

Abstract
INTRODUCTION
Physiologic testosterone continuously stimulates prostate stromal cell secretion of paracrine growth factors (PGFs), which if unopposed would induce hyperplastic overgrowth of normal prostate epithelial cells (PrECs).
METHODS
Lentiviral shRNA stable knock down of c-MYC, β-catenin, or TCF-4 completely inhibits normal (i.e., non-transformed) human PrECs growth. c-MYC enhancer driven reporter expression and growth is inhibited by two chemically distinct molecules, which prevent β-catenin signaling either by blocking TCF-4 binding (i.e., toxoflavin) or by stimulating degradation (i.e., AVX939). Recombinant DKK1 protein at a dose, which inhibits activation of canonical Wnt signaling does not inhibit PrEC growth. Nuclear β-catenin translocation and PrEC growth is prevented by both lack of PGFs or Akt inhibitor-I. Growth inhibition induced by lack of PGFs, toxoflavin, or Akt inhibitor-I is overcome by constitutive c-MYC transcription.
RESULTS
In the presence of continuous PGF signaling, PrEC hyperplasia is prevented by androgen binding to AR suppressing c-MYC transcription, resulting in G0 arrest/terminal differentiation independent of Rb, p21, p27, FoxP3, or down regulation of growth factors receptors and instead involves androgen-induced formation of AR/β-catenin/TCF-4 complexes, which suppress c-MYC transcription. Such suppression does not occur when AR is mutated in its zinc-finger binding domain.
DISCUSSION
Proliferation of non-transformed human PrECs is dependent upon c-MYC transcription via formation/binding of β-catenin/TCF-4 complexes at both 5′ and 3′ c-MYC enhancers stimulated by Wnt-independent, PGF induced Akt signaling. In the presence of continuous PGF signaling, PrEC hyperplasia is prevented by androgen-induced formation of AR/β-catenin/TCF-4 complexes, which retains binding to 3′ c-MYC enhancer, but now suppresses c-MYC transcription.
Keywords: androgen receptor; human prostate epithelial cells; c-MYC, β-catenin; TCF-4
INTRODUCTION
The prostate is the most common site of neoplastic transformation in the human body. Such transformation can be either benign [i.e., benign prostatic hyperplasia (BPH)] or malignant [i.e., prostate cancer (PCa)]. By the age of 50, half of males throughout the world have histologically detectable BPH with eventually a quarter developing clinical symptoms of BPH [1]. One in six American males will develop PCa during their lifetime [2]. Thus, it is remarkable that despite these staggering demographic facts, the mechanism(s) for neoplastic transformation of prostatic epithelium are not established. It is established that the prostate is dependent upon sufficient level of circulating testosterone for its development, growth, and maintenance. Circulating testosterone is converted in prostate tissue to dihydrotestosterone (DHT), which is the major intracellular ligand, which binds to androgen receptor (AR) initiating it’s signaling. The critical importance of chronically maintaining a sufficient level of androgen is documented by the fact that androgen deprivation induces the regression of the prostatic epithelium [3]. Such regression is fully reversible, however, since androgen replacement results in complete epithelial cell regeneration, which is self-limiting so that prostatic epithelial hyperplasia does not normally occur [4]. In fact, the prostate can undergo more than 30 successive cycles of androgen deprivation and replacement without diminishing its ability for full epithelial regeneration and without inducing hyperplasia [4]. A large number of independent groups have clarified that the prostate epithelium is organized into adult epithelial stem cell units and how this organization allows such profound cyclic regenerative growth capacity without hyperplasia [4–9].
In these adult prostate epithelial stem units, AR-negative adult prostate epithelial stem cells are located in the basal epithelial layer in niches, which stimulates their survival but limits their proliferation. The androgen independence of the adult prostate epithelial stem cells is documented by the fact that epithelial morphogenesis occurs even when androgen receptor is not expressed by prostate epithelial cells as long as there is expression and signaling of AR in the supporting stromal cells [10]. The mechanism for how such epithelial morphogenesis involves the hierarchical expansion/maturation of adult prostate epithelial stem cells and their progeny [5]. Under the appropriate conditions, AR-negative adult prostate epithelial stem cells divide asymmetrically to self-renew and to give rise to progeny, which differentiate into either non-proliferating AR-negative neuroendocrine cells or ΔNp63-positive/AR-negative transient amplifying (TA) cells. The basal located AR negative TA cells undergo a limited number of amplifying rounds of proliferation before maturing into ΔNp63-negative/prostate stem cell antigen (PSCA)-positive intermediate cells [5]. Such TA proliferation requires androgen dependent AR signaling within AR expressing prostate stromal cells, which stimulates production and secretion of diffusible stromal-derived peptide growth factors collectively termed “andromedins” [11–14]. These paracrine-secreted andromedins diffuse from the stroma into the epithelial compartment where their binding to cognate receptors stimulates AR negative TA cell proliferation and maturation into intermediate cells. Intermediate cells migrate to the luminal layer where they express AR protein, which when occupied by DHT induces their terminally differentiate into prostate specific antigen (PSA) positive secretory-luminal cells whose survival is critically dependent upon adequate levels of andromedins [5]. Due to the hierarchically expanding nature of this process, secretory-luminal cells are the most numerous cell type within an adult prostate epithelial stem cell unit, even though they eventually terminally differentiate (i.e., terminally arrested in G0).
In a non-castrated adult male, high level of circulating testosterone continuously stimulates andromedin production, which if unopposed would induce hyperplastic overgrowth of the gland. Since in the adult human male, only a small fraction [i.e., <1% per day] of prostate epithelial cells (PrECs) are proliferating [15], there is a mechanism to suppress andromedin-stimulated proliferation. Based upon the fact that the majority of epithelial proliferation is located in the basal cells not expressing AR protein and that the AR positive secretory-luminal cells are proliferatively quiescent [16,17], attention has focused on the role of the AR as a suppressor of proliferation of prostate epithelial cells. For example, it has been demonstrated experimentally that AR signaling activated by androgen binding in PrECs induces their growth arrest [18–21] and eventual terminal differentiation into secretory-luminal cells [21,22]. Likewise, transgenic mouse studies have documented that when the AR gene is selectively knocked-out in secretory-luminal cells within the prostate, these AR deficient cells become hyperplastic and do not terminally differentiate [23,24]. These combined data demonstrate that androgen-dependent AR-signaling within AR expressing PrECs suppresses their growth thus preventing prostatic epithelial hyperplasia even in the presence of continuous andromedins production. The present studies focused upon identifying the mechanism of androgen-dependent AR-mediated growth suppression in normal AR-expressing PrECs.
MATERIALS AND METHODS
Reagents
The synthetic androgen R1881 was purchased from Perkin Elmer (Boston, MA). The AR antagonist, Casodex was purchased from LKT laboratories (St. Paul, MN). Toxoflavin [i.e., (1-methyl-6-methylpyrimido[5,4-e]-1,2,4-triazine-5,7(1H,6H)-diones) also known as PKF-118-310] was obtained from the Drug Synthesis and Chemistry Branch, Developmental Therapeutics Program, Division of Cancer Treatment and Diagnosis, National Cancer Institute (Bethesda, MD). XAV939 was obtained from Cellagen Technology (San Diego). AKT-inhibitor I was obtained from EMD (La Jolla, CA). Recombinant DKK-1(cat#5439-DK/CF) was purchased from R&D Systems (Minneapolis, MN). All other chemicals were purchased from JT Baker (Phillipsburg, NJ) or Sigma–Aldrich (St. Louis, MO). PrEC and PrEC-hTERT human prostate lines were obtained with informed consent according to Johns Hopkins Medicine-IRB approved protocol NA_00001575 as previously described [25]. Human normal prostate epithelial cells (PrECs) from young donors were obtained commercially from Lonza (Lonza/Cambrex, Walkersville, MD). Human normal prostate stromal cells [PrSC] from older donors undergoing radical prostatectomy were established according to a Johns Hopkins Medicine-IRB approved protocol NA_00001575 and grown in RPMI-1640 plus 10% fetal bovine serum (FBS) as previously described [26]. PrEC and all its derivatives were grown in Keratinoctye Serum Free defined media supplemented with growth factors (GFs) [standard K-SFM] as supplied by manufacture (Invitrogen Life Technologies, Carlsbad, CA). The WPMY-1 (cat# CRL-2854) immortalized human prostate stromal cell line was obtained commercially from the ATCC (Manassas, VA) and serially passaged in RPMI-1640 media plus 10% FBS. All cells were routinely screened for the absence of mycoplasma contamination.
In Vitro Growth Assays and Time Lapse Microscopy
Cell growth over a 1-week period was measured by a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay [CellTiter 96 Non-Radioactive Cell Proliferation Assay from Promega Corp. (Madison, WI)] as previously described [27]. Time lapse digital microscopy was performed using a TE2000 (Nikon, Melville, NY) inverted microscope with a heated stage and the Live Cell (Pathology Devices) CO2 chamber using an ELWD 20× objective and the Photometric CoolSnap ES digital camera; images were captured using Elements AR software program (Nikon).
Vectors
Creation of lentiviral-GFP control vector and the expression vector containing GFP and wild type or mutant AR flanked by loxP sites (lenti-GFP/AR vector) were described previously [28]. Isolation of GFP-expressing populations of cells following transduction with these vectors was via a Becton Dickinson FACS Aria machine as described previously [21]. The Lenti-GFP/AR(A573D) mutant vector was created by replacing the Bsu36I/Tth111I fragment with the same fragment from an A573D mutant vector generously provided by Dr. Hetty van der Korput [29]. The Lenti-GFP/AR(A573D) vector was sequenced to confirm the presence of the DNA-binding domain mutation. The pWZL-Blast-MYC plasmid was obtained from Addgene (Cambridge, MA; Plasmid 10674) as described previously [21]. This c-Myc plasmid was packaged using the LinX retroviral expression system and contains the blasticidin resistance gene. Drug selection of transduced cells was via growth in 2.5 μg/ml of blasticidin. This same system was used to package the V2HS_130611 shRNA construct coding for siRNA for Rb, which was purchased from Open Biosystems (Huntsville, AL). Lentiviral shRNA vectors targeting p21 and p27 were constructed using the pRNATin-H1.4-Lenti vector according to the manufacturer’s specifications (Genscript, Piscataway, NJ). Targeted sequences were p21 (AACTTCGACTTTGTCACCGAG) and p27 (AACCCGGGACTTGGAGAAGCA) as previously reported [30]. A shRNA lentiviral vector for c-Myc was purchased from Addgene (Plasmid 29435). A series (N = 3–5) of shRNA lentiviral vectors for TCF-4 and β-catenin were obtained from the Sigma–Aldrich as part of the mission-shRNA library. The best vector for TCF-4 was clone ID: TRCN0000061893 and the best for β-catenin was clone ID: TRCN0000003843. Each of these lentiviral vectors contains the puromycin resistance gene and was packaged and as previously described [21]. PrECs were transduced with these package lenti-particles and cell selection preformed using puromycin.
Western Blotting
Western blotting was performed as previously described [7]. Whole-cell lysates collected from 100,000 cells were used per lane. Antibodies used were: anti-AR (N-20, Santa Cruz; Santa Cruz, CA); anti-β-Actin (Cell Signaling; Beverly, MA); anti-ΔNp63 (4A4, Santa Cruz); anti-p21 (Cell Signaling); anti-p27 (BD Transduction Labs; San Diego, CA); anti-RB (4H1, Cell Signaling); anti-phospho-RB (Ser 608, Cell Signaling); anti-SKP2 (Zymed; San Francisco, CA); anti EGF receptor (#2232, Cell Signaling); anti-IGF-type 1 receptor (#3018; Cell Signaling); anti-CDK-2 (H-298; Santa Cruz); anti-Cyclin D1 (Upstate Biotechnology; Lake Placid, NY); anti-c-MYC (Calbiochem; San Diego, CA); anti-TCF-4 (05-511, Millipore; Billerica, MA); anti-active β-Catenin (05-665, Millipore); anti-phospho-S552 β-catenin (#9566; Cell Signaling); anti-FOXP3 (mAbcam 450, Abcam; Cambridge, MA). All secondary horseradish peroxidase-conjugated antibodies and chemiluminescent detection reagents (ECL) were purchased from Amersham Biosciences (Piscataway, NJ).
c-MYC Enhancer Driven Luciferase Reporter Assays
The canonical wild type and loss of function 5′ c-MYC enhancer elements described by He et al. [31] and wild type and loss of function 3′ c-MYC enhancer elements identified by Yochum et al. [32] alone and in combination were used to drive SV40-promoter-firefly luciferase constructs as described previously [32]. Cells were transiently transfected with these vectors in combination with a control SV40-driven promoter-Renilla luciferase construct. Twenty-four hours later, luciferase assays were performed using a dual luciferase kit (Promega), and firefly and Renilla luciferase activities measured using a luminometer. The results were initially normalized to the Renilla luciferase expression and these normalized results were then divided by the normalized results for the construct containing the loss of function mutated 5′ enhancer in front of the luciferase gene followed by the loss of function mutated 3′ enhancer to obtain the fold increase in specific expression.
Chip Assay for TCF-4 Binding to c-MYC Enhancer Elements
The assay was performed using the Magna-ChIP kit from Upstate (Temecula, CA) according to the recommended protocol using 4 μg of either non-specific mouse IgG or TCF-4 mouse monoclonal antibody (Cat. # 05-511 from Millipore). To reverse the cross-linking, the modification of Nelson et al. [33] was used. PCR was performed using primers for both 5′ and 3′ c-MYC enhancer elements as described previously [32].
Immunofluoresence (IF), Immunohistochemical (IHC), and SA β-Galactosidase Staining
Single and dual AR, c-MYC, and Ki67, IF staining of human prostate tissue obtained under and IRB approved protocol were performed as described previously [34]. Senescence associated β-galactosidase staining of cells in culture was performed as described previously [35].
Statistics
All of the values are presented as means ± SE. Statistical analysis was performed by a one-way ANOVA with the Newman–Keuls test for multiple comparisons.
RESULTS
Androgen-Dependent AR-Mediated Growth Suppression of Prostate Epithelial Cells Is Not Due to Senescence and Is Cell-Context Dependent
To study the mechanism for androgen-dependent AR-mediated growth suppression of human prostate epithelium, cultures were initiated from non-transformed adult human prostate tissue using a low (<300 nM) calcium, serum-free paracrine growth factor (i.e., EGF, IGF, FGF) defined (SFD) media lacking androgen [25]. Even without added paracrine growth factors, normal prostatic stem and TA cells survive in this low calcium media [7,27] since in this media notch-1 signaling is constitutively activated while e-cadherin signaling is inhibited [36]. In this media, prostatic stem and TA cells secrete and adhere to their own laminin 5 rich extracellular matrix initiating an integrin-mediated, ligand-independent activation of epidermal growth factor receptor [37]. The addition of paracrine growth factors (i.e., EGF, IGF, FGF) to the low calcium media induces the proliferation of prostate epithelial stem and TA cells neither of which expresses AR protein [27]. Under these conditions, stem cells undergo asymmetric division to self-renew and also give rise to either a minor population of progeny, which terminally differentiate into neuroendocrine cells or more commonly into transit-amplifying (TA) cell progeny [5]. These TA cells undergo several rounds of proliferation before eventually maturing into intermediate cells [5,25,27]. Such intermediate cells, however, do not complete their full maturation into AR-positive/PSA-positive secretoryluminal cells in this low Ca2+ SFD medium [25]. Such full maturation into secretory-luminal cells requires the addition of another stromally produced andromedin, Keratinocyte growth factor [i.e., KGF aka FGF-7], and high cell density [22] Under these conditions, the epithelium stratifies resulting in expression of AR in the upper layer of cells, which in the presence of added androgen stop proliferating and produce PSA [22]. In contrast, in low Ca2+ SFD medium containing paracrine growth factors but lacking KGF, these normal human prostate epithelial cell (PrEC) remain AR negative and proliferate rapidly [i.e., 45 hr doubling time] [25]. This situation is very different from that in vivo where despite high level of paracrine growth factors present in the prostate of a non-androgen ablated male, <1% of prostate epithelial cells are proliferating per day [15]. These results suggest that the lack of androgen dependent AR signaling could be responsible for the high proliferation rate of PrECs in vitro in low Ca2+ SFD medium without KGF.
To test this, both normal non-immortalized and telomerase-immortalized (i.e., hTERT) PrECs were transduced with lentiviral (LV) GFP/AR constructs and GFP expressing population isolated by FACs. The expression of AR protein in these cells was documented by Western blots [21]. Immunocyochemical staining (IC) also documented that >98% of the GFP positive cell co-expressed AR, which is present in both the cytoplasm and nuclei of these transduced cells even without the addition of androgen to the serum free media [21]. Addition of a physiologic concentration of the synthetic androgen R1881 (i.e., 1 nM) increases nuclear AR [21] and induces growth arrest of both AR-expressing non-immortalized (i.e., PrEC-LVAR) and immortalized (i.e., PrEC-hTERT-LV-AR) cells, but has no effect on such cells, which do not express AR (i.e., PrEC-LV-Control or PrEC-hTERT-LV-Control) cells, Figure 1. Co-administering R1881 with the AR antagonist Casodex blocks growth arrest of PrEC-hTERT-LV-AR cells demonstrating that this growth inhibition is AR-dependent, Figure 1. Time-lapse microscopy demonstrates that androgen-dependent AR signaling in PrEC-LV-AR and PrEC-hTERT-LV-AR cells results in irreversible proliferative quiescence (i.e., terminal growth arrest). This androgen-dependent AR-induced growth arrest is not due to cellular senescence as confirmed by the lack of expression of senescence associated (SA) β-galactosidase. The observed androgen-dependent AR signaling-induced growth arrest is consistently observed with a series (n = 4) of PrEC-LV-AR cultures initiated from both a commercial source (i.e., donors less than 40 years of age) as well as derived within our institution from patients undergoing radical prostatectomy (i.e., donors greater than 50 years of age).
Fig. 1.
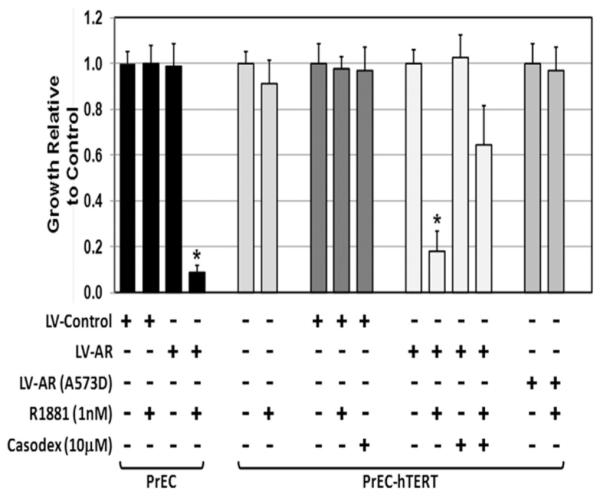
Androgen mediated AR inhibition of the growth of human prostate epithelial cells (PrECs) and PrEC-hTERT cells in response to AR stimulation using physiologic levels of the synthetic androgen R1881 (i.e., 1nM). Expression and ligand activation of AR in both PrEC-LV-AR and PrEC-hTERT-LV-AR cells results in a significant growth suppression over a 1-week observation period, which is inhibited by co-administration of the anti-androgen Casodex (10 μM). An AR DNA-binding mutant (A573D) failed to inhibit growth when ligand-activated [* indicates a P-value < 0.05].
To test whether AR-induced growth arrest is cell-context dependent and not a ubiquitous cellular response, cultures of AR-positive normal non-immortalized human prostate stromal cells (termed PrSCs) containing smooth muscle cells and fibroblasts were established from fresh surgical material [26]. In addition, the immortalized but not transformed stromal WPMY-1 cell line was also evaluated [38]. As expected, all of these prostate derived stromal cells express detectable endogenous AR protein, which is stabilized by ligand [21]. In contrast to PrEC cells, none of these independently derived stromal cell cultures is growth inhibited by androgen-induced AR signaling [21]. These results document that AR-mediated growth arrest of PrECs is cell-context dependent and not a ubiquitous cellular response.
Androgen-Dependent AR-Mediated Terminal G0 Growth Arrest and Differentiation of Prostate Epithelial Cells Does Not Require Rb, p21, p27, Nor Down-Regulation of Growth Factors Receptors
Flow cytometric DNA analysis documents that androgen-exposure of AR-expressing PrEC cells induces their growth arrest in G0 [21]. Despite androgen-dependent growth arrest, R1881-treated cells continue to remain viable with no increase in apoptosis as documented both with flow cytometric DNA analysis (i.e., no sub-G0 population detected) as well as time lapse microscopy. Such androgen-induced growth inhibition is associated with a significant (P < 0.05) increase in the proportion of cells exiting cycle and arresting in G0, as monitored by their lack of nuclear Ki67 expression, and undergoing differentiation, as monitored both by down regulation of the basal marker ΔNp63 and an increase (P < 0.05) in the mRNA expression for PSA, an androgen-regulated gene whose expression is characteristic of differentiated prostatic secretory-luminal cells [21]. Androgen-dependent AR-mediated growth arrest is distinctly different from growth arrest induced by growth factor restriction, however, since growth factor restricted cells do not differentiate as monitored by a lack of change in their ΔNp63 expression even though they growth arrest in G0 as monitored by a decrease in nuclear Ki67 expression [21]. Such GF restriction-induced growth arrest is reversible, however, since the re-addition of GFs stimulates subsequent growth [27]. The decreased expression of the transit-amplifying specific marker ΔNp63 following androgen-dependent AR signaling induced growth arrest is significant for two reasons. First, ΔNp63 is a marker of normal but not malignant prostate epithelial cells [39] validating that these cells are derived from normal not malignant prostate epithelial cells. Second, ΔNp63 protein expression decreases as PrECs differentiate into intermediate cells [40].
Coincident with this androgen induced terminal growth arrest is a time dependent increase of Cyclin-dependent kinase (Cdk) inhibitors p21 and p27 proteins coupled with a decrease in Cyclin D1 and phospho-Rb [21]. Furthermore, decreased expression of the S-phase specific protein p150 as well as the DNA licensing factor cdc6 is also observed [21]. RNAse protection assays demonstrate, however, that the steady state level of p21, p27, and AR mRNA remain unchanged throughout the growth arrest induced during the first 48 hr of exposure to androgen [21]. In addition, shRNA knockdown documented that neither RB, p21, p27 alone or in combination are required for such AR induced G0 growth arrest [21].
Removal of EGF, IGF-1, and FGFs from the serum free defined media arrests PrECs in G0 [27]. These ligands signal via their cognate receptors; EGFR or Erb1 for EGF and IGF-Type1 receptor (IGF-1R) for IGFs and FGFR2 for FGFs. Western blot analysis documented that androgen-dependent AR signaling in PrEC-hTERTLV-AR cells does not decrease the expression of EGFR or IGFR proteins and instead elevates the expression of FGFR2-IIIB protein [i.e., KGF receptor] without isotype switching [21]. Thus, androgen dependent AR-mediated growth arrest of PrEC-AR cells does not involve repression of growth factor receptor expression.
Androgen-Dependent AR-Mediated G0 Growth Arrest of Prostate Epithelial Cells Is Dependent Upon Suppression of c-MYC Expression and Does Not Involve Nuclear FoxP3
Associated with the androgen induced growth arrest in PrEC-hTERT-LV-AR is a decrease in c-MYC mRNA [21]. This transcriptional down-regulation results in a total loss of detectable c-MYC protein from the nucleus and cytosol within 24 hr of androgen treatment [21]. Such a decrease in c-MYC transcription could be either a consequence of growth arrest or causal, inducing such growth arrest. To resolve this, PrEC-hTERT-LV-AR cells were transduced (RV-c-MYC) and drug selection (i.e., blastacidin) used to isolate cells, which stably transcribe c-MYC m-RNA constitutively even when androgen is added to the media [21]. Indeed, stable constitutive c-MYC transcription abrogated the cell proliferation block induced by androgen-activated AR. This is demonstrated by an increase in cell growth, Figure 2. These data demonstrate that down regulation of c-MYC transcription is required for androgen-dependent AR mediated growth arrest of normal prostate epithelial cells.
Fig. 2.
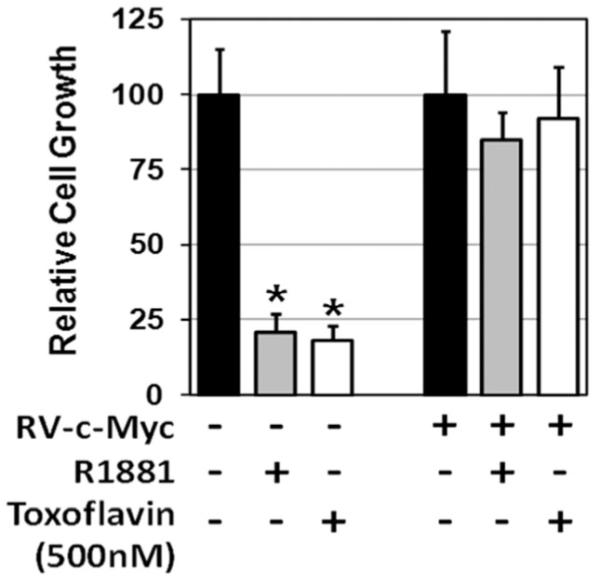
Exogenous c-MYC expression (RV-c-MYC) overrides growth inhibition of PrEC-hTERT-LV-AR cells induced by both AR-signaling and toxoflavin induced β-catenin inhibition [* indicates a P-value < 0.05].
To test the causal relationship between nuclear c-MYC transcription and proliferation of basal PrECs induce in low Ca2+-serum free/growth factor supplemented media, lentiviral c-MYC shRNA was used to stably knock down c-Myc by >90%, Figure 3A. c-MYC knock down results in essentially complete inhibition (P < 0.05) of PrECs growth, Figure 3B.
Fig. 3.
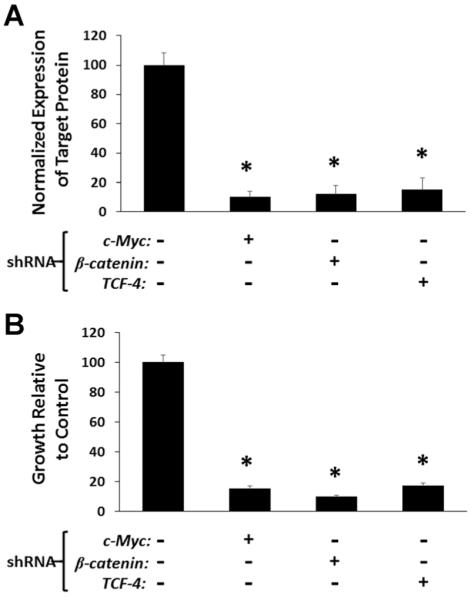
(A) Western blot determination of efficiency of lentiviral shRNA knockdown of c-MYC, β-catenin, or TCF-4 protein expression in human PrECs. (B) Growth response to such specific protein knock down[* indicates a P-value < 0.05].
Previous studies reported that FoxP3 is a tumor suppressor for prostate epithelial cells via its ability to bind to the c-MYC promoter repressing its transcription [41]. Therefore, PrEC-hTERT-LV-AR cells were treated with androgen and the cells analyzed for their nuclear level of FoxP3 protein. The results of these studies documented that when PrEC-hTERT-LV-AR cells are growing without androgen, FoxP3 is present only in the cytosol and that androgen treatment does not result in nuclear localization of FoxP3 even though these androgen treated cells down regulate their c-MYC transcription and arrest in G0, Figure 4.
Fig. 4.
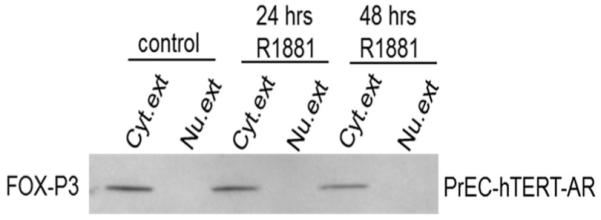
Western blot documents a lack of FOXP3 nuclear translocation in response to AR-mediated growth suppression of PrEC-hTERT-LV-AR cells. Cyt. ext, cytoplasmic extract; Nu. ext, nuclear extract.
β-Catenin/TCF-4 Complex Binding to 5′ Plus 3′ c-MYC Enhancers are Required for c-MYC Transcription and Growth of Prostate Epithelial Cells
In a cell context dependent manner, β-catenin enters the nucleus, binds to TCF-4 complexes displacing corepressors (e.g., Groucho), thus converting β-catenin/TCF-4 complexes from a repressor to an activator of c-MYC transcription [31]. In PrECs, the majority (i.e., ~90%) of β-catenin is cytoplasmic, however, ~10% is within the nucleus, Figure 5A. These Western blot results are consistent with IHC analysis, which documents that besides its plasma membrane localization, a fraction of the β-catenin is also present within the nuclei of growing PrECs, Figure 5B. To test whether nuclear β-catenin/TCF-4 complexes are formed during PrEC growth, co-immunoprecipitation (co-IP) was performed on nuclear extracts using an antibody specific for TCF-4. These studies document that in growing PrEC cells, β-catenin/TCF-4 complexes are detectable in the nuclei of these cells, Figure 5A. Once formed, such nuclear β-catenin/TCF-4 complexes can bind to genomic DNA at enhancers located at close proximity to both the 5′ and 3′ c-MYC gene boundaries [32] and form a chromatin loop between the 5′ and 3′ enhancer driving c-MYC transcription [42]. To determine whether such nuclear β-catenin/TCF-4 complexes bind 5′ and/or 3′ c-MYC enhancers, chromatin-IP (ChIP) analyses using TCF-4 antibody was performed. These studies document that TCF-4 binds to the 5′ and 3′ c-MYC enhancer in growing PrECs, Figure 6A.
Fig. 5.

(A) β-Catenin and ligand (R1881)-bound wild type AR, but not DNA-binding AR mutant A573D, co-immunoprecipitate (IP) with nuclear TCF-4 in PrEC-hTERT-LV-AR cells. Non-specific IgG antibody was used as a negative control. (B) Immuno-histochemical staining of active β-catenin in PrEC-hTERT-LV-AR cells.
Fig. 6.
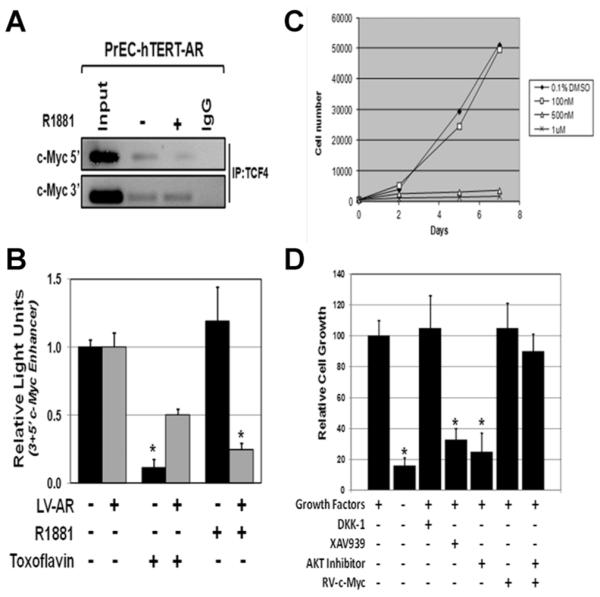
(A) Chromatin immunoprecipitation (ChIP) documents that TCF-4 continues to bind the 3′ c-MYC enhancer elements of the c-MYC gene even in the presence of ligand-activated AR, but there is a decrease binding to the 5′ c-MYC enhancer element. (B) Ligand-activated AR and toxoflavin inhibition of β-catenin/TCF-4 complex formation inhibits the transcriptional activity of the 5′ plus 3′ c-MYC enhancer element using appropriate luciferase reporter constructs. (C) Toxoflavin dose-dependent growth inhibition of non-immortalized PrEC cells. (D) Growth response of PrEC-hTERT-LVAR cells to growth factor removal, Wnt inhibition (DKK1), or AKT inhibition (AKT-inhibitor) alone and in combination with constitutive c-MYC expression (RV-c-MYC), and to XAV939 induced inhibition of β-catenin signaling.
To determine if such binding of β-catenin/TCF-4 complexes induces c-MYC transcription, PrECs were transiently transfected with a luciferase construct driven by either 5′ c-MYC enhancer, 3′ c-MYC enhancer, 5′ enhancer in front of the luciferase gene followed by the 3′ enhancer (i.e., 5′ plus 3′ c-MYC enhancers), or loss of function mutated 5′ enhancer in front of the luciferase gene followed by the loss of function mutated 3′ enhancer, and the bioluminescence determined for each construct. These results documented that in growing PrECs, there is no enhancement of luciferase expression in the cells transfected with wild type 5′ c-Myc. In contrast, there is a 25.8 ± 3.5 fold (P < 0.05) increase in luciferase expression in cells transfected with 3′ c-Myc enhancer compared to the loss of function mutated 5′ enhancer in front of the luciferase gene followed by the loss of function mutated 3′ enhancer construct. Interestingly, when cells were transfected with a construct containing the 5′enhancer in front of the luciferase gene followed by the 3′ enhancer, luciferase expression was increased (P < 0.05) even more [i.e., 38.4 ± 5.5 fold compared to loss of function mutated 5′ enhancer in front of the luciferase gene followed by the loss of function mutated 3′ enhancer construct]. These results are consistent with formation of a chromatin loop between the 5′ and 3′ enhancer being required for maximal c-MYC transcription in prostate epithelial cells.
To determine whether such 5′ plus 3′ c-MYC enhancer driven expression requires binding of β-catenin/TCF-4 complexes, the 5′ plus 3′ c-MYC enhancer driven luciferase reporter response was evaluated with or without co-treatment with a small molecule inhibitor (i.e., toxoflavin), which specifically prevents β-catenin binding to TCF-4 [43]. These studies document that 500 nM toxoflavin decreases (P < 0.05) 5′ 3′ c-MYC enhancer driven reporter expression in PrECs, Figure 6B. Co-incidentally, this dose of toxoflavin inhibited the growth of PrECs, Figure 6C. Such toxoflavin growth inhibition was confirmed in a series of five independently derived non-immortalized PrECs. Importantly, such toxoflavin-induced growth inhibition is overcome when the cells are transduced to constitutive express c-MYC (i.e., RV-c-MYC) in a β-catenin/TCF-4 independent manner, Figure 2. To confirm that toxoflavin’s ability to inhibit c-MYC transcription and growth is not an off-target effect, these responses were evaluated on PrECs exposed to a second small molecule inhibitor, XAV939, which selectively restricts β-catenin-mediated transcription via a mechanism distinct from that of toxoflavin. XAV939 stimulates β-catenin degradation by inhibiting tank-yrases, which stabilizes axin, the rate-limiting component of β-catenin degradation [44]. Like the situation with toxoflavin, treatment of PrECs with XAV939 profoundly inhibits their growth, Figure 6D.
As an additional validation of the causal role of the β-catenin/TCF-4 complex in c-MYC driven proliferation of PrECs, appropriate lentiviral shRNAs was used to stably knock down either β-catenin or TCF-4 by >85%, Figure 3A. Knock down of either genes results in essentially complete inhibition (P < 0.05) of PrECs growth, Figure 3B.
Nuclear Translocation of β-Catenin in PrECs Is Induced by a Wnt-Independent Akt Signaling Pathway
Nuclear translocation of β-catenin needed for β-catenin/TCF-4 complex formation can occur via both Wnt-dependent and independent pathways [45]. In the absence of growth factor signaling, cytoplasmic β-catenin interacts with axin, glycogen synthase kinase-3β (GSK-3β), and the adenomatous polyposis coli (APC) protein. Within this complex, β-Catenin is phosphorylated in its N-terminal domain at serines 31 and 33 and threonine 41 by GSK-3β leading to its degradation via the ubiquitin/proteasome pathway. Wnt receptor activation inhibits GSK-3β-dependent phosphorylation of β-catenin allowing N-terminal hypophosphorylated β-catenin to translocates to the nucleus, where it interacts with transcription factors of the TCF-4 [46]. To resolve whether a Wnt-dependent pathway is involved, PrEC-hTERT-ARs were treated with recombinant DKK1 protein at a dose, which totally inhibits activation of the canonical Wnt signaling pathway [47]. The results documented that growth of these cells is not inhibited by DKK1 treatment, Figure 6C.
Feng et al. [45] documented that as an alternative to Wnt activation, β-catenin translocation can be induced by other growth factors, such as EGF, IGF, etc., via activation of Akt since activated Akt can elevate the level of N-terminal hypophosphorylated β-catenin through phosphorylation and thus inhibition of GSK-3β and/or by directly phosphorylating β-catenin at serine 552, resulting in its disassociation from cell to cell contacts and nuclear translocation. Previously, we documented that removal of EGF, IGF-1, and FGFs from the serum free defined media arrests PrECs in G0 [25,27,48]. Associated with this growth arrest is loss of activated (i.e., phosphorylated) Akt [48]. Conversely, when PrECs growing in serum free media containing these growth factors are treated with a small molecule AKT inhibitor, they are growth arrested [48]. Like PrEC cell, PrEC-hTERT-AR cells are also growth inhibited even without the addition of androgen by both removal of the growth factors from the media and by treatment with the 20 μM Akt inhibitor, which is overcome when the cells are transduced to constitutive express RV-c-Myc in a β-catenin/TCF-4 independent manner, Figure 6C.
To determine whether β-catenin/TCF-4 signaling is Akt dependent, nuclear extracts from PrEC-hTERT-AR cells growth arrested by either treatment with Akt inhibitor or by maintenance in serum free media without added growth factors versus growing in media containing growth factors were evaluated by Western blots. For these analyses, two different anti-β-catenin antibodies were used. The first antibody (termed anti-ABC) only binds β-catenin when it is non-phosphorylated in serine 31 and 33 and threonine 41 [46]. The second antibody (termed anti-phos-S552) binds β-catenin when it is phosphorylated at serine 552 [45]. These results document that β-catenin is present within the nucleus of paracrine growth factor simulated, growing PrEC-hTERT-AR cells (i.e., denoted either as control or +GF) where it is non-phosphorylated at serine 31 and 33 and threonine 41, but phosphorylated at serine 552, Figure 7. In contrast, β-catenin is reduced by >90% within the nucleus of growth arrested PrEC-hTERT-AR cells induced by either Akt inhibitor (i.e., denoted AIN) or removal of the growth factors (i.e., denoted-GF) using either antibodies, Figure 7. These results document that activated Akt is the major regulator of β-catenin nuclear trans-location in PrECs induced by paracrine growth factors.
Fig. 7.
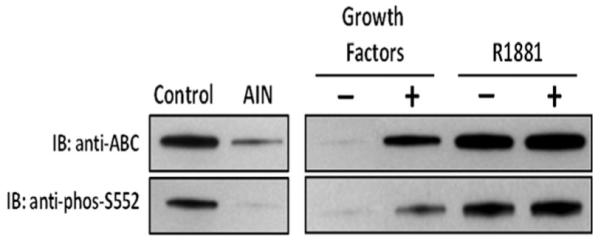
Phosphorylation status of β-catenin in nuclear extracts of PrEC-hTERT-LV-AR cells growth arrested by treatment with AKT-inhibitor (AIN), removal of pararcrine growth factors (−GF), or addition of androgen (+R1881) compared to growing cells (i.e., growth factor +).
Androgen-Dependent Suppression of c-MYC Transcription in Prostate Epithelial Cells Requires Binding of β-Catenin/TCF-4 Complexes to the Zinc-Finger Domain of AR
To determine whether the mechanism for androgen dependent c-Myc down regulation in AR expressing PrECs involves a decrease in β-catenin/TCF-4 complex formation, TCF-4 co-IP was performed. These studies document that exposure to androgen did not prevent formation of nuclear β-catenin/TCF-4 complexes in androgen induced growth arrested PrECs expressing wild type AR, Figure 4. Also, β-catenin is still non-phosphorylated at serine 31 and 33 and threonine 41, but phosphorylated at serine 552 within the nucleus of these androgen-induced growth arrested PrEC-hTERT-AR cells [i.e., denoted +R1881], Figure 7. Such androgen exposure, however, did result in AR binding to nuclear β-catenin/TCF-4 complexes in growth arrested PrECs expressing AR, Figure 4. These results are consistent with the previous documentation that both β-catenin and TCF-4 bind AR in an androgen-dependent manner [49–51]. AR binds via its ligand-binding domain (LBD) to sequences encoded by exon-3 of β-catenin [52]. Binding toTCF-4 is via the zinc finger domain of AR [53].
To test whether AR binding to TCF-4 is required to inhibit the growth of PrECs, cells expressing a loss of function zinc-finger mutant AR [i.e., AR(A573D)] were tested for their growth response to androgen and ability to form AR/β-catenin/TCF-4 complexes. These PrEC-hTERT-AR(A573D) cells express a loss of function zinc-finger domain mutant AR, which still translocates into the nucleus upon exposure to androgen, Figure 4. In contrast to the situation for PrECs expressing wild type AR, androgen treatment of PrEC-hTERT-AR(A573D) cells does not result in formation of AR/β-catenin/TCF-4 complexes, Figure 4, nor does it inhibit growth, Figure 1. These data document that AR-mediated growth arrest of prostate epithelial cells requires ligand dependent AR binding via its zinc-finger domain to TCF-4 to suppress c-Myc transcription.
ChIP analysis documented that this androgen-dependent suppression of c-MYC transcription is not due to inhibition of AR/β-catenin/TCF-4 complexes binding to the 3′ c-Myc enhancer in androgen treated cells, but is associated with a decrease of binding to the 5′ c-MYC enhancer, Figure 6A. This latter effect is associated with suppression of 5′ plus 3′ c-MYC reporter expression in androgen treated cells, Figure 6A. These results are consistent with binding of AR to the β-catenin/TCF-4 complex retarding the formation of the chromatin loop between the 5′ and 3′ enhancers thus suppressing c-MYC transcription.
In Vivo β-Catenin Is Present in Nuclei of Terminally Differentiated Prostatic Secretory-Luminal Cells, Which Co-Express Nuclear AR, But Not c-MYC
Previous studies have documented that in the prostate of an adult male, cell proliferation is very low [i.e., <1% of epithelial cells are Ki67 positive] [15] and predominantly occurring in basal epithelial cell despite the continuous presence of high levels of andromedins [16,17]. Thus, the results of the present studies predicts that in prostate of such an adult male, only the small number of proliferating basal cells should express nuclear c-MYC protein and these cells should not be express nuclear AR. In contrast, the terminally differentiated, proliferatively quiescent, prostatic secretory-luminal epithelial cells should express nuclear β-catenin plus AR and, but not nuclear c-MYC protein. To test these predictions, prostate tissues harvested from non-androgen ablated patients without culturing were analyzed by IHC or IF staining using appropriately validated antibodies. As predicted, these analyses documented that only a rare basal cell expresses active β-catenin, Figure 8-upper panel, and c-MYC Figure 8-lower left panel denoted by arrow in their nuclei and that these positive basal cells are AR negative, Figure 8-lower middle panel denoted by arrow with the vast majority of basal cells being negative for all three markers as predicted, Figure 8 lower right panel denoted by arrow heads. In contrast, essential all of the terminally differentiated, proliferatively quiescent, secretory-luminal cells express activated nuclear β-catenin, Figure 8-upper panel, and nuclear AR, Figure 8-lower middle panel, but not express c-MYC, Figure 8-lower left panel also as predicted.
Fig. 8.
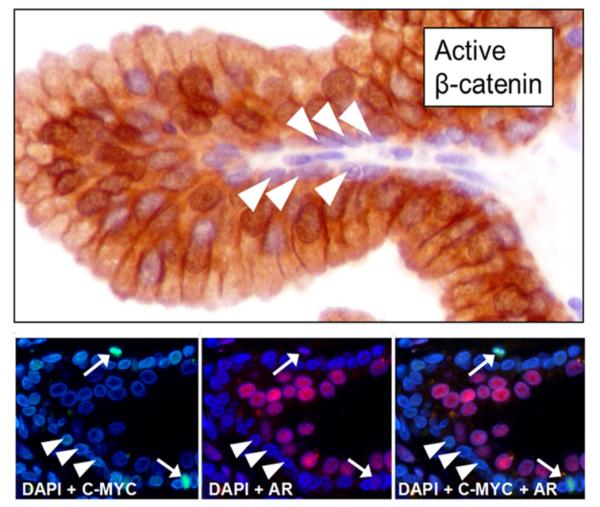
Nuclear AR, c-MYC, and β-catenin are expressed in different populations in the normal human prostate. Upper Panel-IHC staining for active (i.e., N-terminal hypo-phosphorylated) nuclear β-catenin. Arrowheads indicated basal cells, which are usually negative for active nuclear β-catenin. Secretory-luminal cells are nearly universally positive for nuclear active β-catenin. Lower Left Panel-Immuno-fluorescent (IF) staining for nuclear c-MYC and DAPI counter-stain. Arrows indicate occasionally c-MYC positive basal cells; arrowheads usual c-MYC negative basal cells. Lower Middle Panel-IF staining for AR and DAPI counter-stain. Arrows indicate occasionally c-MYC positive basal cells, which are AR negative; arrowheads usual c-MYC negative basal cells, which are also AR negative. Lower Right Panel-Dual IF staining for c-MYC plus AR and DAPI counter-stain. Arrows and arrowheads are as described in left and middle panels.
DISCUSSION
The present studies documents that the growth of non-transformed adult human prostate epithelial cells is dependent upon c-MYC transcription, which is stimulated by nuclear β-catenin/TCF-4 complex binding to 5′ and 3′ enhancer elements in the c-MYC gene. Such formation of nuclear β-catenin/TCF-4 complex is dependent upon Wnt-independent Akt kinase dependent phosphorylation of β-catenin at serine 552 induced by paracrine growth factors. In an adult male with a physiological normal level of circulation androgen, these paracrine factors are continuously produced by the prostatic stromal cells and are thus chronically present at high levels within the gland. Prostatic epithelial hyperplasia is prevented despite chronic high levels of these paracrine growth factors by androgen dependent AR signaling within the epithelial cells, which suppresses their proliferation. Such androgen dependent AR signaling induces the G0 growth arrest of non-transformed human prostate epithelial cells and directs their differentiation along a pathway to ΔNp63 negative, PSA-expressing secretory-luminal cells. The mechanism for such G0 growth arrest involves androgen dependent binding of AR to β-catenin/TCF-4 complexes suppressing c-MYC transcription.
Besides the present prostate studies, there are additional reports demonstrating that androgen dependent AR signaling induces growth suppression and terminal differentiation in other normal human epithelial cell types. For example, ligand-dependent endogenous AR signaling suppresses growth of adrenocortical and thyroid epithelial cells, which is likewise associated with a down regulation of c-MYC transcription [54,55]. Such AR signaling induced growth arrest is cell-context dependent, however, and not a ubiquitous cellular response. This is documented by the observations of the present studies that androgen dependent signaling is not growth inhibitory to prostate stromal cells.
The conclusion that c-MYC transcriptional down regulation induced suppression of proliferation is a major normal function of androgen-dependent AR signaling in prostatic epithelial cells is supported by additional observations. For example, constitutive targeted expression of floxed exon-3 deletion, dominant activating (DA), in AR-positive secretory-luminal epithelial cells via AR-driven CRE robustly induces prostatic hyperplasia in mice [56–58]. These results are supportive since AR does not bind β-catenin if exon 3 encoded sequences are deleted [53] and thus AR cannot down regulate c-MYC transcription in these secretory-luminal cells expressing DA mutant β-catenin. This inability of AR to down regulate c-MYC results in the development of high-grade intraepithelial neoplasia (HGPIN), but not invasive cancer in the mouse [56–58]. In contrast to the hyperplasia/HGPIN induced in these exon-3 deleted DA β-catenin transgenic mice models in which constitutive c-MYC transcription is maintained in an AR independent manner, invasive adenocarcinomas are produced when c-MYC transcription is transgenically driven in rodent prostatic secretoryluminal cells by androgen occupied AR [59] These latter results suggest that malignant transformation of prostatic secretory-luminal cells requires additional events besides c-Myc expression, which androgen occupied AR stimulates.
Using IHC analysis of human prostate tissues, elevated c-MYC protein expression occurs very early during prostatic carcinogenesis (i.e., detectable in PIA and PIN lesions in addition to frank cancer) in cells that co-express AR and cell proliferation marker Ki67 [34]. Elevation of c-MYC expression in AR expressing transformed prostate cells is thus paradoxical to the situation in the normal prostate where c-MYC protein is undetectable in Ki67 negative/AR expressing luminal cells even though AR is signaling as documented by these cells expressing the AR dependent protein, PSA. These results are consistent with studies demonstrating that for prostate cancer to develop; malignant cells must loss AR dependent growth suppression and that this can occur independently from acquiring oncogenic addiction to AR induced growth stimulation [60]. During prostate carcinogenesis, however, the majority of human prostate cancers characteristically not only loose AR dependent growth suppression, but acquire oncogenic addiction to androgen dependent AR signaling for their growth [21,61]. This gain of oncogenic function is the basis for the use of androgen ablation (i.e., castration) therapy for metastatic prostate cancer [62]. Associated with this addiction, androgen-dependent AR signaling no longer inhibits but instead stimulates c-Myc expression; however, this is via a non-transcriptional effect upon c-MYC protein stability [63]. While these combined results document that AR signaling is characteristically subverted from a growth suppressor to a growth stimulatory function in prostate cancers [21], they do not clarify at what point in the carcinogenic process this occurs. There are clinical data, which emphasize the critical importance of resolving this issue since the answer has significant clinical implications for the use of androgen deprivation as therapy for prevention verses treatment of prostate cancer.
For example, male dogs are often neutered at a young age and such long-term androgen deprivation is associated with an increase incidence of developing symptomatic prostate cancer and death [64]. Two large, randomized controlled human trials, the prostate cancer prevention trial (PCPT) [65] and the reduction by dutasteride of prostate cancer events (REDUCE) trial [66] evaluated daily use of 5α-reductase inhibitors for the reduction in the risk of prostate cancer in men at least 50 years of age. The trials demonstrated an overall 25% reduction in prostate cancer diagnoses with 5α-reductase inhibitor treatment. This overall reduction was due to a decreased incidence of lower risk (i.e., Gleason score 6) prostate cancers. However, both trials showed an increased incidence of high-grade (i.e., Gleason score >7) prostate cancer with 5α-reductase inhibitor treatment. These data suggest that: (1) Gleason 6 prostate cancer cells are still dependent upon AR mediated stromal production of andromedin and therefore androgen ablation therapy by inhibiting andromedin production inhibits their growth, and (2) in contrast, higher Gleason score cancer cells acquire AR mediated ability to produce their own autocrine growth factors, but a subset of these high grade cancers still retain the AR-dependent negative regulation of c-MYC transcription and thus androgen ablation removes this growth constraint. Presently, this concept is being tested experimentally in pre-clinical models, which form the basis of a clinical trial using rapid cycling of super-physiologic androgen to stimulate growth constraint followed by rapid androgen ablation to restrict autocrine growth factor production in men with castration resistant metastatic prostate cancer [67,68].
ACKNOWLEDGMENTS
We wish to acknowledge the expert technical assistance of Lee Blosser and Ada Tam of the Johns Hopkins School of Medicine Flow Sorting Facility (i.e., supported by NIH Grant U54CA091409) for their assistance. We also wish to thank Angelo De Marzo and Alan Meeker, Department of Pathology and Helen Fedor and the Brady Urological Institute Prostate Specimen Repository supported by the NIH-Prostate SPORE Grant (P50 CA058236) for Tissue Microarrays and Jessica Hicks of the Comprehensive Cancer Center Histology Core (i.e., supported by NIH Grant U54CA091409) for immuno-cytochemical staining. We wish to thank Gregory S. Yochum, Pennsylvania State University, College of Medicine for generously supplying the c-Myc enhancer reporter constructs and Dr. Hetty van der Korput, Department of Pathology, Erasmus University Medical Centre, Rotterdam, The Netherlands for generously providing the A573D AR mutant vector.
Grant sponsor: NIH; Grant numbers: R01DK52645; U54CA091409; Grant sponsor: NIH-Prostate SPORE; Grant number: P50 CA058236; Grant sponsor: Maryland Stem Cell Research Fund; Grant number: MSCRFII-0428-00.
Abbreviations
- AR
androgen receptor
- ARE
androgen response element
- DHT
dihydrotestosterone
- FBS
fetal bovine serum
- FACS
fluorescence activated cell sorting
- GFP
green fluorescence protein
- hTERT
human telomerase
- PrEC
prostate epithelial cell
- PSA
prostate specific antigen
REFERENCES
- 1.Berry SJ, Coffey DS, Walsh PC, Ewing LL. The development of human benign prostatic hyperplasia with age. J Urol. 1984;132:474–479. doi: 10.1016/s0022-5347(17)49698-4. [DOI] [PubMed] [Google Scholar]
- 2.Siegel R, Ward E, Brawley O, Jemal A. Cancer statistics. 2011:the impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J Clin. 2011;61:212–236. doi: 10.3322/caac.20121. [DOI] [PubMed] [Google Scholar]
- 3.Kyprianou N, Isaacs JT. Activation of programmed cell death in the rat ventral prostate after castration. Endocrinology. 1988;122:552–562. doi: 10.1210/endo-122-2-552. [DOI] [PubMed] [Google Scholar]
- 4.Isaacs JT. Control of cell proliferation and cell death in the normal and neoplastic prostate: A stem cell model. U.S. Dept. of Health and Human Services; 1987. NIH Publication # 87-2881. [Google Scholar]
- 5.Isaacs JT. Prostate stem cells and benign prostatic hyperplasia. Prostate. 2008;68:1025–1034. doi: 10.1002/pros.20763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leong KG, Wang BE, Johnson L, Gao WQ. Generation of a prostate from a single adult stem cell. Nature. 2008;456:804–808. doi: 10.1038/nature07427. [DOI] [PubMed] [Google Scholar]
- 7.Vander Griend DJ, Karthaus WK, Dalrymple S, Meeker AK, De Marzo AM, Isaacs JT. The role of CD133 in normal human prostate stem cells and malignant cancer initiating cells. Cancer Res. 2008;68:9703–9711. doi: 10.1158/0008-5472.CAN-08-3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang X, Julio MK, Economides KD, Walker D, Yu H, Halili MV, Hu YP, Price SM, Abate-Shen C, Shen MM. A luminal epithelial stem cell that is a cell of origin for prostate cancer. Nature. 2009;461:495–500. doi: 10.1038/nature08361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goldstein AS, Huang J, Guo C, Garraway IP, Witte ON. Identification of a cell of origin for human prostate cancer. Science. 2010;329:568–571. doi: 10.1126/science.1189992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cunha GR, Chung LW, Shannon JM, Taguchi O, Fujii H. Hormone-induced morphogenesis and growth: Role of mesenchymal–epithelial interactions. Recent Prog Horm Res. 1983;39:559–598. doi: 10.1016/b978-0-12-571139-5.50018-5. [DOI] [PubMed] [Google Scholar]
- 11.Yan G, Fukabori Y, Nikolaropoulos S, Wang F, McKeehan WL. Heparin-binding keratinocyte growth factor is a candidate stromal-to-epithelial-cell andromedin. Mol Endocrinol. 1992;6:2123–2128. doi: 10.1210/mend.6.12.1491693. [DOI] [PubMed] [Google Scholar]
- 12.Lu W, Luo Y, Kan M, McKeehan WL. Fibroblast growth factor-10. A second candidate stromal to epithelial cell andromedin in prostate. J Biol Chem. 1999;274:12827–12834. doi: 10.1074/jbc.274.18.12827. [DOI] [PubMed] [Google Scholar]
- 13.Le H, Arnold JT, McFann KK, Blackman MR. DHT and testosterone, but not DHEA or E2, differentially modulate IGF-I, IGFBP-2, and IGFBP-3 in human prostatic stromal cells. Am J Physiol Endocrinol Metab. 2006;290:E952–E960. doi: 10.1152/ajpendo.00451.2005. [DOI] [PubMed] [Google Scholar]
- 14.Ohlson N, Bergh A, Persson ML, Wikstrom P. Castration rapidly decreases local insulin-like growth factor-1 levels and inhibits its effects in the ventral prostate in mice. Prostate. 2006;66:1687–1697. doi: 10.1002/pros.20368. [DOI] [PubMed] [Google Scholar]
- 15.Berges RR, Vukanovic J, Epstein JI, CarMichel M, Cisek L, Johnson DE, Veltri RW, Walsh PC, Isaacs JT. Implication of cell kinetic changes during the progression of human prostatic cancer. Clin Cancer Res. 1995;1:473–480. [PMC free article] [PubMed] [Google Scholar]
- 16.Bonkhoff H, Srein U, Remberger K. The proliferative function of basal cells in the normal and hyperplastic human prostate. Prostate. 1994;24:114–118. doi: 10.1002/pros.2990240303. [DOI] [PubMed] [Google Scholar]
- 17.De Marzo AM, Meeker AK, Epstein JI, Coffey DS. Prostate stem cell compartments: Expression of the cell cycle inhibitor p27Kip1 in normal, hyperplastic, and neoplastic cells. Am J Pathol. 1998;153:911–919. doi: 10.1016/S0002-9440(10)65632-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ling MT, Chan KW, Choo CK. Androgen induces differentiation of a human papillomavirus 16 E6/E7 immortalized prostate epithelial cell line. J Endocrinol. 2001;170:287–296. doi: 10.1677/joe.0.1700287. [DOI] [PubMed] [Google Scholar]
- 19.Whitacre DC, Chauhan S, Davis T, Gordon D, Cress AE, Miesfeld RL. Androgen induction of in vitro prostate cell differentiation. Cell Growth Differ. 2002;13:1–11. [PubMed] [Google Scholar]
- 20.Berger R, Febbo PG, Majumder PK, Zhao J, Mukherjee S, Signoretti S, Campbell K, Sellers WR, Roberts TM, Loda M, Golub TR, Hahn WC. Androgen-induced differentiation and tumorigenicity of human prostate epithelial cells. Cancer Res. 2004;64:8867–8875. doi: 10.1158/0008-5472.CAN-04-2938. [DOI] [PubMed] [Google Scholar]
- 21.Vander Greind DJ, Litvinov IV, Isaacs JT. Conversion of androgen receptor signaling from a growth suppressor in normal prostate epithelial cells to an oncogene in prostate cancer cells involves a gain in c-Myc regulation. Int J Biol Sci. 2014 doi: 10.7150/ijbs.8756. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lamb LE, Knudsen BS, Miranti CK. E-cadherin-mediated survival of androgen-receptor-expressing secretory prostate epithelial cells derived from a stratified in vitro differentiation model. J Cell Sci. 2010;123:266–276. doi: 10.1242/jcs.054502. [DOI] [PubMed] [Google Scholar]
- 23.Simanainen U, McNamara K, Gao YR, Handelsman DJ. Androgen sensitivity of prostate epithelium is enhanced by postnatal androgen receptor in activation. Am J Physiol Endocrinol Metab. 2009;296:E1335–E1343. doi: 10.1152/ajpendo.00017.2009. [DOI] [PubMed] [Google Scholar]
- 24.Wu CT, Altuwaijri S, Ricke WA, Huang SP, Yeh S, Zhang C, Niu Y, Tsai MY, Chang C. Increased prostate cell proliferation and loss of cell differentiation in mice lacking prostate epithelial androgen receptor. Proc Natl Acad Sci USA. 2007;104:12679–12684. doi: 10.1073/pnas.0704940104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Litvinov IV, Vander Griend DJ, Xu Y, Antony L, Dalrymple SL, Isaacs JT. Low-calcium serum-free defined medium selects for growth of normal prostatic epithelial stem cells. Cancer Res. 2006;66:8598–8607. doi: 10.1158/0008-5472.CAN-06-1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gao J, Arnold JT, Isaacs JT. Conversion from a paracrine to an autocrine mechanism of androgen-stimulated growth during malignant transformation of prostatic epithelial cells. Cancer Res. 2001;61:5038–5044. [PubMed] [Google Scholar]
- 27.Uzgare AR, Xu Y, Isaacs JT. In vitro culturing and characteristics of transit amplifying epithelial cells from human prostate tissue. J Cell Biochem. 2004;91:196–205. doi: 10.1002/jcb.10764. [DOI] [PubMed] [Google Scholar]
- 28.Litvinov IV, Vander Griend DJ, Antony L, Dalrymple SL, Becker R, Cheng L, Isaacs JT. PC3, but not DU145, human prostate cancer cells retain the coregulators required for tumor suppressor ability of androgen receptor. Prostate. 2006;66:1329–1338. doi: 10.1002/pros.20483. [DOI] [PubMed] [Google Scholar]
- 29.Farla P, Hersmus R, Trapman J, Houtsmuller AB. Antiandrogens prevent stable DNA-binding of the androgen receptor. J Cell Sci. 2005;118:4187–4198. doi: 10.1242/jcs.02546. [DOI] [PubMed] [Google Scholar]
- 30.Halfter H, Friedrich M, Resch A, Kullmann M, Stogbauer F, Ring’elstein EB, Hengst L. Oncostatin M induces growth arrest by inhibition of Skp2, Cks1, and cyclin A expression and induced p21 expression. Cancer Res. 2006;66:6530–6539. doi: 10.1158/0008-5472.CAN-04-3734. [DOI] [PubMed] [Google Scholar]
- 31.He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW. Identification of c-MYC as a target of the APC pathway. Science. 1998;281:1509–1512. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- 32.Yochum GS, Cleland R, Goodman RH. A genome-wide screen for beta-catenin binding sites identifies a downstream enhancer element that controls c-Myc gene expression. Mol Cell Biol. 2008;28:7368–7379. doi: 10.1128/MCB.00744-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nelson JD, Denisenko O, Bomsztyk K. Protocol for fast chromatin immunoprecipitation (ChIP) method. Nat Protoc. 2006;1:179–185. doi: 10.1038/nprot.2006.27. [DOI] [PubMed] [Google Scholar]
- 34.Gurel B, Iwata T, Koh CM, Jenkins RB, Lan F, Van Dang C, Hicks JL, Morgan J, Cornish TC, Sutcliffe S, Isaacs WB, Luo J, De Marzo AM. Nuclear MYC protein over expression is an early alteration in human prostate carcinogenesis. Mod Pathol. 2008;21:1156–1167. doi: 10.1038/modpathol.2008.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jarrard DF, Sarkar S, Shi Y, Yeager TR, Magrane G, Kinoshita H, Nassif N, Meisner L, Newton MA, Waldman FM, Reznikoff CA. p16/pRb pathway alterations are required for bypassing senescence in human prostate epithelial cells. Cancer Res. 1999;59:2957–2964. [PubMed] [Google Scholar]
- 36.Dalrymple S, Antony L, Xu Y, Uzgare AR, Arnold JT, Savaugeot J, Sokoll LJ, De Marzo AM, Isaacs JT. Role of notch-1 and e-cadherin in the differential response to calcium in culturing normal versus maligannt prostate cells. Cancer Res. 2005;65:9269–9279. doi: 10.1158/0008-5472.CAN-04-3989. [DOI] [PubMed] [Google Scholar]
- 37.Edick MJ, Tesfay L, Lamb LE, Knudsen BS, Miranti1 CK. Inhibition of integrin-mediated crosstalk with epidermal growth factor receptor/Erk or Src signaling pathways in autophagic prostate epithelial cells induces caspase-independent death. Mol Biol Cell. 2007;18:2481–2490. doi: 10.1091/mbc.E06-04-0261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Webber MM, Trakul N, Thraves PS, Bello-DeOcampo D, Chu WW, Storto PD, Huard TK, Rhim JS, Williams DE. A human prostatic stromal myofibroblast cell line WPMY-1: A model for stromal–epithelial interactions in prostatic neoplasia. Carcino-genesis. 1999;20:1185–1192. doi: 10.1093/carcin/20.7.1185. [DOI] [PubMed] [Google Scholar]
- 39.Parsons JK, Gage WR, Nelson WG, De Marzo AM. p63 protein expression is rare in prostate adenocarcinoma: Implications for cancer diagnosis and carcinogenesis. Urology. 2001;58:619–624. doi: 10.1016/s0090-4295(01)01311-5. [DOI] [PubMed] [Google Scholar]
- 40.Tran CP, Lin C, Yamashiro J, Reiter RE. Prostate stem cell antigen is a marker of late intermediate prostate epithelial cells. Mol Cancer Res. 2002;1:113–121. [PubMed] [Google Scholar]
- 41.Wang L, Liu R, Li W, Chen C, Katoh H, Chen GY, McNally B, Lin L, Zhou P, Zuo T, Cooney KA, Liu Y, Zheng P. Somatic single hits inactivate the X-linked tumor suppressor FOXP3 in the prostate. Cancer Cell. 2009;16:336–346. doi: 10.1016/j.ccr.2009.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yochum GS, Sherrick CM, Macpartlin M, Goodman RH. A beta-catenin/TCF-coordinated chromatin loop at MYC integrates 5′ and 3′ Wnt responsive enhancers. Proc Natl Acad Sci USA. 2010;107:145–150. doi: 10.1073/pnas.0912294107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lepourcelet M, Chen YN, France DS, Wang H, Crews P, Petersen F, Bruseo C, Wood AW, Shivdasani RA. Small-molecule antagonists of the oncogenic TCF/beta-catenin protein complex. Cancer Cell. 2004;5:91–102. doi: 10.1016/s1535-6108(03)00334-9. [DOI] [PubMed] [Google Scholar]
- 44.Huang SM, Mishina YM, Liu S, Cheung A, Stegmeier F, Michaud GA, Charlat O, Wiellette E, Zhang Y, Wiessner S, Hild M, Shi X, Wilson CJ, Mickanin C, Myer V, Fazal A, Tomlinson R, Serluca F, Shao W, Cheng H, Shultz M, Rau C, Schirle M, Schlegl J, Ghidelli S, Fawell S, Lu C, Curtis D, Kirschner MW, Lengauer C, Finan PM, Tallarico JA, Bouwmeester T, Porter JA, Bauer A, Cong F. Tankyrase inhibition stabilizes axin and antagonizes Wnt signaling. Nature. 2009;461:614–620. doi: 10.1038/nature08356. [DOI] [PubMed] [Google Scholar]
- 45.Fang D, Hawke D, Zheng Y, Xia Y, Meisenhelder J, Nika H, Mills GB, Kobayashi R, Hunter T, Lu Z. Phosphorylation of β-catenin by AKT promotes β-catenin transcriptional activity. J Biol Chem. 2007;282:11221–11229. doi: 10.1074/jbc.M611871200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Staal FJ, Noort MM, Strous GJ, Clevers HC. Wnt signals are transmitted through N-terminally dephosphorylated beta-carenin. EMBO Rep. 2002;3:63–68. doi: 10.1093/embo-reports/kvf002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bafico A, Liu G, Yaniv A, Gazit A, Aaronson SA. Novel mechanism of Wnt signalling inhibition mediated by Dickkopf-1 interaction with LRP6/arrow. Nat Cell Biol. 2001;3:683–686. doi: 10.1038/35083081. [DOI] [PubMed] [Google Scholar]
- 48.Uzgare AR, Isaacs JT. Enhanced redundancy in Akt and mitogen-activated protein kinase-induced survival of malignant versus normal prostate epithelial cells. Cancer Res. 2004;64:6190–6199. doi: 10.1158/0008-5472.CAN-04-0968. [DOI] [PubMed] [Google Scholar]
- 49.Truica CI, Byers S, Gelmann EP. Beta-catenin affects androgen receptor transcriptional activity and ligand specificity. Cancer Res. 2000;60:4709–4713. [PubMed] [Google Scholar]
- 50.Chesire DR, Isaacs WB. Ligand-dependent inhibition of beta-catenin/TCF signaling by androgen receptor. Oncogene. 2002;21:8453–8469. doi: 10.1038/sj.onc.1206049. [DOI] [PubMed] [Google Scholar]
- 51.Mulholland DJ, Read JT, Rennie PS, Cox ME, Nelson CC. Functional localization and competition between the androgen receptor and T-cell factor for nuclear beta-catenin: A means for inhibition of the Tcf signaling axis. Oncogene. 2003;22:5602–5613. doi: 10.1038/sj.onc.1206802. [DOI] [PubMed] [Google Scholar]
- 52.Yang F, Li X, Sharma M, Sasaki CY, Longo DL, Lim B, Sun Z. Linking beta-catenin to androgen-signaling pathway. J Biol Chem. 2002;27:11336–11344. doi: 10.1074/jbc.M111962200. [DOI] [PubMed] [Google Scholar]
- 53.Amir A, Barua M, McKnight N, Cheng S, Yuan X, Balk SP. A direst beta-catenin-independent interaction between androgen receptor and T cell factor 4. J Biol Chem. 2003;278:30828–30834. doi: 10.1074/jbc.M301208200. [DOI] [PubMed] [Google Scholar]
- 54.Rossi R, Zatelli MC, Franceschetti P, Maestri I, Magri E, Aguiari G, Cavazzini P, degli Uberti EC, del Senno L. Inhibitory effect of dihydrotestosterone on thyroid cell growth. J Endocrinol. 1996;151:185–194. doi: 10.1677/joe.0.1510185. [DOI] [PubMed] [Google Scholar]
- 55.Rossi R, Zatelli MC, Valentini A, Cavazzini P, Fallo F, del Senno L, degli Uberti EC. Evidence for androgen receptor gene expression and growth inhibitory effect of dihydrotestosterone on human adrenocortical cells. J Endocrinol. 1998;159:373–380. doi: 10.1677/joe.0.1590373. [DOI] [PubMed] [Google Scholar]
- 56.Gounari F, Signoretti S, Bronson R, Klein L, Sellers WR, Kum J, Siermann A, Taketo MM, von Boehmer H, Khazaie K. Stabilization of β-catenin induces lesions reminiscent of prostatic intraepithelial neoplasia, but terminal squamous transdifferentiation of other secretory epithelia. Oncogene. 2002;21:4099–4107. doi: 10.1038/sj.onc.1205562. [DOI] [PubMed] [Google Scholar]
- 57.Bierie B, Nozawa M, Renou JP, Shillingford JM, Morgan F, Oka T, Taketo MM, Cardiff RD, Miyoshi K, Wagner KU, Robinson GW, Hennighausen L. Activation of β-catenin in prostate epithelium induces hyperplasias and squamous differentiation. Oncogene. 2003;22:3875–3887. doi: 10.1038/sj.onc.1206426. [DOI] [PubMed] [Google Scholar]
- 58.Yu X, Wang Y, Jiang M, Bierie B, Roy-Burman P, Shen MM, Taketo MM, Wills M, Matusik RJ. Activation of β-catenin in mouse prostate causes HGPIN and continuous prostate growth after castration. Prostate. 2009;69:249–262. doi: 10.1002/pros.20877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ellwood-Yen K, Graeber TG, Wongvipat J, Iruela-Arispe ML, Zhang J, Matusik R, Thomas GV, Sawyers CL. Myc-driven murine prostate cancer shares molecular features with human prostate tumors. Cancer Cell. 2003;4:223–238. doi: 10.1016/s1535-6108(03)00197-1. [DOI] [PubMed] [Google Scholar]
- 60.D’Antonio JM, Vander Griend DJ, Antony L, Ndikueze G, Dalrymple SL, Koochekpour S, Isaacs JT. Loss of androgen receptor-dependent growth suppression by prostate cancer cells can occur independently from acquiring oncogenic addiction to androgen receptor signaling. PLoS ONE. 2010;5:e11475. doi: 10.1371/journal.pone.0011475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vander Griend DJ, D’Antonio J, Gurel B, Antony L, De Marzo AM, Isaacs JT. Cell-autonomous intracellular androgen receptor signaling drives the growth of human prostate cancer initiating cells. Prostate. 2010;70:90–99. doi: 10.1002/pros.21043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Litvinov IV, De Marzo AM, Isaacs JT. Is the Achilles’ heel for prostate cancer therapy a gain of function in androgen receptor signaling? J Clin Endocrinol Metab. 2003;88:2972–2982. doi: 10.1210/jc.2002-022038. [DOI] [PubMed] [Google Scholar]
- 63.Bernard D, Pourtier-Manzanedo A, Gil J, Beach DH. Myc confers androgen-independent prostate cancer cell growth. J Clin Invest. 2003;112:1724–1731. doi: 10.1172/JCI19035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bryan JN, Keeler MR, Henry CJ, Bryan ME, Hahn AW, Caldwell CW. A population study of neutering status as a risk factor for canine prostate cancer. Prostate. 2007;67:1174–1181. doi: 10.1002/pros.20590. [DOI] [PubMed] [Google Scholar]
- 65.Thompson IM, Goodman PJ, Tangen CM, Lucia MS, Miller GJ, Ford LG, Lieber MM, Cespedes RD, Atkins JN, Lippman SM, Carlin SM, Ryan A, Szczepanek CM, Crowley JJ, Coltman CA. The influence of finasteride on the development of prostate cancer. N Engl J Med. 2003;349:215. doi: 10.1056/NEJMoa030660. [DOI] [PubMed] [Google Scholar]
- 66.Andriole GL, Bostwick DG, Brawley OW, Gomella LG, Marberger M, Montorsi F, Pettaway CA, Tammela TL, Teloken C, Tindall DJ, Somerville MC, Wilson TH, Fowler IL, Rittmaster RS, REDUCE Study Group Effect of dutasteride on the risk of prostate cancer. N Engl J Med. 2010;362:1192–1202. doi: 10.1056/NEJMoa0908127. [DOI] [PubMed] [Google Scholar]
- 67.Denmeade SR, Isaacs JT. Bipolar androgen therapy: The rationale for rapid cycling of supraphysiologic androgen/ablation in men with castration resistant prostate cancer. Prostate. 2010;70:1600–1607. doi: 10.1002/pros.21196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Isaacs JT, D’Antonio JM, Chen S, Antony L, Dalrymple SP, Ndikuyeze GH, Luo J, Denmeade S. Adaptive auto-regulation of androgen receptor provides a paradigm shifting rationale for bipolar androgen therapy (BAT) for castrate resistant human prostate cancer. Prostate. 2012;72:1491–1505. doi: 10.1002/pros.22504. [DOI] [PMC free article] [PubMed] [Google Scholar]