

Abstract
Background and Objectives:
46 XY disorders of sexual development (DSD) cover a wide spectrum of phenotypes ranging from unambiguous female genitalia to ambiguous male genitalia with hypospadias or dysgenetic gonads. Management of these patients depends on the cause of DSD, degree of feminization, age at presentation, and gender orientation. The aim of this study was to evaluate the presentation and management of patients with 46XY DSD at our center.
Patients and Methods:
All new and old patients of 46XY DSD attending the endocrine OPD in a period of 16 months were included in this study. Clinical, cytogenetic, hormonal, and radiological evaluation were done to identify the cause of DSD.
Results:
Among 19 patients, eight were diagnosed with disorders of gonadal development (one with complete gonadal dysgenesis, four with partial gonadal dysgenesis, two with congenital bilateral anorchia, and one with ovotesticular DSD) and eight with disorders of androgen synthesis and action (one with complete androgen insensitivity syndrome [AIS], three with partial AIS and four with 5α reductase deficiency). In three patients, a definitive diagnosis could not be made.
Conclusions:
Management of patients with DSD depends on etiology, gender assignment, gender orientation, hormonal treatment, genital surgery, and consequent psychosocial implications. Due to the overlapping clinical and biochemical parameters in different subsets of DSD, only a preliminary etiological diagnosis can be made in some cases. Genetic studies with long-term follow-up are required for an accurate diagnosis.
Keywords: 46 XY disorders of sexual development, ambiguous genitalia, disorder of sexual differentiation
INTRODUCTION
Sexual development in males is a multistep process, implying a delicate network of molecular events that directs the bipotential gonad to differentiate into testis (sex determination) and consequent differentiation of internal and external genitalia in the presence of testicular hormones (sex differentiation). Alteration of any genetic or endocrine factors involved in testicular development may lead to 46 XY disorders of sexual development (DSD). Genital ambiguity in patients with 46 XY DSD may either occur due to the disorders in gonadal development or due to the disruption in androgen synthesis or action.[1]
Patients with disorders of gonadal development may present with either complete or partial forms of gonadal dysgenesis. Swyer's syndrome or complete gonadal dysgenesis (CGD) is characterized by unambiguous female genitalia, bilateral streak gonads, and elevated gonadotropins. Partial gonadal dysgenesis (PGD) may have a wide spectrum of phenotypes associated with ambiguous genitalia, varying degree of hypospadias and testis of variable size and echotexture present along the path of decent. Ovotesticular DSD (OT-DSD) is a rare condition marked by the presence of both ovarian and testicular tissue in an individual while congenital bilateral anorchia (CBA) is characterized by the complete absence of testicular tissue in genotypic males.
Disorders of androgen synthesis may occur due to deficiency of various enzymes involved in the steroidogenesis. Congenital adrenal hyperplasia (CAH) due to 17β-hydroxysteroid dehydrogenase 3 (17β-HSD3) is caused by the mutations in HSD17B3 gene which is required for the conversion of androstenedione to testosterone. Deficiency of this enzyme results in feminized genitalia in newborns due to the low testosterone levels.
Mutations in SRD5A2 gene impairs the activity of enzyme 5α-reductase and hence the conversion of testosterone to its more potent form dihydrotestosterone (DHT). Individuals with 5α-reductase deficiency (5-αRD) are often raised as females due to undermasculinized genitalia but the gender role changes soon after attaining puberty.
Mutations in androgen receptor gene may lead to the androgen insensitivity syndrome (AIS) which can be partial androgen insensitivity syndrome (PAIS) or complete androgen insensitivity syndrome (CAIS) and is the most common cause of 46XY DSD. The spectrum of phenotypes associated with AIS may range from completely female through mixed male/female to completely male type. In 1995, Charmian Quigley and Frank French proposed new grading system for the phenotypic features in AIS based on Prader classification for CAH.[2]
Individuals diagnosed with 46XY DSD show an overlap in clinical and biochemical parameters which emphasizes the need for comprehensive evaluation with a multidisciplinary approach.
PATIENTS AND METHODS
This is an ambispective study where all new and old follow-up patients diagnosed with 46XY DSD, being managed at endocrine OPD in a period of 16 months (June 2014–October 2015) were included.
External masculinization score (EMS) were calculated based on the external genitalia of patients.[3] Abdominopelvic ultrasound was done for the localization of gonads (magnetic resonance imaging (MRI) also, when required). For patients on follow-up, previous records were reviewed to establish a diagnosis as they were on medication while in new patients, samples were collected and hormonal measurements included serum luteinizing hormone, follicle-stimulating hormone, testosterone, androstenedione (A), cortisol, dehydroepiandrostenedione, and plasma adrenocorticotropic hormone levels by electrochemiluminescence immunoassay using commercial kits (Roche, Germany). Radioimmunoassay kit-based method was used for evaluating 17-hydroxyprogesterone (17-OHP) levels (Diagnostic Systems Laboratories, Inc., Webster, TX, USA). 5αDHT was measured using radioimmunoassay (Immunotech; Prague, Czech Republic) after extraction from other hormones with diethyl ether and Celite chromatography.
The short-term human chorionic gonadotropin (hCG) stimulation test was carried out to check the functioning of testicular tissue. Testosterone levels were measured before and 24–48 h after the last injection of a series of three intramuscular injections of hCG on alternate days (<1-year-old, 500 units; 1–10 years, 1000 units; >10 years, 2000 units).[4] Testosterone: Androstenedione (T:A) and testosterone: 5α DHT (T:5αDHT) ratios were calculated as and when required to rule out 17 beta HSD deficiency and 5 alpha reductase deficiency respectively. A detailed review of literature was done.
RESULTS
On first evaluation, 11 patients had presented with the primary complaint of ambiguous genitalia, three (who were being reared as female) with primary amenorrhea, three with undescended gonads and one with fever and vomiting. The presenting complaints, clinical, hormonal, and radiological findings are summarized in Table 1. Five (26%) of them were reared females and 14 (74%) as males. All had genital abnormalities with an EMS <11. Ten patients (S.5, S.7, S.8, S.9, S.10, S.14, S.15, S.16, S.17, and S.18) had undergone corrective surgeries for ambiguous genitalia.
Table 1.
Clinical profile of patients
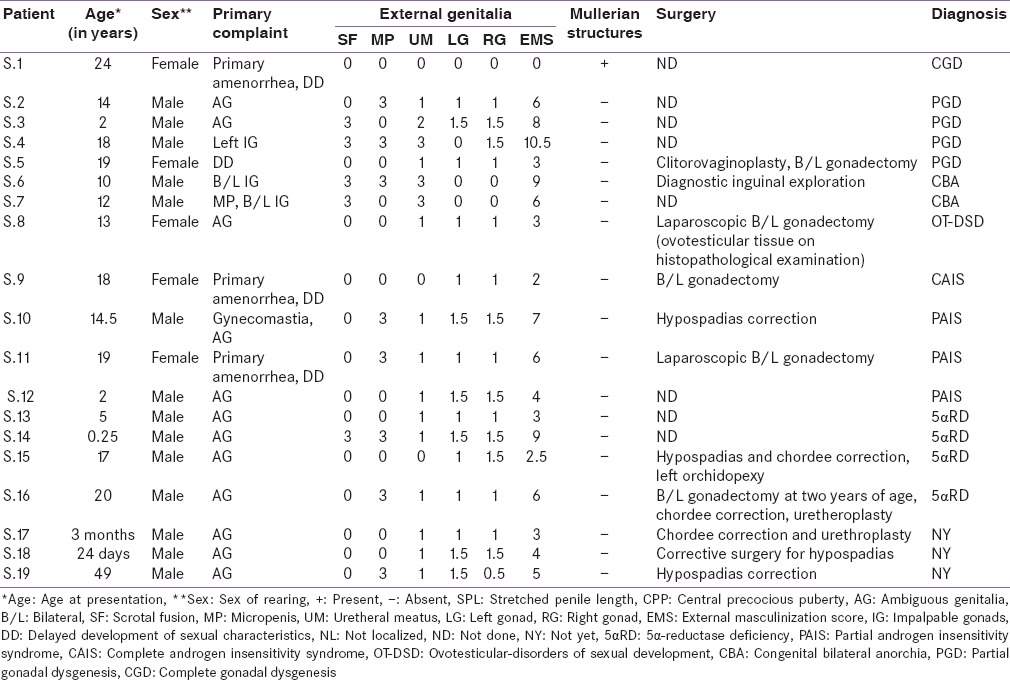
Patient S.1 reared as female presented with primary amenorrhea, delayed development of secondary sexual characteristics and unambiguous female genitalia. Radiological studies showed streak gonads and presence of hypoplastic uterus. Baseline gonadotropins were high; she was diagnosed with CGD.
Patients S.2, S.3, and S.5 had ambiguous genitalia, undescended gonads, raised FSH and low testosterone levels while patient S.4 had unilateral undescended testis with raised FSH and low testosterone levels, all three were suspected to be cases of PGD.
Patients S.6 and S.7 sought medical attention for impalpable gonads. Abdominopelvic ultrasonography (USG) and MRI could not locate gonads in S.7 while in patient S.6, a nubbin was seen in the scrotal area. Inguinal exploration was done to locate testis but it revealed the absence of any testicular tissue. FSH was raised in both [Table 2] and there was no testosterone response to hCG stimulation. They both were diagnosed with CBA.
Table 2.
Hormonal profile of patients
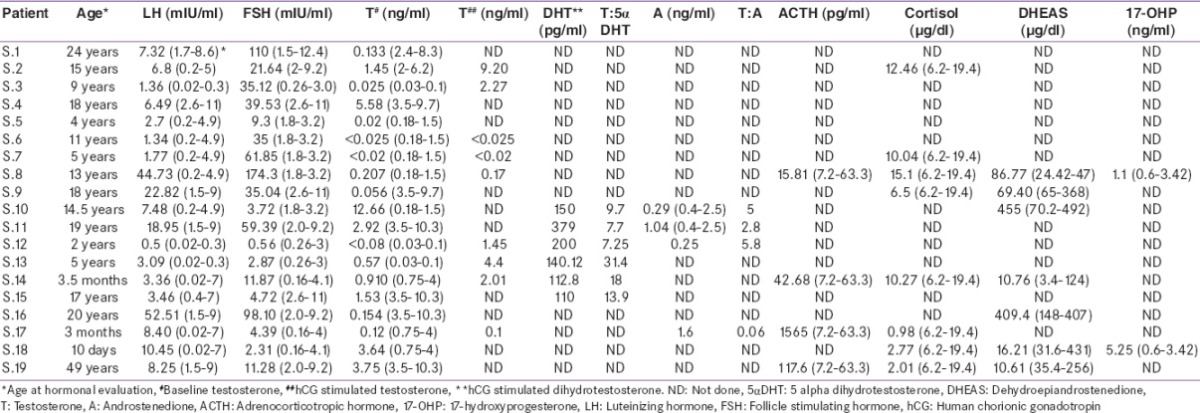
Patient S.8 being reared as female had presented with ambiguous genitalia and raised gonadotropins. She had undergone bilateral gonadectomy and on histological evaluation OT tissue was found, she was diagnosed with OT-DSD.
Patient S.9 being reared as female presented with primary amenorrhea, genital ambiguity and delayed development of secondary sexual characteristics. She underwent bilateral gonadectomy, histopathological examination revealed testicular tissue and was diagnosed with CAIS.
Patient S.10 presented with gynecomastia at 14 years of age. Hormonal profile showed normal testosterone levels with T:DHT ratio of 9.7 and T:A ratio of 5.8. He was diagnosed with PAIS. Patient S.11 being reared as female came with a complaint of hirsutism and delayed development of secondary sexual characteristics. Physical examination revealed genital ambiguity with inguinal gonads and was referred to surgery. Bilateral gonadectomy was done and histological studies showed the presence of testicular tissue which confirmed the diagnosis of PAIS as T:DHT and T:A ratios were also normal. Parents of patient S.12 sought medical attention due to ambiguous genitalia. Gonads were palpable in labioscrotal folds. Testosterone levels were undetectable, but showed a good response to hCG stimulation with T:DHT ratio of 7.25, T:A ratio of 5.8 and was diagnosed with PAIS.
Patients S.13, S.14, S.15, and S.16 came with ambiguous genitalia and low baseline testosterone levels. Testosterone to DHT ratio in S.13, S.14, and S.15 were 31.4, 18, and 13.9, respectively. Due to high T:DHT ratio post-hCG stimulation, all three were diagnosed with 5αRD while in S.16, T:DHT ratio was not calculated as b/l gonadectomy was done at 2 years of age. Molecular analysis of SRD5A2 gene in this patient revealed R246Q mutation[5] commonly seen in 5-αRD.
Patient S.17 presented at 3 months with micropenis with penoscrotal hypospadias and chordee. Records show that his post hCG serum T:A ratio was 0.06, he was thought to be a case of 17βHSD deficiency. At 2 years of age he had fever, vomiting and loose stools with hyponatremia, hyperkalemia and hypoglycemia. His serum ACTH was 1565 pg/ml and cortisol 0.98 µg/dl. We could not find a report of 17 OHP done at that time. Patient was started on steroids at the time of this salt losing crisis. He is now 6 years old and continues on steroid replacement. Subsequent 17 OHP levels were low. A definitive diagnosis could not be made in this patient.
Patient S.18 was admitted in pediatrics with a complaint of poor feeding and poor activity since day one of life. On evaluation, he was found to have hypernatremia, hypokalemia, and genital ambiguity with bifid scrotum and hypospadias. In view of electrolyte imbalance, incomplete masculinization, low cortisol, and elevated 17-OHP, he was suspected to be a case of CAH and treatment was started. An accurate diagnosis could not be established when he came to us as he was already on steroids and had a history of salt wasting, so steroids could not be stopped for evaluation of the cause of CAH.
Patient S.19 is now at 49 years of age. He has been on dexamethasone, sustanon, and antihypertensives. He had undergone three genital surgeries for hypospadias correction but still had difficulty in urination. The patient is kept on antihypertensives, but no definitive diagnosis could be made as the baseline hormonal reports are missing. With the help of clinical, biochemical and radiological evaluation, accurate diagnosis was made in 16 patients while rest 2 are on follow-up and managed in our clinic.
DISCUSSION
Discordance between genetic (XX or XY), gonadal (testis or ovaries), external genital (vulva or penis), and internal sex (Wolffian or Mullerian ducts) results in DSD.[1] In humans, the bipotential gonad differentiates into testis in the presence of SRY gene present on Y-chromosome.[6] Testicular differentiation is followed by the development of internal sex ducts and external genitalia in the presence of fetal hormones produced by the testis. Sertoli cells secrete anti-Mullerian hormone, which causes irreversible regression of paramesonephric or Mullerian ducts around the sixth week of gestational age.[7] Testosterone produced by Leydig cells of fetal testis promotes the development of mesonephric or Wolffian ducts and hence male internal genitalia; epididymis, vas deferens, and vesicular seminalis. The development of external male genitalia depends on the conversion of testosterone to a more potent steroid DHT in the presence of 5α-reductase enzyme.
In 2006, the term “intersex” or “hermaphrodite” was replaced by “DSD” and a new taxonomy was recommended based on the karyotype (genetic sex). 46XY DSD was subcategorized as disorders of testicular development and disorders of androgen synthesis or action. First-line testing in newborns includes karyotyping, imaging (USG/MRI pelvis), serum electrolytes, and serum gonadotropins. Molecular diagnosis has been made in approximately 20% of cases with 46XY DSD, although confirmatory, but is limited by cost and accessibility.[1]
The inability of bipotential gonad to differentiate into testis results in testicular dysgenesis or agenesis which occurs due to the disruption of various genetic factors involved in the process of sex determination.[8] In our study, four patients were diagnosed with PGD, one with CGD, one with OT-DSD, and two with testicular agenesis or CBA.
In the absence of gonadal development, patients with CGD usually present with unambiguous female genitalia due to the absence of gonadal steroid production while in PGD, the external phenotype depends upon the degree of testicular tissue present. In our study, out of four patients with PGD, three (S.2, S.3, and S.5) presented with ambiguous genitalia and one with undescended gonad (S.4). In such cases, corrective surgeries are done if required, and testosterone supplementations are given at pubertal age. Patient S.5 being reared as female had presented with ambiguous genitalia at 4 years of age. With parents’ consent clitorovaginoplasty and bilateral orchidectomy was done and histopathological examination revealed the presence of bilateral dysgenetic gonads. She is now on estrogen replacement therapy for breast development. Orchidopexy and biopsy are usually recommended for undescended gonads as the risk of malignancy ranges from 8.3% to 54% in patients with XY PGD.[9] In 46XY CGD gonadectomy is strongly recommended as the risk of malignancy is 15-30%.[9]
CBA is a rare condition and is marked by the absence of testis since birth. Patients may present with well-developed scrotum and bilateral nonpalpable gonads. The absence of gonads was confirmed by hCG stimulation and inguinal exploration. Previous studies have shown the occurrence of pure or partial forms of 46XY gonadal dysgenesis in families of patients with CBA, but the genetic cause is still not known.[10] In our study, two patients (S.6 and S.7) with CBA were born out of nonconsanguineous marriage and had no family history of genital ambiguity but the father of patient S.7 gave history of oligospermia and assisted reproduction.
46XY OT-DSD is a very rare condition marked by the presence of OT tissue and is confirmed by histopathological examination. Parents of patient S.8 had sought medical attention for ambiguous genitalia at the age of 13 years. She had been assigned female gender at birth. Bilateral gonadectomy was done, and the corrective surgeries for hypospadias and phallus are awaited. The genetic etiology of 46 XY OT-DSD is not known.
The complete or partial resistance to the action of androgens may result in CAIS or PAIS in XY individuals. Patients with CAIS may present with primary amenorrhea in adolescence or inguinal swellings in infants. Patient S.9 was being reared as female, parents sought medical attention at the age of 18 years for primary amenorrhea and delayed development of secondary sexual characteristics. Bilateral gonadectomy was recommended after confirming the gender identity of the patient to be female. A recent study done on 102 phenotypic women with Y-chromosome showed an incidence of 17.6% malignancy. Of these 9 out of 30 patients (30%) of CAIS with a mean age of 20.7 years were found to have gonadoblastoma on histological analysis.[11] Therefore, adult CAIS patients who receive late intervention are recommended to undergo immediate gonadectomy in order to avoid the risk of malignancy.
The phenotype of patients presenting with PAIS depends on the responsiveness of genital tissues to androgens. Infants with PAIS present with the genital ambiguity of varying degrees while adults may present with gynecomastia.[12] However, the risk of malignancy in individuals with PAIS is 15% if the testis is not scrotal in position.[13]
Patient S.10 presented with gynecomastia at 14 years of age. He also had hypospadias. Hormonal profile showed normal testosterone levels with T:DHT ratio of 9.7 and T:A ratio of 5.8. He was diagnosed with PAIS. He underwent surgical correction for hypospadias and gynecomastia.
Patient S.11 being reared as female sought medical attention for hirsutism at the age of 19 years. Biochemical examination showed normal T:DHT and T:A ratios with testosterone levels in male range. Gonadectomy was done and histopathological examination showed the presence of testicular tissue, she was then diagnosed with PAIS. She had female gender orientation and is now on estrogen replacement therapy for breast development. Patient S.12 came with ambiguous genitalia, bilateral palpable gonads. His T:DHT ratio was 7.25 and T:A ratio was 5.8 post-hCG stimulation, which excluded the diagnosis of 5αRD and 17βHSD. The presumptive etiological diagnosis for this patient is PAIS. Though the presumptive diagnosis of patients S.10, S.11, and S.12 was PAIS, as T:DHT ratio and T:A ratio were normal, [Figure 1] diagnosis with needs further confirmation by molecular studies.
Figure 1.

Flow chart for diagnosis of 46XY disorders of sexual development
The interrupted conversion of testosterone to DHT due to the mutations in gene encoding 5α-reductase enzyme may lead to 5αRD. The differentiation of internal and external male genitalia occurs in the presence of testosterone (T) and DHT. The T:DHT ratio is used to diagnose this condition but interpreting these results is not always straightforward. The diagnosis of 5αRD is suspected in newborns with undermasculinized genitalia (depending upon the activity of enzyme 5α-reductase), low serum testosterone levels, good response to hCG stimulation and high testosterone to DHT ratio (>10–12).[14] A good response to hCG stimulation (rise by more than twice the baseline value) is observed in these patients. The patients may present with microphallus, inguinal gonads or gonads in labioscrotal folds and varying degree of hypospadias. Treatment with percutaneous DHT increases the size of the phallus in infants and children with 5α-reductase 2 deficiency.[15] Most of them are reared as males and gender reassignment is required in those who are reared as females due to the spontaneous virilization at puberty. The decisions regarding management of masculinizing puberty need to be taken before puberty with a multidisciplinary approach.[16] Female gender assignment in these patients by surgery should be undertaken postpuberty with full caution and counseling with parents.[17] In our study, patient S.16 had severely undervirilized genitalia, bilateral gonadectomy had been done at 2 years of age and female gender had been assigned. At the age of 17 years, he sought medical attention for gender dysphoria. At this time, the presence of common R246Q mutation on molecular analysis of SRD5A2 gene confirmed the diagnosis of 5αRD.[5]
17β-HSD enzyme catalyzes the conversion of androstenedione to testosterone. 46XY patients with deficiency of 17β-HSD enzyme usually present with female-like genitalia at birth with clitoromegaly and blind-ending vagina or male type genitalia with micropenis and hypospadias.[18] At puberty, these patients experience significant virilization due to the abnormally elevated levels of androstenedione or extragonadal synthesis of testosterone, the exact cause of which is unknown.[19] The diagnosis of 17β-HSD3 can be suspected if testosterone to androstenedione ratio is <0.8.[20] The overlapping phenotype of individuals with 17β-HSD and PAIS can be differentiated by molecular analysis or on the basis of hCG stimulated T:A ratio, which is <0.8 in cases of 17β-HSD.[21] Patient S.17 initially predicted to have 17β HSD deficiency, but later presented with features of mineralocorticoid and glucocorticoid deficiency at 2 years of age. Patient was then kept on steroid replacement and is on follow-up. A definitive diagnosis could not be made in this patient.
Parents of patient S.18 had sought medical attention for salt wasting crisis and ambiguity. He was diagnosed with CAH and then managed on steroid replacement therapy. He is now on follow-up for definitive diagnosis. Patient S.19 is presently 49 years of age and gave a history of ambiguous genitalia. He was on dexamethasone, sustanon and had undergone three corrective surgeries for hypospadias but still has a problem in urination. Routine examination revealed raised blood pressure for which he is on hypertensives. An accurate diagnosis was not made as his baseline hormonal reports were missing.
Limitations of this study are that, being an ambispective study, all tests were not done in all patients and baseline records/tests to make a diagnosis could not be obtained in two patients. Nonetheless, this gives an insight into how to diagnose and manage 46XY DSD, who may present at different ages, with different etiologies and different gender of rearing in our set up.
There is overlap in the clinical, hormonal and anatomic profile of different subsets of 46XY DSD. The diagnosis of 46XY DSD needs to be confirmed by cytogenetic, hormonal, radiological, and genetic tests. Phenotype varies depending on the degree of genetic, hormonal and anatomic defect. At times, in spite of all the tests a definitive diagnosis may not be possible. The pediatrician, endocrinologist, surgeon, psychologist, and clinical geneticist need to work as a team for multifaceted management of these patients.
Financial support and sponsorship
This study was supported by Department of Biotechnology, Ministry of Science and Technology (BT/PR7681/MED/12/597/2013) and Indian Council of Medical Research (DHR/GIA/1/2014), New Delhi, India.
Conflicts of interest
There are no conflicts of interest.
Acknowledgment
We acknowledge the intellectual support of late Prof. A.C. Ammini at All India Institute of Medical Sciences, New Delhi, India.
REFERENCES
- 1.Lee PA, Houk CP, Ahmed SF, Hughes IA. International Consensus Conference on Intersex Organized by the Lawson Wilkins Pediatric Endocrine Society and the European Society for Paediatric Endocrinology. Consensus statement on management of intersex disorders. International Consensus Conference on Intersex. Pediatrics. 2006;118:e488–500. doi: 10.1542/peds.2006-0738. [DOI] [PubMed] [Google Scholar]
- 2.Quigley CA, De Bellis A, Marschke KB, el-Awady MK, Wilson EM, French FS. Androgen receptor defects: Historical, clinical, and molecular perspectives. Endocr Rev. 1995;16:271–321. doi: 10.1210/edrv-16-3-271. [DOI] [PubMed] [Google Scholar]
- 3.Ahmed SF, Khwaja O, Hughes IA. The role of a clinical score in the assessment of ambiguous genitalia. BJU Int. 2000;85:120–4. doi: 10.1046/j.1464-410x.2000.00354.x. [DOI] [PubMed] [Google Scholar]
- 4.Dixon J, Wallace AM, O’Toole S, Ahmed SF. Prolonged human chorionic gonadotrophin stimulation as a tool for investigating and managing undescended testes. Clin Endocrinol (Oxf) 2007;67:816–21. doi: 10.1111/j.1365-2265.2007.02968.x. [DOI] [PubMed] [Google Scholar]
- 5.Sahu R, Boddula R, Sharma P, Bhatia V, Greaves R, Rao S, et al. Genetic analysis of the SRD5A2 gene in Indian patients with 5alpha-reductase deficiency. J Pediatr Endocrinol Metab. 2009;22:247–54. doi: 10.1515/jpem.2009.22.3.247. [DOI] [PubMed] [Google Scholar]
- 6.Lovell-Badge R. The role of Sry in mammalian sex determination. Ciba Found Symp. 1992;165:162–79. doi: 10.1002/9780470514221.ch10. [DOI] [PubMed] [Google Scholar]
- 7.Rey R, Lukas-Croisier C, Lasala C, Bedecarrás P. AMH/MIS: What we know already about the gene, the protein and its regulation. Mol Cell Endocrinol. 2003;211:21–31. doi: 10.1016/j.mce.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 8.Eggers S, Sinclair A. Mammalian sex determination-insights from humans and mice. Chromosome Res. 2012;20:215–38. doi: 10.1007/s10577-012-9274-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McCann-Crosby B, Mansouri R, Dietrich JE, McCullough LB, Sutton VR, Austin EG, et al. State of the art review in gonadal dysgenesis: Challenges in diagnosis and management. Int J Pediatr Endocrinol 2014. 2014:4. doi: 10.1186/1687-9856-2014-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vinci G, Anjot MN, Trivin C, Lottmann H, Brauner R, McElreavey K. An analysis of the genetic factors involved in testicular descent in a cohort of 14 male patients with anorchia. J Clin Endocrinol Metab. 2004;89:6282–5. doi: 10.1210/jc.2004-0891. [DOI] [PubMed] [Google Scholar]
- 11.Liu AX, Shi HY, Cai ZJ, Liu A, Zhang D, Huang HF, et al. Increased risk of gonadal malignancy and prophylactic gonadectomy: A study of 102 phenotypic female patients with Y chromosome or Y-derived sequences. Hum Reprod. 2014;29:1413–9. doi: 10.1093/humrep/deu109. [DOI] [PubMed] [Google Scholar]
- 12.Hughes IA, Davies JD, Bunch TI, Pasterski V, Mastroyannopoulou K, MacDougall J. Androgen insensitivity syndrome. Lancet. 2012;380:1419–28. doi: 10.1016/S0140-6736(12)60071-3. [DOI] [PubMed] [Google Scholar]
- 13.Cools M, Drop SL, Wolffenbuttel KP, Oosterhuis JW, Looijenga LH. Germ cell tumors in the intersex gonad: Old paths, new directions, moving frontiers. Endocr Rev. 2006;27:468–84. doi: 10.1210/er.2006-0005. [DOI] [PubMed] [Google Scholar]
- 14.Maimoun L, Philibert P, Cammas B, Audran F, Bouchard P, Fenichel P, et al. Phenotypical, biological, and molecular heterogeneity of 5a-reductase deficiency: An extensive international experience of 55 patients. J Clin Endocrinol Metab. 2011;96:296–307. doi: 10.1210/jc.2010-1024. [DOI] [PubMed] [Google Scholar]
- 15.Ahmed SF, Achermann JC, Arlt W, Balen AH, Conway G, Edwards ZL, et al. UK guidance on the initial evaluation of an infant or an adolescent with a suspected disorder of sex development. Clin Endocrinol (Oxf) 2011;75:12–26. doi: 10.1111/j.1365-2265.2011.04076.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cohen-Kettenis PT. Gender change in 46, XY persons with 5alpha-reductase-2 deficiency and 17beta-hydroxysteroid dehydrogenase-3 deficiency. Arch Sex Behav. 2005;34:399–410. doi: 10.1007/s10508-005-4339-4. [DOI] [PubMed] [Google Scholar]
- 17.Rangecroft L. British Association of Paediatric Surgeons Working Party on the Surgical Management of Children Born with Ambiguous. Genitalia. Surgical management of ambiguous genitalia. Arch Dis Child. 2003;88:799–801. doi: 10.1136/adc.88.9.799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ulloa-Aguirre A, Bassol S, Poo J, Mendez JP, Mutchinick O, Robles C, et al. Endocrine and biochemical studies in a 46, XY phenotypically male infant with 17-ketosteroid reductase deficiency. J Clin Endocrinol Metab. 1985;60:639–43. doi: 10.1210/jcem-60-4-639. [DOI] [PubMed] [Google Scholar]
- 19.Werner R, Kulle A, Sommerfeld I, Riepe FG, Wudy S, Hartmann MF, et al. Testosterone synthesis in patients with 17ß-hydroxysteroid dehydrogenase 3 deficiency. Sex Dev. 2012;6:161–8. doi: 10.1159/000336605. [DOI] [PubMed] [Google Scholar]
- 20.Faisal Ahmed S, Iqbal A, Hughes IA. The testosterone: Androstenedione ratio in male undermasculinization. Clin Endocrinol (Oxf) 2000;53:697–702. doi: 10.1046/j.1365-2265.2000.01166.x. [DOI] [PubMed] [Google Scholar]
- 21.Boehmer AL, Brinkmann AO, Sandkuijl LA, Halley DJ, Niermeijer MF, Andersson S, et al. 17Beta-hydroxysteroid dehydrogenase-3 deficiency: Diagnosis, phenotypic variability, population genetics, and worldwide distribution of ancient and de novo mutations. J Clin Endocrinol Metab. 1999;84:4713–21. doi: 10.1210/jcem.84.12.6174. [DOI] [PubMed] [Google Scholar]