

Abstract
Epigenetic mechanisms are able to alter gene expression, without altering DNA sequence, in a stable manner through cell divisions. They include, among others, the methylation of DNA cytosines and microRNAs and allow the cells to adapt to changing environmental conditions. In recent years, epigenetic association studies are providing new insights into the pathogenesis of complex disorders including prevalent skeletal disorders. Unlike the genome, the epigenome is cell and tissue specific and may change with age and a number of acquired factors. This poses particular difficulties for the design and interpretation of epigenetic studies, particularly those exploring the association of genome-wide epigenetic marks with disease phenotypes. In this report, we propose a framework to help in the critical appraisal of epigenetic association studies. In line with previous suggestions, we focus on the questions critical to appraise the validity of the study, to interpret the results and to assess the generalizability and relevance of the information.
Introduction
Genetic information stored in DNA sequence provides stability through generations to animal and vegetal species. Genetic changes are also considered as a major driver of evolution by improving the adaptation to the environment. However, genetic-driven evolution is a slow process. It takes generations and does not allow cells to adapt to fast changing environmental conditions. On the contrary, epigenetic mechanisms include a number of processes that permit reversible modulation of gene expression, without altering DNA sequence, to rapidly adjust cellular processes to constantly changing environmental conditions.1 These can be transmitted through cell divisions (mitosis) and, in some cases, through generations (meiosis).2,3,4 Among epigenetic mechanisms, the methylation of cytosines in DNA and the microRNAs (miRNAs) have received much attention in human studies, and they are the focus of this article.5 Other epigenetic mechanisms include long noncoding RNAs, post-transcriptional changes of histones and other factors influencing chromatin conformation.6 The modifications of histone cores influence chromatin structure by affecting histone–histone and histone–DNA interactions (reviewed in Tessarz and Kouzarides7). The modifications of histone tails are considered as important mechanisms in the activation and repression of gene transcription.
Many, but not all, cytosines followed by guanines in DNA (the so called CG or CpG dinucleotides) are methylated. The methylation of intergenic regions and gene bodies likely provides stability to DNA, maintains repetitive and transposable elements silenced and reduces spurious gene transcription and transcriptional noise. However, the methylation of cytosines in gene-regulatory regions is involved in the regulation of canonical gene expression.8,9,10 CpG islands are CpG-rich regions that are present in the promoters of many genes. The classical concept was that the methylation of cytosines in promoters' CpG islands was associated with gene repression, whereas the demethylation of those regions was associated with active gene expression.11,12 However, recent studies have delineated a more complex picture. On the one hand, the inverse correlation between DNA methylation and gene expression is not constant; on the other hand, the methylation of distant regulatory regions appears to have an important role in the modulation of gene expression.13,14
miRNAs are endogenous noncoding ribonucleic acids, usually 21–25 nucleotides long, that modulate gene expression at a post-transcriptional level.15 miRNAs are able to inhibit the translation of mRNA and promote their degradation by binding to the 3′ untranslated region (3′-UTR).16 Each miRNA targets hundreds of genes and at the same time each individual gene is targeted by multiple miRNAs. miRNAs are transcribed in the nucleus and afterward they are exported to the cytoplasm. Once there, they undergo a maturation process before becoming functionally active. They can act locally in the cytoplasm or be secreted into exosomes and sent away to other cells or tissues.17
There is a complex interplay between the various epigenetic mechanisms that contribute to the regulation of gene expression. For example, histone lysine methylation and DNA methylation are closely related. Thus, the methylation of lysine 4 (H3K4me, Box 1) is generally associated with gene promoters with unmethylated cytosines and active gene transcription, as recently demonstrated in mesenchymal stem cells.18 On the other hand, H3K9me3 marks are associated with DNA methylation and gene repression.19 Other interactions between epigenetic mechanisms are schematically depicted in Figure 1. More details can be found in recent reviews.5,6,19,20,21,22,23
Definitions in epigenomics.
Locus: Is a specific location of a gene or other genetic mark on a chromosome. In plural it is called Loci.
Enhancers: Enhancers are cis-acting DNA located up to 1Mbps up or downstream from the start site of a given gene. They modulate the transcription.
Promoters: DNA sequences that define where transcription of a gene has to be started and modulate it. They are commonly located upstream of the transcription initiation site.
Untranslated regions (UTR): Regions defined in each side of a coding sequence on a strand of mRNA. These regions are not translated into proteins. The region in the side 5′ is known as 5′-UTR, and in the other side it is called the 3′-UTR.
Transcription factors: Transcription factors are proteins involved in the process of transcribing DNA into RNA. Hence, they regulate gene expression.
Transcription factor-binding sites (TFBSs): Short segments of DNA where the transcription factors bind. They are common in promoter and enhancer regions.
DNA methylation: DNA methylation is an epigenetic modification in the DNA strand, which consists in an addition of a methyl group to DNA. The most common DNA methylation process is the incorporation of a methyl group at the 5-carbon of the Cytosine (5-methylcytosine).
Transcriptional noise: It refers to the variability of gene expression between cells of the same type. It may occur due to several factors including transcriptional bursting of promoters, as well as spurious transcription within coding sequences. The methylation of gene bodies is associated with reduced transcriptional noise.
CpG islands (CpGi): Short interspersed DNA sequences that deviate significantly from the average genomic pattern by being CpG rich and predominantly nonmethylated.
CpG Island shores: Regions flanking CpG islands (up to 2000 base pairs).
Bisulfite conversion: Use of bisulfite treatment of DNA to determine its pattern of methylation. Bisulfite converts unmethylated cytosines to uracil but leaves methylated cytosines unaffected. Thus, bisulfite treatment introduces specific changes in the DNA sequence that depend on the methylation status of individual cytosines. Various techniques can be applied to retrieve the information on the bisulfite-converted DNA sequence, thus inferring the methylation status of the cytosines in the original DNA (arrays, standard sequencing, next-generation sequencing, pyrosequencing, etc.).
Reduced representation bisulfite sequencing (RRBS): Technique used to analyze the genome-wide methylation profiles on a single nucleotide level. This technique combines restriction enzymes and bisulfite sequencing in order to enrich for the regions of the genome that have a high CpG content, and thus reducing the number of nucleotides that need to be sequenced.
Histones: Proteins bound to the DNA that influence chromatin conformation and gene transcription.
Nucleosome: Basic functional unit of the chromatin, composed by 147 basepairs of DNA wrapped around a histone octamer that includes two copies of histones H2A, H2B, H3 and H4.
Histone post-translational modifications: Covalent modifications of histone aminoacids by phosphorylation, methylation, acetylation, ubiquitylation or sumoylation. The modified residues act as docking sites for other proteins that specifically recognize these changes and form molecular complexes that influence chromatin structure and remodeling, as well as DNA repair and gene activity. They are named with letters and numbers stating the histone itself, the single-letter aminoacid code (for example, K stands for lysine, R for arginine, H for histidine, etc. ) and position, and the type of modification. Thus, H3K4me1 stands for histone 3 with methylated lysines at position 4; H3K9me2 stands for histone 3 with dimethylated lysines at position 9.
Noncoding RNAs (ncRNA): Noncoding RNAs are functional RNA molecules that are transcribed but not translated into proteins. They are classified as short noncoding RNAs (<200 nucleotides), including microRNAs, and long noncoding RNAS (>200 nt).
Homeo box: DNA segment characteristic for homeotic genes (Hox genes) that regulate developmental morphogenetic processes.
Figure 1.
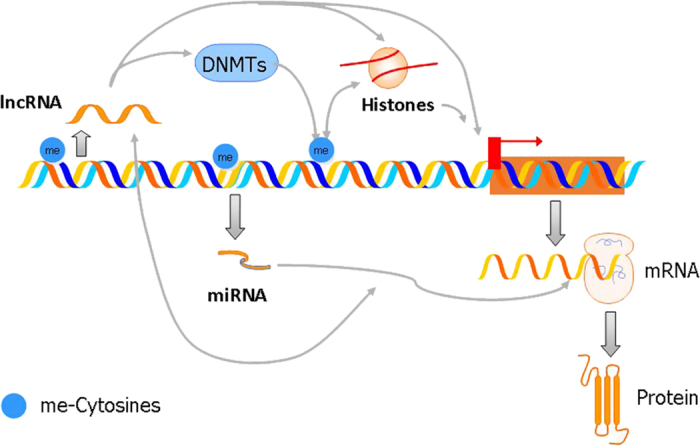
Schematic view of the interplay between epigenetic mechanisms. The transcription of the coding regions of the protein-coding genes originates, after a splicing and maturation of precursors, mature mRNAs that lead the synthesis of the protein chains on the ribosomes. DNA methyltransferases (DNMTs) are a family of enzymes that methylate cytosines in CpG dinucleotides. The methylation/demethylation of CpGs influences gene expression by several mechanisms including changing the binding of transcription factors and the interaction with other enzymes that, in turn, modify histone tails. Chemical modifications of histones (such as methylation and acetylation) influence chromatin conformation and the binding of protein complexes needed for gene transcription. Long noncoding RNAs (lncRNAs) may modulate the activity of DNMTs and interact with the histone machinery. Precursors of microRNAs (miRNAs) are transcribed and processed into mature miRNAs. These miRNAs bind to mRNAs bearing in their 3′ untranslated region sequences complementary to the miRNAs. The binding of miRNAs to their targets mRNAs can stop protein translation or even induce the degradation of the mRNA. Some lncRNAs may also modulate the stability of miRNAs. These, in turn, appear to modulate the transcription of some lncRNA. Thick arrows represent DNA-encoded RNA transcription and RNA-encoded protein translation. Thin arrows represent direct or indirect interactions.
In view of the overwhelming amount of biomedical literature, critical appraisal is an absolute need both for the researcher and the clinician. Thus, a structured approach has been postulated to help analyzing different types of papers such as those concerned with therapy, diagnostic tests or, more recently, genome-wide association studies.24,25,26 In this view, we propose a similar framework to interpret the epigenetic literature—in particular, human epigenetic association studies. We will focus on studies about DNA methylation and miRNAs because they are the most common so far. Following other schemas, we will divide the appraisal into three parts: internal validity, interpretation of the results, and external validity and relevance. The key items to check are based on a previous report20 and are summarized in Table 1.
Table 1. Main issues to consider when appraising the validity and importance of epigenetic association studies (based on Riancho20).
| Domain | Key items |
|---|---|
| Study validity | 1. Subjects |
| •Age and sex | |
| •Diagnostic criteria, disease stage and prior therapy | |
| •Appropriateness of controls | |
| 2. Tissue | |
| •Relevance for disease pathogenesis | |
| •Heterogeneity of cell composition | |
| 3. Technology and data analysis | |
| •Epigenome coverage | |
| •Precision and accuracy | |
| •Statistical errors | |
| Main results | 1. Analysis of association with phenotype |
| •Single locus | |
| •Genomic regions (promoters, enhancers, TFBSs etc.) | |
| 2. Gene network analysis | |
| •Molecular interaction networks | |
| •Pathways | |
| •Other (GSEA, miRNA families etc.) | |
| 3. Functional assays | |
| •In silico | |
| •In vitro | |
| •In vivo | |
| Study importance | 1. Biological rationale |
| 2. Replication in independent populations | |
| 3. Direction of effect (causality and reverse causality) | |
| 4. Practical relevance | |
| •Biomarkers | |
| •Disease pathogenesis | |
| •Direct and indirect therapeutic targets |
Abbreviations: GSEA, gene set enrichment analysis; miRNA, microRNA; TFBS, transcription factor-binding site.
Evaluating study validity
Subjects
It should be stressed that, unlike the genome, the epigenome is not stable. It depends on the sex of the individual and changes with age and the internal and external environmental conditions.9 This poses particular difficulties for carrying out and interpreting epigenetic studies. Therefore, a precise description of the subjects included in the study is a critical issue in order to assess its internal and external validity. For example, DNA methylation changes with aging (Figure 2). In general, there is a trend for decreased methylation with aging, but this is not a universal phenomenon throughout the genome. In fact, the methylation signature at some CpGs has been proposed as a method to estimate the age of an individual.27 Therefore, a clear statement of the age of the subjects included and a proper matching between patients and controls are needed in order to make meaningful comparisons between the study groups. Likewise, the age of the patients needs to be taken into consideration when the results of different studies are compared.
Figure 2.
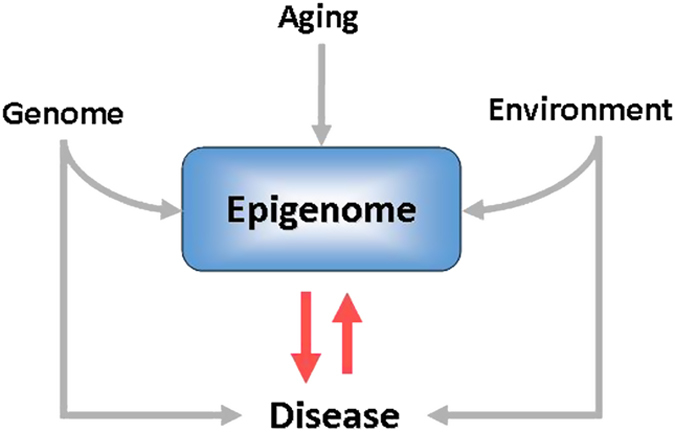
Determinants of the epigenome. The epigenome is the consequence of the interaction between genomic variants and environmental influences, both external and internal (within the body or even within the cell). Aging itself is associated with some epigenomic changes including modifications of the methylation status of DNA. Epigenomic changes induce modifications of gene expression that may lead to disease's development. On the other hand, the changes in cell environment induced by the disease may modify the epigenetic marks. In addition, some epigenomic signatures depend on the cell lineage, which determines the pattern of gene expression characteristic of each cell type. Since the influence of these factors, other than the genome, varies across tissues, the epigenome is tissue and cell specific.
Of course, the phenotype is to be clearly defined. If the study included patients and controls, the diagnostic criteria, and eventually the disease stage or severity, must be precisely established and the appropriateness of controls has to be checked. In addition, the possible influence of previous therapies or interventions on the epigenome needs to be considered. This does not cause any concern in genetic studies but may have an important influence in the results of epigenetic association studies. In fact, several drugs and hormones are known to influence DNA methylation.28,29
In some cases, especially when the study material includes solid tissues, practical and ethical reasons may preclude from obtaining tissue samples from normal controls. Then, comparing patients with different conditions may be an alternative. As an example, Delgado-Calle et al. analyzed DNA methylation in bone from patients with hip fractures and with hip osteoarthritis.30 These between-disease comparisons may produce meaningful results, but it may be difficult to know which disorder is actually different from normal and, consequently, which are the disease-driving changes.
Tissue
One of the convenient characteristics of genetic-association studies is that almost any tissue or fluid can be used as the source of DNA. However, unlike the genetic sequence, epigenomic marks are tissue (or cell) specific.14,31 In other words, the epigenome differs across tissues. Consequently, the consideration of the tissue analyzed is critical for data interpretation. In many human studies, epigenomic data are obtained by analyzing blood cells (that is, in DNA methylation analysis) or plasma (that is, for miRNA studies). Using peripheral blood certainly has some advantages: it can be obtained easily and repeated sampling over time is possible. However, blood-derived results may or may not be relevant to the disease's pathogenesis. For instance, blood data are likely relevant for immune-mediated disorders. On the contrary, in studies concerned with skeletal diseases, the DNA methylation signatures of blood cells may not reflect the methylation pattern of the skeletal cells, which are more directly involved in determining skeletal homeostasis. Thus, as it was the case in some recent studies, bone samples are preferable in principle to study osteoporosis-related epigenetic markers, whereas cartilage samples or synovial samples are preferable in studies of osteoarthritis or rheumatoid arthritis.30,32,33,34 Nevertheless, some intriguing similarities in the methylation of some genes in cartilage and bone have been recently reported.35
There are very scarce data about the correlation of epigenetic marks in blood and the skeleton, but, in view of other studies, caution is recommended before extrapolating blood signatures to bone signatures. Investigators exploring the correlation of DNA methylation between blood and solid tissues found that the dominant difference (>70%) in DNA methylation across samples depends on the source tissue,36 and <3% of CpG sites show r2>0.5 between blood and solid tissues such as brain or muscle.37,38 Therefore, the reader should not assume that epigenetic marks in blood reflect the skeletal epigenome, unless such a correlation is confirmed experimentally. The minority of CpG sites showing correlated methylation across tissues may reflect genetic influences on DNA methylation or aging effects.38,39 When the results in blood samples are comparable to those in bone samples, then there is an opportunity for using blood/serum epigenomic features as biomarkers, as it was suggested in a recent study that explored the association of several miRNAs with osteoporotic fractures.40
Cell heterogeneity is another source of potential bias. For example, in some cases, the apparent differences in epigenomic marks in blood samples may merely reflect differences in the distribution of white blood cell subtypes across individuals. Several algorithms have been proposed to adjust DNA methylation data for differences in blood cell composition.41,42 However, such kind of adjustments may be considerably more difficult in solid tissues such as the bone, where the cell proportions are harder to determine. Fortunately, several mathematical strategies are emerging that allow adjusting the results for differences in cell composition, even in the absence of a reference epigenome.43,44 Therefore, in general, researchers should take care in using comparable tissue samples. If they are to draw conclusions about disease-related epigenomic patterns, they must confirm that the differences between groups do not merely depend on cell distributions but indeed reflect different epigenomic signatures. However, these concerns have not been usually addressed in studies about skeletal epigenetics.30,32
The analysis of a single type of cells, including immortalized cell lines and primary cell cultures (for example, primary osteoblasts grown from bone pieces), could emerge as an interesting alternative to avoid the problem of heterogeneity. However, the results of those studies must be interpreted with caution, because of the modifications of epigenetic signatures that experience cells in culture.45 When feasible, the separation of cell populations from tissue samples, followed by the analysis of the epigenetic marks without the need of culturing the cells, would be an appealing alternative.
In some studies, both target and control samples come from the same individual (typically, in studies with cancer patients, where the tumor tissue is compared with normal tissue of the same subject). Some comparisons may be easier in this situation. For example, it is easier to avoid the influence of DNA sequence (genetics) on DNA methylation when the samples come from the same individual.46,47 However, the issues of tissue relevance and cell composition heterogeneity are still pertinent.
Technology and data analysis
As in any research paper, the methodological aspects of the study should be clearly explained. In addition, some questions specifically apply to epigenomic studies. Genome-wide data analyses, such as arrays and next-generation sequencing, have a number of intricacies regarding technical aspects such as background correction, normalization, probe design bias, annealing and so on.48,49 Authors must describe the analysis pipeline with detail, so that it can be assessed and eventually reproduced. However, these issues may be difficult to appraise fully for the non-expert reader.
Genome coverage
The degree of coverage (that is, the proportion of CpGs in the genome that are actually analyzed, or the proportion of miRNAs that are actually explored) is often the result of a compromise between desirability and feasibility, and it must be kept in mind when interpreting the results. The number of CpGs in the human genome is about 25 × 106. They can be analyzed by whole-genome sequencing after bisulfite conversion (bisulfite treatment of DNA allows distinguishing methylated from unmethylated cytosines), but the procedure is expensive and hardly applicable to studies with large sample sizes. Many studies follow a candidate gene approach and focus just on a genomic region. As a nice example, Harvey et al.50 reported that the methylation of the eNOS promoter (an enzyme involved in nitric oxide synthesis) in cord blood was associated with bone mass at age 9 years. These studies have small costs, but they miss information from the vast majority of the genome. More recently, Reppe51 focused in the analysis of the methylation of the SOST promoter (the gene encoding sclerostin) and its association with BMD.
Several methods have been developed trying to reach a compromise between coverage and cost. Methylation arrays are a popular one. Earlier designs interrogated no more than 27 000 CpG, but they produced some useful results including the identification of differentially methylated genes of the HOX family in patients with osteoporosis and osteoarthritis.30 However, that technology explored just about 1/1000 of the epigenome and consequently missed many regions potentially involved in the pathogenesis of the diseases. Many studies of skeletal and non-skeletal disorders have used more modern array designs that explore the methylation status of more than 450 000 CpGs.34,35,52 The most recent arrays allow the analysis of more than 850 000 sites, but this still represents just about 3% of the potentially methylated cytosines. Reduced representation bisulfite sequencing, which explores about 5 × 106 CpGs,53,54,55 allows a more extensive coverage, at a higher cost. This technology has been used to elucidate the methylation signature of mesenchymal stem cells in young and elderly individuals.56
More than 1900 human miRNAs have been well described, but, according to recent data, the actual number may be much higher.57 As with other transcripts, they can be measured individually by using standard real-time quantitative PCR after reverse transcription of the miRNAs into complementary DNA (RT-qPCR), or in a more comprehensive way. Thus, there are available both expression arrays and microplate-based ‘mini-arrays' (based on RT-qPCR) that allow for the simultaneous measuring of several hundred miRNAs. A more extensive coverage can be obtained by using next-generation sequencing after RNA extraction, also called transcriptome analysis. However, some general transcriptome analysis protocols that are mainly devoted to the analysis of mRNAs may not be adequate for analyzing miRNAs; specific methods for miRNAs are usually needed. The concordance between different platforms is far from perfect,58 which may lead to discordant results across studies.
Precision and accuracy
The precision (that is, reproducibility) of the measurements can be easily determined by including several sample replicates within the same run. Between-run precision is assessed by including some samples repeatedly in all runs. If there is suggestion for poor inter-assay reproducibility, some normalization procedure is mandatory to adjust for batch effects.
The accuracy of the measurements is best confirmed by using an alternative procedure to analyze the samples. For example, miRNA array data can be validated by using qPCR. Methylation array data can be confirmed by using procedures such as standard sequencing, pyrosequencing and so on.
Statistical errors
The large amount of data generated with genome-wide analysis brings forward the issue of statistical errors. The type I error occurs when a difference that appeared by chance is considered to be statistically significant; it is a form of ‘false positive' result. It increases rapidly when many statistical tests are performed (as is the case in genome-wide studies), because then, just by chance, it becomes probable to find some ‘statistically significant' results. To avoid this problem of type I error inflation, there is a need for some correction. The Bonferroni's correction is a classical method to take into consideration the multiple tests carried out on non-correlated markers. Thus, in epigenome-wide studies using the popular 450 K arrays, P-values' significance thresholds between 10−7 and 10−6 have been suggested.59,60 However, Bonferroni's correction is frequently regarded as too conservative. The false discovery rate first proposed by Benjamini61 is a popular alternative, and, perhaps, it has the best combination of statistical power (the ability to pinpoint as statistically significant differences or associations that are real) and specificity (the ability to identify as non-significant differences in the database that do not represent real differences in the population). This was the method for correction used in several epigenome-wide studies in the bone field (Box 2).30,32,56
Adjustment of significance thresholds.
Bonferroni correction: A method to correct for the inflation of type I error (the probability of obtaining false-positive associations) when multiple tests are performed. It implies that between-group differences are considered significant only when the P-values are below an ‘adjusted threshold'. This is estimated by dividing the usual significance threshold (P=0.05) by the number of tests performed. For example, if 500 000 DNA sites are analyzed in patients and controls, only those with P-values below 10−7 (0.05/500 000) will be considered as significantly different.
False discovery rate (FDR): Another approach to control the inflation of type I errors in the context of multiple comparisons. It represents the expected proportion of false-positive associations among the associations declared as statistically significant. The FDR-based threshold is usually set at 0.05 or 0.1, which means that a 5% or 10% of the significant results are assumed to be spurious. These procedures are less stringent than those based on the familywise error rate such as Bonferroni correction. Therefore, the FDR-based approach has greater power and also higher rates of type I errors than Bonferroni's approach. Consequently, FDR-set thresholds may be specially suited for exploratory, hypothesis-generating studies.
Nevertheless, any procedure increasing the statistical stringency to avoid the inflation of type I error implicates increasing the chances of type II error. This is the error occurring when true differences or associations are not identified because they do not appear to be ‘significant' in the study. Thus, type II error is a cause of false-negative results. Other factors that increase type II error are a small effect of the epigenetic mark on the phenotype and a small sample size. In short, the reader critically appraising a paper showing significant results should ask whether they are real or just the consequence of inflated type I error, especially if the authors did not consider multiple test issues. On the other hand, if the study does not find an association, the reader should consider whether the power of the study was enough to find significant differences if they actually existed.
Understanding the results
Single locus and region analysis
The results of association studies can be depicted at several levels of complexity. The first step is usually to explore the association of single loci with the phenotype. For example, if miRNAs are being explored, then typically the expression of individual miRNAs is measured across phenotypes.
If the study aims to explore DNA methylation, then the association of individual CpGs with the phenotype is first explored. Changes in the methylation of a single CpG may be functionally relevant (for example, if they block a critical transcription factor-binding site). However, to obtain a deeper insight of the significance of the methylation differences within the genomic context, other analyses are frequently carried out after grouping or stratifying the CpGs. Thus, it is common to explore the overall methylation of CpGs within CpG islands, island shores, gene bodies or regulatory regions such as promoters or enhancers. Indeed, differentially methylated regions (DMRs) are frequently found within enhancer-like regions, outside classic gene-centric genomic locations (such as promoters or exons).14 For these analyses, CpGs are grouped within a given genomic region, and an overall methylation value for the region is obtained.
Network analysis
Most cellular activities depend not only on a single gene but also on several interconnected genes that are activated or repressed in a concerted way. Therefore, many epigenomic changes involve several related loci. Thus, investigators frequently check whether the genes showing methylation differences are connected in some way (network analysis) or whether they are overrepresented in any biochemical or cellular pathway (pathways analysis). There are many software tools to perform these analyses (Box 3). They can be grouped into two general categories. In the simplest one, the researcher checks whether there is an increased proportion of genes with differential methylation in a given pathway (in comparison with the overall proportion in the genome). In other words, the analysis considers a pathway as a simple gene list, disregarding any knowledge of gene or protein interactions, and compares it with the list of differentially methylated loci in the study. More sophisticated approaches consider the direction and the magnitude of the differences found in the study (a procedure often called gene set enrichment analysis) or the topological structure of a pathway. The relative value of each of these methods has not been clearly established yet.62
Selected websites with epigenomic databases and tools.
http://www.ifmrs.org/ifmrs-big-data-website-inventory. Inventory of websites with information about genomics, epigenomics and transcriptomics created by the International Federation of Musculoskeletal Research Societies.
https://www.encodeproject.org/. The ENCODE (Encyclopedia of DNA Elements) Consortium aims to build a comprehensive list of functional elements in the human genome.
https://genome.ucsc.edu. UCSC Genome Browser website. This extensively visited site contains the reference sequence and working draft assemblies for a large collection of genomes. It provides various tracks from other databases to compare with the genomic regions
http://www.ensembl.org. The Ensembl project produces genome databases for vertebrates and other eukaryotic species.
http://epigenomegateway.wustl.edu/browser/roadmap. Genome browser of the Washington University with data from several tissues and species, and links to the NIH Roadmap Epigenomics Mapping Consortium.
http://thebiogrid.org. BioGRID is an interaction repository with curated data.
http://bejerano.stanford.edu/great/public/html. GREAT assigns biological meaning to a set of noncoding genomic regions by analyzing the annotations of the nearby genes.
http://geneontology.org. The gene ontology consortium provides online tools for enrichment analysis and other analyses.
http://babelomics.bioinfo.cipf.es. Babelomics provides online tools for gene expression, genome variation and functional profiling analysis.
https://david.ncifcrf.gov/. A widely used web resource for enrichment analysis and functional annotations.
http://bioinfo.vanderbilt.edu/webgestalt/. WEBGESTALT (WEB-based GEne SeT AnaLysis Toolkit) is a webtool that performs various enrichment and network analysis.
http://www.networkanalyst.ca/NetworkAnalyst/. NetworkAnalyst is designed to support integrative analysis of gene expression from a list of proteins or genes derived from expression experiments.
http://software.broadinstitute.org/gsea/index.jsp. GSEA (gene set enrichment analysis) determines whether a set of genes shows statistically significant, concordant differences between two biological states. It takes into consideration not only the genes but also their relative contributions (as represented by measures such as P-values or fold-changes).
http://www.mirbase.org/. Searchable database of published miRNAs and annotations.
http://mirtarbase.mbc.nctu.edu.tw/index.php. Database of miRNA-target interactions witth experimental validation and curated manually.
http://www.cbrc.kaust.edu.sa/mirnavisa/manual.php. Web application that allows interrogation and comparisons of miRNA families for hypotheses generation and comparison of the per-species chromosomal distribution of miRNA genes in different families.
Similarly, several bioinformatic tools permit the analysis of miRNA families and the interaction of miRNAs with target genes and biological pathways (Box 3).63,64
Network analysis including several layers of epigenomic and transcriptomic signatures may help in indentifying relevant genes and pathways, as exemplified by a recent study that explored gene expression, miRNAs and DNA methylation in relation to bone mineral density.65 Whichever analysis is chosen, in general, if several genes within a given pathway show epigenetic differences between phenotypes, they are more likely to have an important role in determining those phenotypes.
Functional studies
Successful epigenetic epidemiology studies may reveal differences in DNA methylation (or miRNA expression) between individuals with a different phenotype (such as patients and controls). However, they rarely provide evidence for causality. Therefore, functional studies are usually needed to validate the biological relevance of the results. A number of approaches can be useful to obtain mechanistic information. They may include in silico analysis (for example, searching for relationship between DNA methylation and gene expression in publicly available databases), in vitro analysis (for example, exploring the influence of overexpressing or knocking-down a miRNA in cell cultures), or experimental in vivo models.
Assessing study importance
Biological rationale
Epigenetic association data are always more credible when they fit with previous knowledge about the biology of disease. However, genome-wide genetic and epigenetic studies have the potential to uncover new genes involved in the mechanisms of disease. Thus, new discoveries must not be merely disregarded because there is no known biological rationale for them. Rather, they may open new opportunities for a better understanding of the disease or identifying novel therapeutic targets. The relevance of finding DMRs is often based on their correlation with changes in gene expression. However, even if a DMR does not impact transcription and gene expression, it may have some other yet unknown effects or serve as a marker of the exposure (for example, maintaining a memory of exposure over lifetime) or outcome of interest (that is, biomarker of the disease).66
Replication
It is highly desirable confirming the study findings obtained in the initial group of subjects (the ‘discovery' sample) in an independent group of individuals (the ‘replication' sample).This proves that the initial results were real and not merely a chance finding. The replication in a group of individuals from a different geographical region is also desirable in order to confirm that the results are generalizable to different populations. However, the absence of replication in populations from different regions does not necessarily imply that the results were wrong. Epigenomic marks are the consequence of the interaction of genetic variants, nutrition and other environmental factors, aging and stochastic variation.9,67,68,69,70 Therefore, differences in the genetic background, environmental exposures, age and other characteristics of the study subjects may explain why true epigenetic differences present in one study population are not present in other populations.
Direction of effects and causation
One of the convenient features of genetic association studies is that the results do not depend on the time point the genetic data are acquired. As the genome remains stable from the conception (apart from some somatic mutations), the possibility of reverse causation does not hold. In other words, the genome is established first, and therefore the direction of the relationship must go from the genome to the phenotype. However, that is not the case with epigenetic studies, which often raise the possibility of reverse causation. For example, if some epigenetic differences are found between patients and controls, the reader should ask whether the epigenetic differences are the cause or the consequence of the disease. For example, increased methylation of the SOST promoter has been reported in bone samples of patients with osteoporosis.51 As sclerostin is an antianabolic factor, this finding may be counterintuitive; yet, it is in line with studies showing reduced SOST expression in osteoporosis.51,71 Although the involved mechanisms are still unclear, it has been suggested that those changes are a compensatory response, rather than a driver of the bone loss.
Answering the question of the direction of changes may not be important if the epigenetic marks are to be used as biomarkers for diagnostic or prognostic purposes, but it is certainly critical in studies about the pathogenetic mechanisms determining the disease. The response may need longitudinal studies such as cohorts incorporating the analysis of epigenetic signatures in healthy individuals before the onset of disease.72 Biobanking strategies of blood and other body fluids may be very useful in this regard. However, a similar approach may not be feasible to study tissue samples such as bone. Functional studies using experimental models may be needed in those cases.
Practical relevance
Epigenetic studies may be important for a variety of reasons, both in the short term and in the long term. The latter may not be readily evident. However, some studies may clearly have direct relevance for clinical practice.
Epigenetic marks may have a role as biomarkers for disease risk, for diagnosing disease or for tailoring therapy. With those goals in mind, the markers need to be feasible to analyze in readily available samples such as blood, urine or tissue biopsies. miRNAs may be particularly attractive as biomarkers in body fluids because they are quite stable.73
The interference of epigenetic mechanisms may also be of therapeutic interest, particularly in localized disorders. Thus, blocking miRNAs that inhibit osteoblast differentiation or delivering miRNAs that promote bone formation are both actively investigated strategies to promote fracture healing and for curing bone defects.74 As most miRNAs have widespread effects, their use for systemic disorders is less appealing.
Epigenetic association studies may have therapeutical importance for indirect reasons. If the studies pinpoint new genes involved in the pathogenesis of disease, those genes may eventually be targeted by pharmacological methods independent of epigenetic mechanisms.
Acknowledgments
Authors' research is supported by grants (PI12/0615) of the Instituto de Salud Carlos III (Spain) that may be partially financed by the European Regional Development Fund of the EU.
Footnotes
The authors declare no conflict of interest.
References
- Zucchi FC, Yao Y, Metz GA. The secret language of destiny: stress imprinting and transgenerational origins of disease. Front Genet 2012; 3: 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein BE, Meissner A, Lander ES. The mammalian epigenome. Cell 2007; 128: 669–681. [DOI] [PubMed] [Google Scholar]
- Jirtle RL, Skinner MK. Environmental epigenomics and disease susceptibility. Nat Rev Genet 2007; 8: 253–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake AJ, Liu L, Kerrigan D, Meehan RR, Seckl JR. Multigenerational programming in the glucocorticoid programmed rat is associated with generation-specific and parent of origin effects. Epigenetics 2011; 6: 1334–1343. [DOI] [PubMed] [Google Scholar]
- Delgado-Calle J, Garmilla P, Riancho JA. Do epigenetic marks govern bone mass and homeostasis? Curr Genomics 2012; 13: 252–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon JA, Montecino MA, Aqeilan RI, Stein JL, Stein GS, Lian JB. Epigenetic pathways regulating bone homeostasis: potential targeting for intervention of skeletal disorders. Curr Osteoporos Rep 2014; 12: 496–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessarz P, Kouzarides T. Histone core modifications regulating nucleosome structure and dynamics. Nat Rev Mol Cell Biol 2014; 15: 703–708. [DOI] [PubMed] [Google Scholar]
- Huh I, Zeng J, Park T, Yi SV. DNA methylation and transcriptional noise. Epigenetics Chromatin 2013; 6: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvanese V, Lara E, Kahn A, Fraga MF. The role of epigenetics in aging and age-related diseases. Ageing Res Rev 2009; 8: 268–276. [DOI] [PubMed] [Google Scholar]
- Allis CD, Jenuwein T, Reinberg D, Caparros ML. Epigenetics Cold Spring Harbor: Cold Spring Harbor Laboratory Press2007; . [Google Scholar]
- Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet 2012; 13: 484–492. [DOI] [PubMed] [Google Scholar]
- Deaton AM, Webb S, Kerr AR, Illingworth RS, Guy J, Andrews R et al. Cell type-specific DNA methylation at intragenic CpG islands in the immune system. Genome Res 2011; 21: 1074–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddington JP, Pennings S, Meehan RR. Non-canonical functions of the DNA methylome in gene regulation. Biochem J 2013; 451: 13–23. [DOI] [PubMed] [Google Scholar]
- Ziller MJ, Gu H, Muller F, Donaghey J, Tsai LT, Kohlbacher O et al. Charting a dynamic DNA methylation landscape of the human genome. Nature 2013; 500: 477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol 2014; 15: 509–524. [DOI] [PubMed] [Google Scholar]
- Erson AE, Petty EM. MicroRNAs in development and disease. Clin Genet 2008; 74: 296–306. [DOI] [PubMed] [Google Scholar]
- Properzi F, Ferroni E, Poleggi A, Vinci R. The regulation of exosome function in the CNS: implications for neurodegeneration. Swiss Med Wkly 2015; 145: w14204. [DOI] [PubMed] [Google Scholar]
- Fernandez AF, Bayon GF, Urdinguio RG, Torano EG, Garcia MG, Carella A et al. H3K4me1 marks DNA regions hypomethylated during aging in human stem and differentiated cells. Genome Res 2015; 25: 27–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose NR, Klose RJ. Understanding the relationship between DNA methylation and histone lysine methylation. Biochim Biophys Acta 2014; 1839: 1362–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riancho JA. Epigenetics of osteoporosis: critical analusis of epigenetic epidemiology studies. Curr Genomics 2015; 16: 405–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynard LN, Loughlin J. Genetics and epigenetics of osteoarthritis. Maturitas 2012; 71: 200–204. [DOI] [PubMed] [Google Scholar]
- Vrtacnik P, Marc J, Ostanek B. Epigenetic mechanisms in bone. Clin Chem Lab Med 2014; 52: 589–608. [DOI] [PubMed] [Google Scholar]
- Pi C, Li YP, Zhou X, Gao B. The expression and function of microRNAs in bone homeostasis. Front Biosci (Landmark Ed) 2015; 20: 119–138. [DOI] [PubMed] [Google Scholar]
- Pearson TA, Manolio TA. How to interpret a genome-wide association study. JAMA 2008; 299: 1335–1344. [DOI] [PubMed] [Google Scholar]
- Little J, Higgins JP, Ioannidis JP, Moher D, Gagnon F, von Elm E et al. STrengthening the REporting of Genetic Association studies (STREGA)–an extension of the STROBE statement. Eur J Clin Invest 2009; 39: 247–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeschke R, Guyatt G, Sackett DL. Users' guides to the medical literature. III. How to use an article about a diagnostic test. A. Are the results of the study valid? Evidence-Based Medicine Working Group. JAMA 1994; 271: 389–391. [DOI] [PubMed] [Google Scholar]
- Bocklandt S, Lin W, Sehl ME, Sanchez FJ, Sinsheimer JS, Horvath S et al. Epigenetic predictor of age. PLoS ONE 2011; 6: e14821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nugent BM, Wright CL, Shetty AC, Hodes GE, Lenz KM, Mahurkar A et al. Brain feminization requires active repression of masculinization via DNA methylation. Nat Neurosci 2015; 18: 690–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni G, Qin J, Li H, Chen Z, Zhou Y, Fang Z et al. Effects of antiepileptic drug monotherapy on one-carbon metabolism and DNA methylation in patients with epilepsy. PLoS ONE 2015; 10: e0125656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado-Calle J, Fernandez AF, Sainz J, Zarrabeitia MT, Sanudo C, Garcia-Renedo R et al. Genome-wide profiling of bone reveals differentially methylated regions in osteoporosis and osteoarthritis. Arthritis Rheum 2013; 65: 197–205. [DOI] [PubMed] [Google Scholar]
- Schones DE, Zhao K. Genome-wide approaches to studying chromatin modifications. Nat Rev Genet 2008; 9: 179–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano K, Whitaker JW, Boyle DL, Wang W, Firestein GS. DNA methylome signature in rheumatoid arthritis. Ann Rheum Dis 2013; 72: 110–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts SB, Wootton E, De Ferrari L, Albagha OM, Salter DM. Epigenetics of osteoarticular diseases: recent developments. Rheumatol Int 2015; 35: 1293–1305. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Fukui N, Yahata M, Katsuragawa Y, Tashiro T, Ikegawa S et al. Genome-wide DNA methylation profile implicates potential cartilage regeneration at the late stage of knee osteoarthritis. Osteoarthritis Cartilage 2016; 24: 835–843. [DOI] [PubMed] [Google Scholar]
- Jeffries MA, Donica M, Baker L, Stevenson M, Annan AC, Humphrey MB et al. Genome-wide DNA methylation study identifies significant epigenomic changes in osteoarthritic subchondral bone and similarity to overlying cartilage. Arthritis Rheumatol (doi:10.1002/art.39555). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farre P, Jones MJ, Meaney MJ, Emberly E, Turecki G, Kobor MS. Concordant and discordant DNA methylation signatures of aging in human blood and brain. Epigenetics Chromatin 2015; 8: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannon E, Lunnon K, Schalkwyk L, Mill J. Interindividual methylomic variation across blood, cortex, and cerebellum: implications for epigenetic studies of neurological and neuropsychiatric phenotypes. Epigenetics 2015; 10: 1024–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slieker RC, Bos SD, Goeman JJ, Bovee JV, Talens RP, van der BR et al. Identification and systematic annotation of tissue-specific differentially methylated regions using the Illumina 450k array. Epigenetics Chromatin 2013; 6: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath S, Mah V, Lu AT, Woo JS, Choi OW, Jasinska AJ et al. The cerebellum ages slowly according to the epigenetic clock. Aging (Albany NY) 2015; 7: 294–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeliger C, Karpinski K, Haug AT, Vester H, Schmitt A, Bauer JS et al. Five freely circulating miRNAs and bone tissue miRNAs are associated with osteoporotic fractures. J Bone Miner Res 2014; 29: 1718–1728. [DOI] [PubMed] [Google Scholar]
- Jaffe AE, Irizarry RA. Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biol 2014; 15: R31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang L, Cookson WO. Grasping nettles: cellular heterogeneity and other confounders in epigenome-wide association studies. Hum Mol Genet 2014; 23: R83–R88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houseman EA, Kim S, Kelsey KT, Wiencke JK. DNA methylation in whole blood: uses and challenges. Curr Environ Health Rep 2015; 2: 145–154. [DOI] [PubMed] [Google Scholar]
- Houseman EA, Kelsey KT, Wiencke JK, Marsit CJ. Cell-composition effects in the analysis of DNA methylation array data: a mathematical perspective. BMC Bioinformatics 2015; 16: 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestor CE, Ottaviano R, Reinhardt D, Cruickshanks HA, Mjoseng HK, McPherson RC et al. Rapid reprogramming of epigenetic and transcriptional profiles in mammalian culture systems. Genome Biol 2015; 16: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drong AW, Nicholson G, Hedman AK, Meduri E, Grundberg E, Small KS et al. The presence of methylation quantitative trait loci indicates a direct genetic influence on the level of DNA methylation in adipose tissue. PLoS ONE 2013; 8: e55923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertz J, Varley KE, Reddy TE, Bowling KM, Pauli F, Parker SL et al. Analysis of DNA methylation in a three-generation family reveals widespread genetic influence on epigenetic regulation. PLoS Genet 2011; 7: e1002228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan H, Chen L, Dogra S, Teh AL, Tan JH, Lim YI et al. Measuring the methylome in clinical samples: improved processing of the Infinium Human Methylation450 BeadChip Array. Epigenetics 2012; 7: 1173–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock C. Analysing and interpreting DNA methylation data. Nat Rev Genet 2012; 13: 705–719. [DOI] [PubMed] [Google Scholar]
- Harvey NC, Lillycrop KA, Garratt E, Sheppard A, McLean C, Burdge G et al. Evaluation of methylation status of the eNOS promoter at birth in relation to childhood bone mineral content. Calcif Tissue Int 2012; 90: 120–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reppe S, Noer A, Grimholt RM, Halldorsson BV, Medina-Gomez C, Gautvik VT et al. Methylation of bone SOST, its mRNA, and serum sclerostin levels correlate strongly with fracture risk in postmenopausal women. J Bone Miner Res 2015; 30: 249–256. [DOI] [PubMed] [Google Scholar]
- Dick KJ, Nelson CP, Tsaprouni L, Sandling JK, Aissi D, Wahl S et al. DNA methylation and body-mass index: a genome-wide analysis. Lancet 2014; 383: 1990–1998. [DOI] [PubMed] [Google Scholar]
- Laird PW. Principles and challenges of genomewide DNA methylation analysis. Nat Rev Genet 2010; 11: 191–203. [DOI] [PubMed] [Google Scholar]
- Bock C, Tomazou EM, Brinkman AB, Muller F, Simmer F, Gu H et al. Quantitative comparison of genome-wide DNA methylation mapping technologies. Nat Biotechnol 2010; 28: 1106–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu H, Smith ZD, Bock C, Boyle P, Gnirke A, Meissner A. Preparation of reduced representation bisulfite sequencing libraries for genome-scale DNA methylation profiling. Nat Protoc 2011; 6: 468–481. [DOI] [PubMed] [Google Scholar]
- Roforth MM, Farr JN, Fujita K, McCready LK, Atkinson EJ, Therneau TM et al. Global transcriptional profiling using RNA sequencing and DNA methylation patterns in highly enriched mesenchymal cells from young versus elderly women. Bone 2015; 76: 49–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Londin E, Loher P, Telonis AG, Quann K, Clark P, Jing Y et al. Analysis of 13 cell types reveals evidence for the expression of numerous novel primate- and tissue-specific microRNAs. Proc Natl Acad Sci USA 2015; 112: E1106–E1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Howel P, Bruheim S, Ju J, Owen LB, Fodstad O et al. Systematic evaluation of three microRNA profiling platforms: microarray, beads array, and quantitative real-time PCR array. PLoS ONE 2011; 6: e17167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai PC, Bell JT. Power and sample size estimation for epigenome-wide association scans to detect differential DNA methylation. Int J Epidemiol 2015; 44: 1429–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehne B, Drong AW, Loh M, Zhang W, Scott WR, Tan ST et al. A coherent approach for analysis of the Illumina HumanMethylation450 BeadChip improves data quality and performance in epigenome-wide association studies. Genome Biol 2015; 16: 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Yekutieli D. Quantitative trait loci analysis using the false discovery rate. Genetics 2005; 171: 783–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayerlova M, Jung K, Kramer F, Klemm F, Bleckmann A, Beissbarth T. Comparative study on gene set and pathway topology-based enrichment methods. BMC Bioinformatics 2015; 16: 334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu C, Wang J, Cui Q. miR2Gene: pattern discovery of single gene, multiple genes, and pathways by enrichment analysis of their microRNA regulators. BMC Syst Biol 2011; 5(Suppl 2): S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamanu TK, Radovanovic A, Archer JA, Bajic VB. Exploration of miRNA families for hypotheses generation. Sci Rep 2013; 3: 2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JG, Tan LJ, Xu C, He H, Tian Q, Zhou Y et al. Integrative analysis of transcriptomic and epigenomic data to reveal regulation patterns for BMD variation. PLoS ONE 2015; 10: e0138524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michels KB, Binder AM, Dedeurwaerder S, Epstein CB, Greally JM, Gut I et al. Recommendations for the design and analysis of epigenome-wide association studies. Nat Methods 2013; 10: 949–955. [DOI] [PubMed] [Google Scholar]
- Talens RP, Christensen K, Putter H, Willemsen G, Christiansen L, Kremer D et al. Epigenetic variation during the adult lifespan: cross-sectional and longitudinal data on monozygotic twin pairs. Aging Cell 2012; 11: 694–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almen MS, Nilsson EK, Jacobsson JA, Kalnina I, Klovins J, Fredriksson R et al. Genome-wide analysis reveals DNA methylation markers that vary with both age and obesity. Gene 2014; 548: 61–67. [DOI] [PubMed] [Google Scholar]
- Kok DE, Dhonukshe-Rutten RA, Lute C, Heil SG, Uitterlinden AG, van d V et al. The effects of long-term daily folic acid and vitamin B12 supplementation on genome-wide DNA methylation in elderly subjects. Clin Epigenetics 2015; 7: 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feil R, Fraga MF. Epigenetics and the environment: emerging patterns and implications. Nat Rev Genet 2011; 13: 97–109. [DOI] [PubMed] [Google Scholar]
- Delgado-Calle J, Arozamena J, Garcia-Renedo R, Garcia-Ibarbia C, Pascual-Carra MA, Gonzalez-Macias J et al. Osteocyte deficiency in hip fractures. Calcif Tissue Int 2011; 89: 327–334. [DOI] [PubMed] [Google Scholar]
- Richmond RC, Al Amin A, Smith GD, Relton CL. Approaches for drawing causal inferences from epidemiological birth cohorts: a review. Early Hum Dev 2014; 90: 769–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorval V, Nelson PT, Hebert SS. Circulating microRNAs in Alzheimer's disease: the search for novel biomarkers. Front Mol Neurosci 2013; 6: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang S, Deng Y, Gu P, Fan X. MicroRNAs regulate bone development and regeneration. Int J Mol Sci 2015; 16: 8227–8253. [DOI] [PMC free article] [PubMed] [Google Scholar]