CD8 Tregs were induced from older individuals and transfected with a control or NOX2 expression vector. (A and B) Surface expression of NOX2 was evaluated 24 hours after transfection by flow cytometry. (A) One representative image and (B) results (mean ± SD) from 3 independent experiments are shown. (C) CD8 Tregs transfected with NOX2 expression vector or a control vector were analyzed for their suppressive function by mixing them with naive CD4 T cells and quantifying stimulation-induced pZAP70. Frequencies of pZAP70+ CD4 T cells (mean ± SD) from 10 independent experiments are shown. (D and E) Old CD8 Tregs were transfected with a control or NOX2 expression vector and tested for in vivo–suppressive activity by injecting them into NSG mice that had been reconstituted with naive CD4 T cells and monocytes. Nine days after transfer, expansion of CD4 T cells was quantified by enumerating the frequency and total numbers of human CD4 T cells in the murine spleen. Results are shown as mean ± SD from 6 independent experiments. (F–H) Old transfected CD8 Tregs were examined for in vivo function, as above. CD4 T cells were labeled with CFSE prior to the transfer. Proliferation of CD4 T cells was assessed by flow cytometric analysis of CFSE dilution in splenocytes harvested after 9 days. A representative image is shown, and results from 6 independent experiments are summarized as (mean ± SD) division index and proliferation index. Unpaired 2-tailed Student’s t test was used for comparisons.
To determine whether upregulation of NOX2 was sufficient to rebuild the regulatory function of CD8 Tregs in vivo, NOX2-transfected CD8 Tregs originating from older subjects were adoptively transferred into chimeric mice reconstituted with CD4 T cells. Expansion of CD4 T cells in the murine spleen was effectively suppressed by the NOX2-transfected Tregs (Figure 9, D and E), and homeostatic proliferation of CD4 T cells was effectively inhibited in the presence of NOX2-overexpressing CD8 Tregs (Figure 9, F–H). NOX2 overexpression was sufficient to rescue suppressive function, producing division and proliferation indices encountered with highly efficient young CD8 Tregs (Figure 3, G and H, and Figure 9, G and H). In essence, the in vivo and ex vivo defects of old CD8 Tregs are correctable by enforcing sufficient expression of membrane NOX2, highlighting the critical role of the enzyme in Treg function and pinpointing the aging-related Treg deficiency to NOX2 regulation.
Discussion
Like proimmune T cells, Tregs are equally subject to immune aging, leaving the aging host with an inflammation-biased immune system susceptible to inflammatory disease. Older individuals fail to generate functionally intact CD8 Tregs, a defect particularly pronounced in patients with the age-dependent inflammatory vasculopathy GCA. The molecular defect lies in the inefficient induction of NADPH oxidase, a critical element in the suppressive machinery of CD8 Tregs, which function by shedding NOX2-containing exosomes that are taken up by interacting CD4 T cells. The transfer of NADPH oxidase is fast, effective, and produces lasting effects, allowing CD8 Tregs to fundamentally shape the intensity of CD4 T cell responses. Overexpression of NOX2 reinstates the suppressive activity of old CD8 Tregs, identifying the oxidase as a potential therapeutic target in restoring immune homeostasis in the older individuals.
Transferring membrane NOX2 to regulate activation of neighboring T cells is a novel mechanism of T-T cell suppression to our knowledge. Listed below are known mechanisms of Treg-mediated suppression. Tregs release inhibitory cytokines (IL-10, TGF-β, IL-35) (5), use granzyme B and perforin as effector molecules (5, 35, 36), and kill target cells via granzyme A (5, 37). Tregs express the ectoenzymes CD39 and CD73 to generate pericellular adenosine, a strong suppressor of adenosine A2A receptor–expressing effector T cells (23, 38, 39). TIGIT+ Tregs inhibit proinflammatory Th1 and Th17 cell responses through fibrinogen-like protein 2 (40). Alternatively, Tregs can downregulate the costimulatory molecules CD80 and CD86 on DCs (5, 41) or condition DCs to express indoleamine 2,3-dioxygenase, a potent inducer of tryptophan catabolism (42, 43).
In the current study, we have pinpointed extracellular vesicle transfer as a highly efficient means of T-T cell communication. Extracellular vesicles, including exosomes and microvesicles, participate in intercellular communication through transferring proteins, lipids, and RNAs (44, 45). Here, the suppressive principle could be linked to ROS activity, empowering CD8 Tregs to rapidly disable CD4 cells in their immediate environment. Live-cell microscopy studies indicated that transfer of a small-membrane particle was sufficient to lastingly inhibit the TCR activation network in CD4 T cells. CD8 Tregs have a large cell size and carry multiple NADPH clusters on their surface, enabling them to regulate numerous CD4 T cells. Accordingly, even at a ratio of 1 suppressor cell to 8 effector cells, the CD8 Tregs were highly efficient in inhibiting surrounding CD4 T cells.
The NADPH oxidase complex catalyzes electron transfer from NADPH to molecular oxygen and thus generates superoxide, the precursor of H2O2 and other ROS (29, 46). Genetic defects in NADPH oxidase cause chronic granulomatous disease, an inherited primary immunodeficiency associated with autoinflammation and autoimmunity (47–49). Nox2–/– mice are prone to hyperinflammatory responses and spontaneously develop arthritis (50). NOX2-dependent ROS production in DCs negatively regulates IL-12 expression by constraining p38-MAPK activity (51). Adoptive transfer of NOX2-deficient CD4 T cells into RAG-deficient mice increases arthritic inflammation (50), suggesting that CD4 T cells are subject to the antiinflammatory effect of NOX2. Deletion of NOX2/gp91phox can limit the suppressive function of granulocytic myeloid–derived suppressor cells and contribute to antitumor effects (52, 53). In studies of mice deficient in Ncf1 (also known as p47phox), both the CD4 Treg and targeted T effector cell require functional p47phox for optimal suppression (54). Cumulatively, loss of NADPH oxidase worsens inflammation, identifying the enzyme as an immunosuppressive mediator, which stands in contrast to the concept that ROS-imposed oxidative stress gives rise to injury-induced inflammation. A common mechanism may lie in preventing immune cell activation, as ROS can rapidly inactivate kinases and phosphatases that are necessary to propagate cellular signaling pathways (55, 56). In support, NOX2 overexpression in CD4 T cells efficiently abrogates their activation in response to TCR signaling (Supplemental Figure 9). Data reported here identify NADPH oxidase as a highly effective immune modulator, particularly when delivered in membrane fragments that are easily absorbed by CD4 T cells.
The suppressive mechanism used by CD8 Tregs is strongly dependent on ROS (Figure 5). We did consider the possibility that ROS sources other than NOX contribute to the T-T cell suppression. With almost all intracellular ROS sensitive to DPI or gp91ds-tat, NOX2 was identified as by far the most important ROS generator in intact CD8 Tregs (Supplemental Figure 4). Remarkably, part of CD8 Treg-mediated suppression was preserved after gp91ds-tat treatment, and the suppressive activity of CD8 Treg-derived exosomes was unaffected by gp91ds-tat. These data support the concept that multiunit NOX2 complexes in exosomes are firmly assembled and long-lived, whereas such complexes are constantly reassembled in intact cells.
Data presented here demonstrate that the upregulation of NOX2 membrane clusters is a selective feature of CD8 Tregs, providing an excellent biomarker for this functional T cell subset. Besides its usefulness as a biomarker, the oxidase is also directly involved in the inhibitory function of CD8 Tregs. Further studies are needed to understand how NOX2 expression in CD8 Tregs is regulated. Available data are limited but suggest that transcription factors, coactivators/corepressors, and nuclear receptors all participate in this process. Epigenetic regulation is now surfacing as an important mechanism in governing NOX function in cardiovascular disease (57). Proinflammatory transcription factors, such as NF-κB, activator protein 1 (AP-1), and members of the STAT family, have been implicated in inducing NOX expression and thus oxidative stress (58–61). In addition, NOX expression is subject to negative regulation involving members of the PPAR family (62).
Most of the interest in NOX expression has been related to attempts to avoid oxidative stress; the current project sheds light on a beneficial immunoregulatory role of NOX2. Failure of NOX2 expression in CD8 T cells is obviously detrimental to the host, who is increasingly susceptible to this defect with progressive age. CD8 T cells are known to age faster than their CD4 counterparts (63). Given the expression of CCR7, the naive CD8 T cell compartment may actually contain the precursor cell for CD8 Tregs. Aging imposes particular stress on the naive CD8 compartment, with multiple mechanisms converging to impair CD8 immunity in the elderly (64).
In summary, CD8 Tregs mediate their immunosuppressive functions by directly inhibiting membrane-proximal signaling in CD4 T cells. The inhibitory mechanism involves the membrane transfer of NOX2, which identifies this enzyme as a therapeutic target in immunomodulation. Clinical conditions characterized by excessive T cell responses, specifically autoimmune disease, may be amenable to enhancing the induction and assembly of NADPH oxidase in CD8 Tregs. Functional insufficiency of CD8 Tregs in older individuals, a possible risk factor for the chronic inflammatory syndrome associated with immune aging (65), is associated with age-related loss of NOX2. Here, optimizing NOX2 induction should be pursued as a means to treat inflammaging. Vice versa, hindering the ability of CD4 T cells to absorb the membrane fragments from contacting CD8 T cells or dismantling the process underlying exosome production may provide intersection points to enhance CD4 T cell immunity. Equipped with a lymphoid homing receptor, CD8 Tregs meet CD4 T cells in the microenvironment of secondary lymphoid tissues and thus can regulate antigen dependent as well as homeostatic CD4 T cell proliferation. CD8 Treg hyperactivity could therefore undermine T cell replenishment, and modulating the activity of CD8 Tregs would make this process therapeutically accessible.
Methods
Study subjects and patients.
Blood samples from 64 healthy adult individuals (20 to 85 years of age) were obtained from the Stanford Blood Center. Healthy donors were without a personal or family history of cancer or autoimmune disease. Young donors were 20 to 30 years old, and older donors had a minimal age of 60 years. Thirteen patients (11 women, mean age of 72.3 years) with biopsy-positive GCA were included in the study. Seven patients were untreated at the time of testing. Patients on corticosteroids (n = 6) had received 10 to 50 mg of prednisone for at least 4 weeks. At initial diagnosis, the average erythrocyte sedimentation rate was 59 mm/hr (range 14–138 mm/h). Two patients in the study cohort had visual loss, 7 patients had polymyalgia rheumatica, one patient had fever, and 5 patients had involvement of the aorta or subclavian/axillary arteries, diagnosed by noninvasive imaging. Patients with PsA (mean age of middle-aged group, 41.8 years, 80% men; mean age of older group, 64.6 years, 60% men) were selected due to active disease at the time of enrollment. Patients with SVV had biopsy-positive disease, lacked autoantibodies to PR-3 or MPO, and were on corticosteroid doses of less than 10 mg/d (mean age, 44.2 years, 56% men).
PBMCs were isolated by gradient centrifugation with Lymphocyte Separation Medium (Lonza) and cultured in RPMI 1640 medium supplemented with 10% FCS (Hyclone) plus penicillin/streptomycin/glutamine. Naive CD4 T cells were purified from PBMCs using a naive CD4 T Cell Isolation Kit (Miltenyi Biotec). Fresh CD8 T cells were derived from unstimulated, freshly isolated PBMCs and were purified with a CD8 T Cell Isolation Kit (Miltenyi Biotec). To isolate CD8 Tregs directly without culture, CD8 T cells were purified from PBMCs using a CD8 T Cell Isolation Kit (Stemcell Technologies) and stained with FITC-labeled CD39 plus PE-Cy7–labeled CD26 (Biolegend) (23, 24). CD8+CD39+CD26– cells were sorted with a BD FACSAria cell sorter. Purity of cell populations was consistently >90% by flow cytometry.
Reagents.
Dynabeads Human T-Activator CD3/CD28, CellTrace CFSE Cell Proliferation Kit, CellTracker Red CMTPX, and CellROX Deep Red Reagent for oxidative stress detection were from Life Technologies. PKH67 Green Fluorescent Cell Linker, rotenone, and DPI were obtained from Sigma-Aldrich. Brefeldin A was purchased from MP Biomedicals. Tempol and mitoTEMPO were purchased from Santa Cruz Biotechnology. gp91ds-tat was purchased from Anaspec.
Induction of CD8 Tregs.
Induction of CD8 Tregs has been previously described (22). Briefly, PBMCs (1 × 106 cells/ml) were incubated with 0.1 ng/ml anti-CD3/OKT3 and 5 ng/ml recombinant IL-15 for 6 days. CD8 Tregs with a naive phenotype were isolated with a naive CD8 T Cell Isolation Kit (Miltenyi Biotec); >90% of purified cells had a CD8+CCR7+ phenotype.
T cell suppressor assay.
Naive CD4 T cells were mixed with CD8+CCR7+ Tregs or freshly isolated CD8 T cells at a 1:1 ratio and stimulated with anti-CD3/CD28 beads at a ratio of 1:1 for 5 to 10 minutes. Phosphorylation of ZAP70 or LAT was quantified by flow cytometry. For some experiments, CD8 Tregs were pretreated with brefeldin A (10 μg/ml), tempol (50 μM), rotenone (2 μM), mitoTEMPO (10 μM), DPI (10 μM), or gp91ds-tat (50 μM) for 18 to 20 hours and then examined in the suppressor assay. CFSE-labeled naive CD4 T cells were incubated with or without CD8+CCR7+ Tregs (CD4/CD8 = 1:1) for 30 minutes, stimulated with anti-CD3/CD28 beads at a ratio of 1:1 for 4 days, and then analyzed for proliferative activity by measuring CFSE dilution by flow cytometry.
Flow cytometry.
For detection of phosphorylated ZAP70 in CD4 T cells, cells were fixed with Fix Buffer I (BD Phosflow), permeabilized with Perm Buffer III (BD Phosflow), and stained with FITC anti-CD4 (BD, RPA-T4), PE anti-p-ZAP70 antibody (BD Phosflow, 17A/p-ZAP70), or PE anti-p-LAT antibody (BD Phosflow, J96-1238.58.93) for 45 minutes at 4°C. Flow cytometry was performed on a LSR II flow cytometer (BD Biosciences), and the data were analyzed with FlowJo software (Tree Star Inc.).
Immunohistochemical staining.
Localization of CD8+CCR7+FoxP3+ Tregs in human tonsils and human lymph nodes was performed with in situ immunohistochemical staining (66). Briefly, human tonsils and lymph nodes embedded in OCT were cut into 5-μm sections, fixed with pre-cold acetone for 15 minutes, and blocked and stained with mouse anti-human FoxP3 antibody (10 μg/ml; Abcam, 236A/E7) plus rabbit anti-human CD8 antibody (1 μg/ml; Abcam, SP239). To visualize antibody binding, sections were incubated with goat anti-mouse IgG biotinylated antibody (1:500; DAKO, E0433) and developed with the VECTASTAIN ABC Kit (Vector) and ImmPACT DAB substrate (Vector). To detect CD8+ cells, Alexa Fluor 488–labeled goat anti-rabbit IgG secondary antibody (20 μg/ml; Life Technologies, A-11008) was applied. Isotype control antibodies were used to control for nonspecific staining. Stained sections were analyzed using a Zeiss LSM 710 confocal microscopy system (Carl Zeiss).
Isolation of exosomes.
CD8 Tregs were labeled with PKH67 and cultured in RPMI-1640 with 10% FBS for 20 hours. CD8 Treg-derived exosomes were isolated from the supernatant with a CD63 Exo-Flow Capture Kit (System Biosciences) and quantified by using a CD63 ExoELISA Kit (System Biosciences).
Real-time PCR.
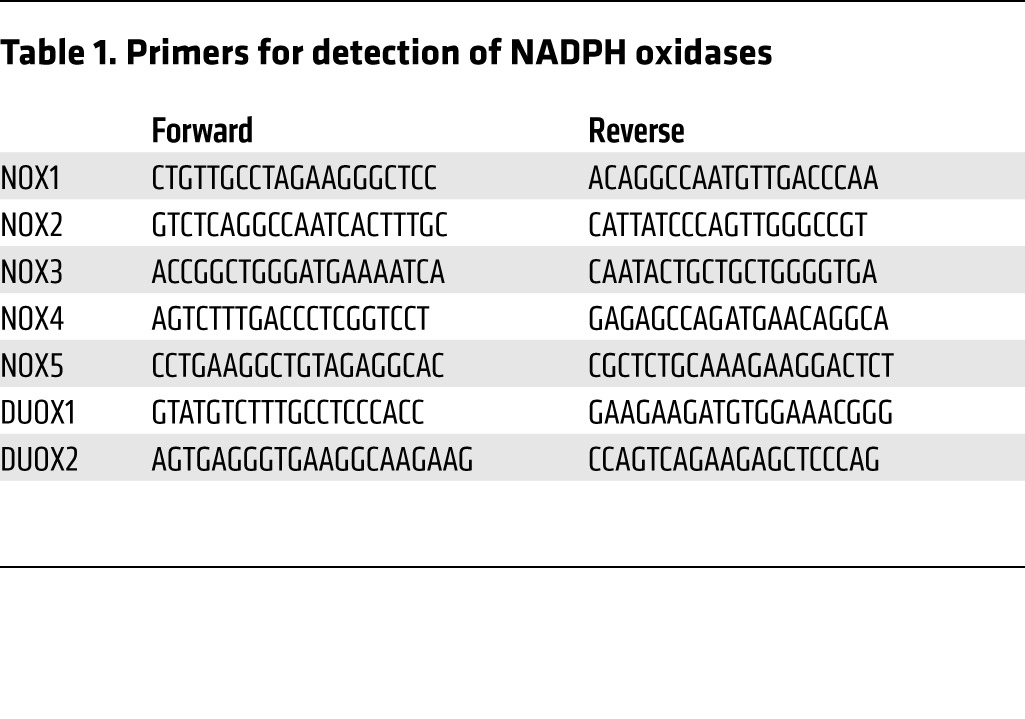
Total RNA was extracted using a RNeasy Mini Kit (Qiagen). cDNA was synthesized with Maxima First Strand cDNA Synthesis Kits for RT-qPCR (Thermo Scientific). qPCR analyses were carried out using Maxima SYBR Green qPCR Master Mixes (Thermo Scientific). The primers for detection of NADPH oxidases are presented in Table 1. 18S rRNA was used as an internal control.
Table 1. Primers for detection of NADPH oxidases.
Plasmid construction and cell transfection.
The NOX2 shRNA expression plasmid was constructed using the pSUPER system (Oligoengine). Briefly, sense and antisense oligonucleotides were annealed and ligated into the pSUPER.retro.neo+GFP vector digested with BglII and HindIII. The target sequences were as follows: NOX2-shRNA-250, CTGCATGCTGATTCTCTTG; NOX2-shRNA-648, CCATCCGGAGGTCTTACTT; NOX2-shRNA-1635, GTCAACACCCTAATACCAG. Lonza Nucleofection Kits were used for transfection.
Live-cell imaging.
To visualize the exchange of membrane fragments between CD8 Tregs and CD4 T cells, we performed live-cell analysis with a 3D Zeiss microscopy-imaging system as previously described (67). Cells were placed into 4-well chamber slides (Thermo Scientific). Images were taken at multiple positions using the motorized x-y stage precisely controlled by Metamorph software (Molecular Devices). Analyses were performed using Metamorph software and ImageJ software (NIH).
Confocal microscopy.
Expression of NOX2 on CD8 Tregs and CD4 T cells was visualized by confocal microscopy. CD8 Tregs were incubated with rabbit anti-human NOX2/gp91phox antibody (1 μg/ml, Abcam, ab31092) overnight at 4°C and then stained with Alexa Fluor 488 goat anti-rabbit IgG antibody (10 μg/ml, Life Technologies, A-11034) for 1 hour at 4°C. Images were captured by a Zeiss LSM 710 confocal microscope (Carl Zeiss) using ×40 Plan-Neo/1.3 NA oil objective. LSM software (Carl Zeiss) was used to analyze fluorescence intensities.
In vivo T cell proliferation.
NOD/SCID IL-2 receptor γnull (NSG) mice were purchased from The Jackson Laboratory and maintained at Stanford University’s animal facilities under specific pathogen–free conditions. To investigate how CD8 Tregs affect the proliferation and expansion of CD4 T cells in vivo, NSG mice were reconstituted by intravenous injection of human naive CD4 T cells (5 × 106 cells), monocytes (5 × 105 cells), and CD8+CCR7+ Tregs (5 × 106 cells). Naive CD4 T cells were labeled with CellTrace CFSE (Life Technologies) prior to injection. Nine days later, mice were sacrificed and splenocytes were analyzed for the percentage and proliferation of human CD4 T cells by flow cytometry using division index (division number of original cell population) and proliferation index (division number of responding cells).
Statistics.
Data are presented as the mean ± SD, and unpaired 2-tailed Student’s t tests were used for statistical analyses using PRISM 6.0 (GraphPad Software Inc.). To adjust for multiple comparisons, the Bonferroni method was applied as appropriate. P < 0.05 was considered to be significant.
Study approval.
The human and animal experimental protocols were approved by the Stanford Institutional Review Boards; appropriate informed consent was obtained.
Author contributions
ZW, YS, JJG, and CMW designed the research and analyzed data. ZW, YS, TS, YL, JJ, and ZY were responsible for the experimental work. LT supervised all statistical analyses. CMW, ZW, and JJG wrote the manuscript.
Supplementary Material
Acknowledgments
This work was supported by grants from the NIH (R01 AR042527, R01 AI044142, P01 HL058000, R01 AI108891, R01 HL117913, R01 AG045779, and R01 AI108906). Z. Yang received fellowship support from the Govenar Discovery Fund. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Yasuhiro Shimojima’s present address is: Department of Neurology and Rheumatology, Shinshu University School of Medicine, Nagano, Japan.
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2016;126(5):1953–1967. doi:10.1172/JCI84181.
See the related Commentary beginning on page 1646.
Contributor Information
Zhenke Wen, Email: zhenke@stanford.edu.
Yasuhiro Shimojima, Email: yshimoji@shinshu-u.ac.jp.
Tsuyoshi Shirai, Email: tshirai@stanford.edu.
Yinyin Li, Email: yinyin@stanford.edu.
Jihang Ju, Email: jihang@stanford.edu.
Zhen Yang, Email: zhenyang@stanford.edu.
Lu Tian, Email: lutian@stanford.edu.
Jörg J. Goronzy, Email: jgoronzy@stanford.edu.
Cornelia M. Weyand, Email: cweyand@stanford.edu.
References
- 1.Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol. 2012;30:531–564. doi: 10.1146/annurev.immunol.25.022106.141623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Campbell DJ. Control of regulatory T cell migration, function, and homeostasis. J Immunol. 2015;195(6):2507–2513. doi: 10.4049/jimmunol.1500801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133(5):775–787. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 4.Zheng Y, Rudensky AY. Foxp3 in control of the regulatory T cell lineage. Nat Immunol. 2007;8(5):457–462. doi: 10.1038/ni1455. [DOI] [PubMed] [Google Scholar]
- 5.Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. 2008;8(7):523–532. doi: 10.1038/nri2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jagger A, Shimojima Y, Goronzy JJ, Weyand CM. Regulatory T cells and the immune aging process: a mini-review. Gerontology. 2014;60(2):130–137. doi: 10.1159/000355303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen H, Zheng X, Zheng Y. Age-associated loss of lamin-B leads to systemic inflammation and gut hyperplasia. Cell. 2014;159(4):829–843. doi: 10.1016/j.cell.2014.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boraschi D, et al. The gracefully aging immune system. Sci Transl Med. 2013;5(185): doi: 10.1126/scitranslmed.3005624. [DOI] [PubMed] [Google Scholar]
- 9.Yang S, Fujikado N, Kolodin D, Benoist C, Mathis D. Immune tolerance. Regulatory T cells generated early in life play a distinct role in maintaining self-tolerance. Science. 2015;348(6234):589–594. doi: 10.1126/science.aaa7017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lopes-Carvalho T, Coutinho A. Old dogs and new tricks: defective peripheral regulatory T cell generation in aged mice. Eur J Immunol. 2013;43(10):2534–2537. doi: 10.1002/eji.201344029. [DOI] [PubMed] [Google Scholar]
- 11.Burzyn D, Benoist C, Mathis D. Regulatory T cells in nonlymphoid tissues. Nat Immunol. 2013;14(10):1007–1013. doi: 10.1038/ni.2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith TR, Kumar V. Revival of CD8+ Treg-mediated suppression. Trends Immunol. 2008;29(7):337–342. doi: 10.1016/j.it.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 13.Overacre AE, Vignali DA. Treg stability: to be or not to be. Curr Opin Immunol. 2016;39:39–43. doi: 10.1016/j.coi.2015.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim HJ, Verbinnen B, Tang X, Lu L, Cantor H. Inhibition of follicular T-helper cells by CD8(+) regulatory T cells is essential for self tolerance. Nature. 2010;467(7313):328–332. doi: 10.1038/nature09370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leavy O. Regulatory T cells: CD8+ TReg cells join the fold. Nat Rev Immunol. 2010;10(10): doi: 10.1038/nri2862. [DOI] [PubMed] [Google Scholar]
- 16.Alvarez Arias DA, et al. Disruption of CD8+ Treg activity results in expansion of T follicular helper cells and enhanced antitumor immunity. Cancer Immunol Res. 2014;2(3):207–216. doi: 10.1158/2326-6066.CIR-13-0121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Holderried TA, Lang PA, Kim HJ, Cantor H. Genetic disruption of CD8+ Treg activity enhances the immune response to viral infection. Proc Natl Acad Sci U S A. 2013;110(52):21089–21094. doi: 10.1073/pnas.1320999110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leavenworth JW, Tang X, Kim HJ, Wang X, Cantor H. Amelioration of arthritis through mobilization of peptide-specific CD8+ regulatory T cells. J Clin Invest. 2013;123(3):1382–1389. doi: 10.1172/JCI66938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zheng J, et al. Human CD8+ regulatory T cells inhibit GVHD and preserve general immunity in humanized mice. Sci Transl Med. 2013;5(168): doi: 10.1126/scitranslmed.3004943. [DOI] [PubMed] [Google Scholar]
- 20.Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol. 2010;10(7):490–500. doi: 10.1038/nri2785. [DOI] [PubMed] [Google Scholar]
- 21.Peterson RA. Regulatory T-cells: diverse phenotypes integral to immune homeostasis and suppression. Toxicol Pathol. 2012;40(2):186–204. doi: 10.1177/0192623311430693. [DOI] [PubMed] [Google Scholar]
- 22.Suzuki M, et al. CD8+CD45RA+CCR7+FOXP3+ T cells with immunosuppressive properties: a novel subset of inducible human regulatory T cells. J Immunol. 2012;189(5):2118–2130. doi: 10.4049/jimmunol.1200122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Borsellino G, et al. Expression of ectonucleotidase CD39 by Foxp3+ Treg cells: hydrolysis of extracellular ATP and immune suppression. Blood. 2007;110(4):1225–1232. doi: 10.1182/blood-2006-12-064527. [DOI] [PubMed] [Google Scholar]
- 24.Salgado FJ, Perez-Diaz A, Villanueva NM, Lamas O, Arias P, Nogueira M. CD26: a negative selection marker for human Treg cells. Cytometry A. 2012;81(10):843–855. doi: 10.1002/cyto.a.22117. [DOI] [PubMed] [Google Scholar]
- 25.Adriaensen W, Mathei C, Vaes B, Van Pottelbergh G, Wallemacq P, Degryse JM. Interleukin-6 as a first-rated serum inflammatory marker to predict mortality and hospitalization in the oldest old: A regression and CART approach in the BELFRAIL study. Exp Gerontol. 2015;69:53–61. doi: 10.1016/j.exger.2015.06.005. [DOI] [PubMed] [Google Scholar]
- 26.Weyand CM, Goronzy JJ. Immune mechanisms in medium and large-vessel vasculitis. Nat Rev Rheumatol. 2013;9(12):731–740. doi: 10.1038/nrrheum.2013.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weyand CM, Goronzy JJ. Clinical practice. Giant-cell arteritis and polymyalgia rheumatica. N Engl J Med. 2014;371(1):50–57. doi: 10.1056/NEJMcp1214825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barnas JL, Ritchlin CT. Etiology and pathogenesis of psoriatic arthritis. Rheum Dis Clin North Am. 2015;41(4):643–663. doi: 10.1016/j.rdc.2015.07.006. [DOI] [PubMed] [Google Scholar]
- 29.Panday A, Sahoo MK, Osorio D, Batra S. NADPH oxidases: an overview from structure to innate immunity-associated pathologies. Cell Mol Immunol. 2015;12(1):5–23. doi: 10.1038/cmi.2014.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deffert C, Cachat J, Krause KH. Phagocyte NADPH oxidase, chronic granulomatous disease and mycobacterial infections. Cell Microbiol. 2014;16(8):1168–1178. doi: 10.1111/cmi.12322. [DOI] [PubMed] [Google Scholar]
- 31.Nathan C, Cunningham-Bussel A. Beyond oxidative stress: an immunologist’s guide to reactive oxygen species. Nat Rev Immunol. 2013;13(5):349–361. doi: 10.1038/nri3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Contreras-Ferrat A, et al. Insulin elicits a ROS-activated and an IP(3)-dependent Ca2+ release, which both impinge on GLUT4 translocation. J Cell Sci. 2014;127(pt 9):1911–1923. doi: 10.1242/jcs.138982. [DOI] [PubMed] [Google Scholar]
- 33.Ohnishi Y, Hirano K, Nishimura J, Furue M, Kanaide H. Inhibitory effects of brefeldin A, a membrane transport blocker, on the bradykinin-induced hyperpolarization-mediated relaxation in the porcine coronary artery. Br J Pharmacol. 2001;134(1):168–178. doi: 10.1038/sj.bjp.0704246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mittelbrunn M, et al. Unidirectional transfer of microRNA-loaded exosomes from T cells to antigen-presenting cells. Nat Commun. 2011;2: doi: 10.1038/ncomms1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao DM, Thornton AM, Dipaolo RJ, Shevach EM. Activated CD4+CD25+ T cells selectively kill B lymphocytes. Blood. 2006;107(10):3925–3932. doi: 10.1182/blood-2005-11-4502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cao X, et al. Granzyme B and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity. 2007;27(4):635–646. doi: 10.1016/j.immuni.2007.08.014. [DOI] [PubMed] [Google Scholar]
- 37.Grossman WJ, Verbsky JW, Tollefsen BL, Kemper C, Atkinson JP, Ley TJ. Differential expression of granzymes A and B in human cytotoxic lymphocyte subsets and T regulatory cells. Blood. 2004;104(9):2840–2848. doi: 10.1182/blood-2004-03-0859. [DOI] [PubMed] [Google Scholar]
- 38.Kobie JJ, Shah PR, Yang L, Rebhahn JA, Fowell DJ, Mosmann TR. T regulatory and primed uncommitted CD4 T cells express CD73, which suppresses effector CD4 T cells by converting 5′-adenosine monophosphate to adenosine. J Immunol. 2006;177(10):6780–6786. doi: 10.4049/jimmunol.177.10.6780. [DOI] [PubMed] [Google Scholar]
- 39.Deaglio S, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. 2007;204(6):1257–1265. doi: 10.1084/jem.20062512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Joller N, et al. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity. 2014;40(4):569–581. doi: 10.1016/j.immuni.2014.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cederbom L, Hall H, Ivars F. CD4+CD25+ regulatory T cells down-regulate co-stimulatory molecules on antigen-presenting cells. Eur J Immunol. 2000;30(6):1538–1543. doi: 10.1002/1521-4141(200006)30:6<1538::AID-IMMU1538>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 42.Fallarino F, et al. Modulation of tryptophan catabolism by regulatory T cells. Nat Immunol. 2003;4(12):1206–1212. doi: 10.1038/ni1003. [DOI] [PubMed] [Google Scholar]
- 43.Mellor AL, Munn DH. IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol. 2004;4(10):762–774. doi: 10.1038/nri1457. [DOI] [PubMed] [Google Scholar]
- 44.Bianco NR, Kim SH, Morelli AE, Robbins PD. Modulation of the immune response using dendritic cell-derived exosomes. Methods Mol Biol. 2007;380:443–455. doi: 10.1007/978-1-59745-395-0_28. [DOI] [PubMed] [Google Scholar]
- 45.Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013;200(4):373–383. doi: 10.1083/jcb.201211138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gardiner GJ, Deffit SN, Mcletchie S, Perez L, Walline CC, Blum JS. A role for NADPH oxidase in antigen presentation. Front Immunol. 2013;4: doi: 10.3389/fimmu.2013.00295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schappi MG, Jaquet V, Belli DC, Krause KH. Hyperinflammation in chronic granulomatous disease and anti-inflammatory role of the phagocyte NADPH oxidase. Semin Immunopathol. 2008;30(3):255–271. doi: 10.1007/s00281-008-0119-2. [DOI] [PubMed] [Google Scholar]
- 48.Stasia MJ, Li XJ. Genetics and immunopathology of chronic granulomatous disease. Semin Immunopathol. 2008;30(3):209–235. doi: 10.1007/s00281-008-0121-8. [DOI] [PubMed] [Google Scholar]
- 49.Roos D, De Boer M. Molecular diagnosis of chronic granulomatous disease. Clin Exp Immunol. 2014;175(2):139–149. doi: 10.1111/cei.12202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee K, Won HY, Bae MA, Hong JH, Hwang ES. Spontaneous and aging-dependent development of arthritis in NADPH oxidase 2 deficiency through altered differentiation of CD11b+ and Th/Treg cells. Proc Natl Acad Sci U S A. 2011;108(23):9548–9553. doi: 10.1073/pnas.1012645108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jendrysik MA, et al. NADPH oxidase-2 derived ROS dictates murine DC cytokine-mediated cell fate decisions during CD4 T helper-cell commitment. PLoS One. 2011;6(12): doi: 10.1371/journal.pone.0028198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tacke RS, et al. Myeloid suppressor cells induced by hepatitis C virus suppress T-cell responses through the production of reactive oxygen species. Hepatology. 2012;55(2):343–353. doi: 10.1002/hep.24700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Raber PL, et al. Subpopulations of myeloid-derived suppressor cells impair T cell responses through independent nitric oxide-related pathways. Int J Cancer. 2014;134(12):2853–2864. doi: 10.1002/ijc.28622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Efimova O, Szankasi P, Kelley TW. Ncf1 (p47phox) is essential for direct regulatory T cell mediated suppression of CD4+ effector T cells. PLoS One. 2011;6(1): doi: 10.1371/journal.pone.0016013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.D’autreaux B, Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol. 2007;8(10):813–824. doi: 10.1038/nrm2256. [DOI] [PubMed] [Google Scholar]
- 56.Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012;24(5):981–990. doi: 10.1016/j.cellsig.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hayes P, Knaus UG. Balancing reactive oxygen species in the epigenome: NADPH oxidases as target and perpetrator. Antioxid Redox Signal. 2013;18(15):1937–1945. doi: 10.1089/ars.2012.4895. [DOI] [PubMed] [Google Scholar]
- 58.Gareus R, et al. Endothelial cell-specific NF-κB inhibition protects mice from atherosclerosis. Cell Metab. 2008;8(5):372–383. doi: 10.1016/j.cmet.2008.08.016. [DOI] [PubMed] [Google Scholar]
- 59.Anrather J, Racchumi G, Iadecola C. NF-κB regulates phagocytic NADPH oxidase by inducing the expression of gp91phox. J Biol Chem. 2006;281(9):5657–5667. doi: 10.1074/jbc.M506172200. [DOI] [PubMed] [Google Scholar]
- 60.Ahn JD, et al. Inhibitory effects of novel AP-1 decoy oligodeoxynucleotides on vascular smooth muscle cell proliferation in vitro and neointimal formation in vivo. Circ Res. 2002;90(12):1325–1332. doi: 10.1161/01.RES.0000023200.19316.D5. [DOI] [PubMed] [Google Scholar]
- 61.Manea A, Tanase LI, Raicu M, Simionescu M. Jak/STAT signaling pathway regulates nox1 and nox4-based NADPH oxidase in human aortic smooth muscle cells. Arterioscler Thromb Vasc Biol. 2010;30(1):105–112. doi: 10.1161/ATVBAHA.109.193896. [DOI] [PubMed] [Google Scholar]
- 62.Quintela AM, et al. Activation of peroxisome proliferator-activated receptor-beta/-delta (PPARβ/Δ) prevents endothelial dysfunction in type 1 diabetic rats. Free Radic Biol Med. 2012;53(4):730–741. doi: 10.1016/j.freeradbiomed.2012.05.045. [DOI] [PubMed] [Google Scholar]
- 63.Czesnikiewicz-Guzik M, et al. T cell subset-specific susceptibility to aging. Clin Immunol. 2008;127(1):107–118. doi: 10.1016/j.clim.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Renkema KR, Li G, Wu A, Smithey MJ, Nikolich-Zugich J. Two separate defects affecting true naive or virtual memory T cell precursors combine to reduce naive T cell responses with aging. J Immunol. 2014;192(1):151–159. doi: 10.4049/jimmunol.1301453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Goronzy JJ, Weyand CM. Understanding immunosenescence to improve responses to vaccines. Nat Immunol. 2013;14(5):428–436. doi: 10.1038/ni.2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lim HW, Hillsamer P, Kim CH. Regulatory T cells can migrate to follicles upon T cell activation and suppress GC-Th cells and GC-Th cell-driven B cell responses. J Clin Invest. 2004;114(11):1640–1649. doi: 10.1172/JCI22325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Huang J, et al. A single peptide-major histocompatibility complex ligand triggers digital cytokine secretion in CD4(+) T cells. Immunity. 2013;39(5):846–857. doi: 10.1016/j.immuni.2013.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.