

Abstract
Emerging studies have shown that pharmacological activation of AMPK produces potent analgesic effects in different animal pain models. Currently, the spinal molecular and synaptic mechanism by which AMPK regulates the pain signaling system remains unclear. To address this issue, we utilized the Cre-LoxP system to conditionally knockout the AMPKα1 gene in the nervous system of mice. We demonstrated that AMPKα1 is imperative for maintaining normal nociception, and mice deficient for AMPKα1 exhibit mechanical allodynia. This is concomitantly associated with increased glutamatergic synaptic activities in neurons located in the superficial spinal dorsal horn, which results from the increased glutamate release from presynaptic terminals and function of ligand-gated glutamate receptors at the postsynaptic neurons. Additionally, AMPKα1 knockout mice have increased activities of extracellular signal-regulated kinases (ERK) and p38 mitogen-activated protein kinases (p38), as well as elevated levels of interleukin-1β (IL-1β), reactive oxygen species (ROS), and heme-oxygenase 1 (HO-1) in the spinal dorsal horn. Systemic administration of a non-specific ROS scavenger (phenyl-N-tert-butylnitrone, PBN) or a HO-1 activator (Cobalt protoporphyrin IX, CoPP) attenuated allodynia in AMPKα1 knockout mice. Bath-perfusion of the ROS scavenger or HO-1 activator effectively attenuated the increased ROS levels and glutamatergic synaptic activities in the spinal dorsal horn. Our findings suggest that ROS are the key down-stream signaling molecules mediating the behavioral hypersensitivity in AMPKα1 knockout mice. Thus, targeting AMPKα1 may represent an effective approach for the treatment of pathological pain conditions associated with neuroinflammation at the spinal dorsal horn.
Keywords: AMPK, ROS, cytokines, nociception, glutamatergic synapses, patch clamp
Introduction
Adenosine monophosphate-activated protein kinase (AMPK) is a key serine/threonine kinase that controls cellular energy homeostasis [1]. AMPK is activated when the AMP/ATP ratio is high leading to a reduction in ATP consumption and the increase of ATP production [1]. Accumulating data has demonstrated that AMPK also regulates many physiological and pathological processes in the nervous system, including long term potentiation in the hippocampus [2], aging [3], Alzheimer's disease [4], and stroke [5]. Multiple studies have shown that pharmacological activation of AMPK produces potent analgesic effects in different animal pain models [6-9]. Currently, whether and how endogenous AMPK activity regulates the pain signaling system remains unclear.
Spinal central sensitization, characterized by enhanced neuronal activation in the spinal dorsal horn, is a key process underlying the genesis of pathological pain [10]. Enhanced glutamatergic synaptic activation in the spinal dorsal horn is a critical mechanism leading to enhanced neuronal activities in the spinal dorsal horn under pathological pain conditions [11-13]. Ample evidence supports the findings that neuronal activities in the spinal dorsal horn are regulated by non-excitable glial cells (i.e., microglia and astrocytes) [14-16]. Activation of microglia and astrocytes and the subsequent increased production of inflammatory molecules, a cardinal feature of neuroinflammation, are commonly observed in animal models with inflammatory pain induced by subcutaneous injection of complete Freund’s adjuvant [17] or formalin [18], or animal models with neuropathic pain [15, 16, 19]. Pro-inflammatory cytokines like TNF-α [14], IL-1β [13, 14, 20], or chemokines like CX3CL1 [21] can directly enhance excitatory glutamatergic synaptic activities in spinal dorsal horn sensory neurons. Activation of MAP kinases (like p-38, ERK) in the spinal dorsal horn is associated with the genesis of persistent inflammatory [22] and neuropathic pain [23]. Suppression of p-38 and ERK attenuates inflammatory pain and neuropathic pain via controlling glial activation and production of pro-inflammatory cytokines (like TNF-α , IL-1β, and IL-6), and chemokines in the spinal dorsal horn [23-28]. Recent reports have demonstrated that activation of AMPK reduces the production of pro-inflammatory molecules in multiple cell types (including neutrophils [29], macrophages [30], and astrocytes and microglia [31, 32]). Our current understanding of the role of AMPK in the pain signaling system is mainly derived from studies using pharmacological activation of AMPK via different activators such as 5-Aminoimidazole-4-carboxamide ribonucleotide (AICAR), metformin, A769662, and resveratrol. Systemic or topical administration of one or more of these AMPK activators has shown to attenuate hypersensitive nociceptive behaviors in animals induced by surgical incision [8], nerve injury [9, 33], cancer chemotherapy drugs (cisplatin and paclitaxel) [34], and diabetic neuropathy [35]. The role of endogenous AMPK in nociceptive processing was recently revealed by us. AMPK activities in the spinal dorsal horn were reduced in animals with partial sciatic nerve ligation. Behavioral hypersensitivity was induced when spinal AMPK is knocked down by siRNA [9]. The spinal molecular and synaptic mechanisms regulated by endogenous AMPK which alter spinal nociceptive processing remain to be determined.
Reactive oxygen species (ROS) are free radicals that are mainly generated as byproducts of cellular metabolism. ROS have functions in cellular signaling and regulation [36-38] and have shown to be master signaling molecules controlling inflammatory signaling cascades. ROS plays an important role in the genesis of many pathological pain conditions [39, 40], including neuropathic pain induced by nerve injury [41, 42] or chemotherapy [43], and inflammatory pain induced by carrageenan [44], and morphine-tolerance [45-47]. Currently, it is not fully understood about endogenous molecular signaling pathways regulating the production of ROS and the impacts of ROS on synaptic activities at the spinal dorsal horn.
AMPK has two isoforms, AMPKα1 and AMPKα2 [48]. Studies of different cell types have shown that AMPKα1 and AMPKα2 play different roles in physiological and pathological processes [49-51]. In this study, to better define the molecular signaling pathways and synaptic mechanisms by which AMPKα1 controls the pain signaling system, we utilized the Cre-LoxP system to conditionally knockout the AMPKα1 gene in the nervous system of mice. We demonstrated that AMPKα1 is imperative for maintaining normal nociception, and mice deficient for AMPKα1 exhibit mechanical allodynia and enhanced glutamatergic synaptic activities at the spinal dorsal horn. We also provide compelling evidence that the behavioral hypersensitivity and enhanced glutamatergic synaptic activities in AMPKα1 knockout mice is mediated by elevated spinal ROS.
Material and Methods
Animals
Male and female mice (weight range 25-40 g) were used. FVB-Tg(GFAP-cre)25Mes/J [52] and Prkaa1tm1.1Sjm/J [53] mice were purchased from Jackson Laboratories. All experiments were approved by the Institutional Animal Care and Use Committee at the University of Georgia and were fully compliant with the National Institutes of Health Guidelines for the Use and Care of Laboratory Animals.
Behavior tests
To measure mechanical sensitivity in the animals, mice were placed on a wire mesh, loosely restrained under a plexiglass cage (12 × 20 × 15 cm3) and allowed to acclimate for at least 1 hour and 30 minutes. A series of von Frey monofilaments (bending force ranging from 0.07 to 2.00 g) were tested in ascending order to evoke hind paw withdrawal responses. Each von Frey filament was applied 5 times to the mid-plantar area of each hind paw from beneath for about 1 s. The response-frequency [(number of withdrawal responses of both hind paws/10) × 100%] for each von Frey filament was determined. Withdrawal response mechanical threshold was defined as the lowest force filament that evoked a response-frequency greater than 50%. This value was later averaged across all animals in each group to yield the group response threshold [20, 54]. There were no significant differences between sexes. AMPKα1 knockout mice did not display visible changes in their motor behaviors, which included grooming, postures, and gaits.
Generation of AMPKα1 gene knockout mice and genotyping
Mice with AMPKα1 gene knockout in the nervous system were generated through the Cre-LoxP method. Mice carrying the homozygous floxed AMPKα1 allele (flanking exon 3 of the kinase [53]) were crossed with mice expressing the Cre recombinase under the control of the glial fibrillary acid protein promoter (GFAP) [51] to produce their offspring. Male offspring which carried the Cre-GFAP recombinase and were heterozygous for the AMPKα1 LoxP flanked allele were crossed with homozygous floxed AMPKα1 female mice to generate GFAP-AMPKα1 conditional knockout mice in the nervous system. The GFAP promoter is transiently active in neural progenitor cells/radial glial precursor cells at an early embryonic period [52, 55, 56]. These cells develop into astrocytes, ependymal cells, oligodendroglia, and neurons [52]. Therefore, in our study, the AMPKα1 gene in these cellular types was deleted in the CNS through recombination of the AMPKα1 LoxP site with the GFAP-Cre. It was reported that the peripheral nerve is not affected by this GFAP-Cre transgene [52]. The genotype of GFAP-AMPKα1 conditional knockout mice was confirmed through polymerase chain reaction (PCR) as we reported previously [9]. Genotyping was conducted using the primers described by Jackson Laboratories for the sense strand 5’- CCC ACC ATC ACT CCA TCT -3’ and for the anti-sense strand 5’- AGC CTG CTT GGC ACA CTT AT -3’ to detect the AMPKα1 floxed allele and for the sense strand 5’- ACT CCT TCA TAA AGC CCT -3’ and for anti-sense strand 5’- ATC ACT CGT TGC ATC GAC CG -3’ to detect the GFAP-Cre transgene [52]. Littermates were used as controls in all experiments for genetic background effects.
Western blot experiments
Animals were deeply anesthetized with urethane (1.3-1.5 g/kg, i.p.). The L4 to L5 spinal segment was exposed by surgery and removed from the animals. The dorsal half of the spinal cord at the L4 to L5 segment was removed. The spinal tissue was quickly frozen in liquid nitrogen and stored at -80°C for later use. Following tissue isolation the animals were euthanized. Frozen tissues were homogenized as previously described [9, 57]. Protein concentrations were quantified with the Pierce BCA method (Thermo Scientific). Protein samples were electrophoresed in SDS polyacrylamide gels and transferred to a polyvinylidene difluoride membrane (Millipore, Bedford, MA). The membranes were blocked with milk or BSA and incubated overnight at 4°C with an anti phospho-p38 (1:500, Cell Signaling), anti p-38 (1:1000, Cell Signaling), phospho-ERK (1:1000, Cell Signaling), anti-ERK(1:1000, Cell Signaling), anti-HO-1 (a marker for oxidative stress, 1:500, Enzo Life Sciences), anti -IL-1β (1:1000, Millipore), or a monoclonal anti-β-Actin (1:2000, Sigma-Aldrich, St. Louis, USA) primary antibody as a loading control. The blots were then incubated for 1 hr at room temperature (RT) with the corresponding HRP-conjugated secondary antibody (1:5000; Santa Cruz Biotechnology, CA, USA), visualized in ECL solution (SuperSignal West Chemiluminescent Substrate, Pierce, Rockford, IL, USA), and exposed on the FluorChem HD2 System. The intensity of immunoreactive bands was quantified using ImageJ 1.46 software (NIH). Results were expressed as the ratio of each marker over β-Actin control unless otherwise indicated.
Spinal Slice Preparation
Transverse mouse spinal cord slices (400 µm) of the L4 to L5 segment were prepared as previously described [58]. Briefly, animals were anesthetized via isoflurane inhalation. Surgery was performed to expose and remove the spinal lumbar enlargement segment. The lumbar spinal cord section was then placed in ice-cold sucrose artificial cerebrospinal fluid (aCSF) pre-saturated with 95% O2 and 5% CO2. The sucrose aCSF contained 234 mM sucrose, 3.6 mM KCl, 1.2 mM MgCl2, 2.5 mM CaCl2, 1.2 mM NaH2PO4,12.0 mM glucose, and 25.0 mM NaHCO3. The pia-arachnoid membrane was removed from the section. The L4 to L5 spinal segment was attached with cyanoacrylate glue to a cutting support, which was then glued onto the stage of a vibratome (Series 1000, Technical Products International, St. Louis, MO). Transverse spinal cord slices were cut in the ice-cold sucrose aCSF and then pre-incubated in Krebs solution oxygenated with 95% O2 and 5% CO2 at 35°C. The Krebs solution contained: 117.0 mM NaCl, 3.6 mM KCl, 1.2 mM MgCl2, 2.5 mM CaCl2, 1.2 mM NaH2PO4, 11.0 mM glucose, and 25.0 mM NaHCO3 at 35°C.
Measurements of spinal ROS
ROS production in spinal slices was measured using 5-(and-6)-carboxy-2,7- dichlorodihydrofluorescein diacetate (Carboxy-H2DCFDA, C400, Molecular Probes, Invitrogen) as described by others [59]. Carboxy-H2DCFDA is a dye sensitive to peroxynitrite anions (ONOO-), hydrogen peroxide (H2O2), and hydroxyl radicals (HO·). After a hour recovery period from slicing, spinal slices were loaded in the dark at 30°C for 1 hour with 50 µM Carboxy-H2DCFDA in dimethyl sulfoxide (DMSO, 1 mg in 30 µL). After loading, the slices were transferred to aCSF and recovered for 30-45 min in the dark to allow the acetate groups to be removed by intracellular esterase [59]. ROS imaging was then performed on an Olympus fluorescent microscope under a 60X objective (numerical aperture: 1.2). Images of fluorescence signals (excitation 450–490 nm, emission 510–550 nm) were acquired with a Hamamatsu CCD camera. Metafluor imaging software was used to detect and analyze ROS levels throughout the experiment [60].
Drug Administration
Drugs were systemically administered through a single intraperitoneal injection. PBN was dissolved in sterile saline for a total volume of 0.5 mL. CoPP was dissolved in DMSO and then sterile saline for a < 1% DMSO solution in a total volume of 0.5 mL. Equal volumes of sterile saline or DMSO were used as a vehicle control.
In Vitro whole cell-voltage clamp recordings and data analysis
Transverse mouse spinal cord slices (400 µm) of the L4 to L5 segment were prepared as previously described [58]. Briefly, animals were anesthetized via isoflurane inhalation. Surgery was performed to expose and remove the spinal lumbar enlargement segment. The lumbar spinal cord section was then placed in ice-cold sucrose aCSF pre-saturated with 95% O2 and 5% CO2. The pia-arachnoid membrane was removed from the section. The L4 to L5 spinal segment was attached with cyanoacrylate glue to a cutting support, which was then glued onto the stage of a vibratome (Series 1000, Technical Products International, St. Louis, MO). Transverse spinal cord slices were cut in the ice-cold sucrose aCSF and then pre-incubated in Krebs solution oxygenated with 95% O2 and 5% CO2 at 35°C. The Krebs solution contained: 117.0 mM NaCl, 3.6 mM KCl, 1.2 mM MgCl2, 2.5 mM CaCl2, 1.2 mM NaH2PO4, 11.0 mM glucose, and 25.0 mM NaHCO3 at 35°C. Following pre-incubation for at least 1.5 hrs, a single slice was placed in the recording chamber (volume, 1.5 ml), perfused with Krebs solution at 35°C, and saturated with 95% O2 and 5% CO2. Borosilicate glass recording electrodes (resistance, 3-5 MO.) were pulled and filled with an internal solution containing 135 mM potassium-gluconate; 5.0 mM KCl; 2.0 mM MgCl2; 0.5 mM CaCl2; 5.0 mM HEPES; 5.0 mM EGTA; 5.0 mM ATP-Mg; 0.5 mM Na-GTP; 10 mM QX-314. Live dorsal horn neurons in the spinal lamina I and outer lamina II (IIo) were visualized using a microscope system and approached using a three-dimensional motorized manipulator (Sutter Instrument, Novato, CA, USA), and whole-cell configurations were established by applying moderate negative pressure after electrode contact [61]. Recordings were made from neurons receiving monosynaptic input from the primary afferents using the criteria previously established [62, 63] . Miniature excitatory postsynaptic currents (mEPSCs) were recorded at a membrane potential at −70 mV in the presence of tetrodotoxin (1 µM), bicuculline (10 µM), and strychnine (5 µM) in the external solution to block voltage-gated sodium channels, GABAA, and glycine receptors. Signals were amplified using an Axopatch 700B (Molecular Devices), digitized at 10 kHz, and displayed and stored in a personal computer. The frequency and amplitude of mEPSCs at 3 min before and after perfusion of tested drugs were analyzed and averaged using a peak detection program (MiniAnalysis; Synaptosoft Inc., Decatur, GA).
Materials
Phenyl-N-tert-butylnitrone (PBN) was purchased from Sigma-Aldrich and Cobalt protoporphyrin IX (CoPP) was purchased from Enzo Life Sciences.
Data Analysis
All data are presented as the mean ± standard error (S.E). One or two way analysis of variance (ANOVA) with repeated measures was used to detect differences on nociceptive behaviors between mice receiving different treatments. A Bonferroni post-hoc test was performed to determine sources of the differences. When applicable, Student’s t-tests were used to make comparison between groups (non-paired) or within the same group (paired). Comparisons were run as two-tailed tests. A p value less than 0.05 was considered statistically significant. Statistical analysis was performed using GraphPad Prism 5 (GraphPad Software Inc.).
Results
Selective AMPKα1 deletion induced mechanical allodynia via enhancing spinal glutamatergic synaptic activities in the spinal dorsal horn
To determine the role of the AMPKα1 subtype in the pain signaling system, the AMPKα1 isoform in the CNS was conditionally deleted in mice through Cre-LoxP recombination [52]. We compared mechanical thresholds of the hind paw withdrawal response between AMPKα1 knockout mice and their AMPKα1 littermate controls. We found that mechanical thresholds of the hind paw withdrawal response in AMPKα1 knockout mice were significantly (p < 0.001) lower (0.72 ± 0.09 g, n = 10) in comparison with their AMPKα1 littermate controls (1.50 ± 0.17 g, n = 10)(Fig. 1A). These results indicate that AMPKα1 is necessary to maintain normal nociceptive behaviors in rodents.
Figure 1. Mice with selective AMPKα1 deletion had mechanical allodynia and enhanced spinal glutamatergic synaptic activities in the spinal dorsal horn.

(A) shows that mechanical thresholds of hind paw withdrawal responses were significantly lower in AMPKα1 knockout mice than AMPKα1 littermate controls. (B) shows representative recordings of mEPSCs recorded from spinal superficial dorsal horn neurons of spinal slices obtained from AMPKα1 littermate controls (upper trace) and AMPKα1 knockout mice (lower trace). Bar graphs (C) show the mean (+ S.E.) amplitude and frequency obtained from AMPKα1 littermate controls and AMPKα1 knockout mice. ***, p < 0.001.
To investigate the spinal mechanisms underlying the behavioral hypersensitivity in mice with conditional AMPKα1 knockout, we determined whether glutamatergic synaptic activities in the superficial dorsal horn were altered. Glutamate release from presynaptic terminals and activities of postsynaptic glutamate ligand-gated ion receptors are conventionally determined by analyzing the frequency and amplitude of mEPSCs. An increase of mEPSC frequencies indicates an increase in the release of presynaptic glutamate, while an increase of mEPSC amplitudes indicates an increase in the function of postsynaptic glutamate receptors [13]. We recorded mEPSCs from neurons that received monosynaptic input from primary afferents at the superficial dorsal horn (lamina I and IIo) using whole cell-voltage clamp techniques. As shown in Figure 1B to 1D, neurons recorded from mice with AMPKα1 knockout displayed a significantly higher mEPSC frequency (6.46 ± 0.22 Hz, n = 23, p < 0.001) and amplitude (28.97 ± 0.12 pA, n = 23, p < 0.001) than the AMPKα1 littermate controls (frequency: 2.76 ± 0.07 Hz, n = 24; amplitude: 25.18 ± 0.41 pA, n = 24). These data indicate that the increased release of glutamate from presynaptic terminals and activities of postsynaptic glutamate receptors, and therefore, enhanced neuronal activation in the spinal dorsal horn is ascribed to the behavioral hypersensitivity in mice with AMPKα1 knockout.
Selective AMPKα1 deletion induced neuroinflammation in the spinal dorsal horn
We next investigated the potential molecular mechanisms underlying the changes of the increased glutamatergic synaptic activities in the spinal dorsal horn of AMPKα1 knockout mice. We first examined levels of p-38 and ERK activity, and protein expression of a prototypic proinflammatory cytokine, IL-1β. These molecules are known to be critically implicated in the genesis of neuroinflammation and pathological pain [23, 26, 64]. The dorsal half of the spinal cord was removed and used for Western blotting experiments. The activity levels of p-38 and ERK were determined by the ratios of phosphorylated p-38 (p-p38) versus total p-38 (t-p-38), and phosphorylated ERK (p-ERK) versus total ERK (t-ERK) in addition to the measurement of t-p-38 and t-ERK. In comparison with the AMPKα1 littermate controls (n = 4), AMPKα1 knockout mice (n = 5) had significantly increased ratios of p-p38/t-p-38 (p < 0.05), p-ERK/t-ERK (p<0.05), and IL-1β (p < 0.05) protein expression (Figure 2). The total protein levels of p-38 and ERK in AMPKα1 knockout mice and AMPKα1 littermate controls (n = 4) were similar (data not shown). These data indicate that AMPKα1 deletion results in increased activities of p-38 and ERK and production of IL-1β.
Figure 2. AMPKα1 deletion resulted in increased activities of p-38 and ERK and production of IL-1β in the spinal dorsal horn.
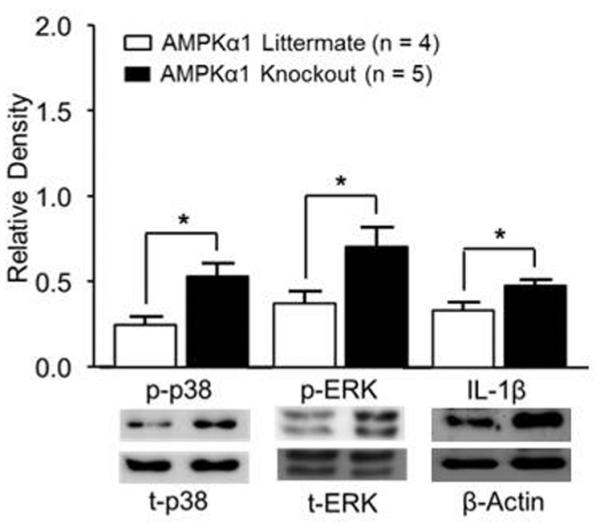
Bar graphs show the mean (+ S.E.) relative density of p-p38 to t-p-38, p-ERK to t-ERK, and IL-1β to β-actin obtained from AMPKα1 littermate controls and AMPKα1 knockout mice. Samples from Western blots of each molecule are shown below. *, p < 0.05.
Studies in the forebrain area and different cell lines have demonstrated that ROS are leading molecules whose over-production triggers a cascade of inflammatory signaling, including activation of p-38, ERK, and production of pro-inflammatory cytokines (such as IL-1β) [65-67]. We then determined whether ROS levels in the spinal dorsal horn of AMPKα1 knockout mice were altered. The ROS-sensitive dye 5-(and-6)-carboxy-2’,7’- dichlorodihydrofleuroscein diacetate (Carboxy-H2DCFDA) [68] was used to measure ROS levels. ROS activity oxidizes the non-fluorescent Carboxy-H2DCFDA into a green fluorescent form which accumulates in cells [69], and fluorescent intensity can be measured [59]. We made live spinal slices (450 µm) [60] and then incubated the slices in artificial CSF with 50 µL of Carboxy-H2DCFDA for 1 hour [59]. The fluorescent intensity in the spinal dorsal horn under a fluorescent microscope was captured with a Hamamatsu CCD camera and analyzed using Metafluor imaging software [60]. We found that AMPKα1 knockout mice had a significant (p < 0.001, n = 17) increase in ROS production in the spinal dorsal horn compared to AMPKα1 littermate controls (n = 22) (Figure 3A and B). These results demonstrate that the selective deletion of AMPKα1 induces over-production of ROS in the spinal dorsal horn, and implies that suppression of ROS levels may reverse the pathological changes induced by AMPKα1.
Figure 3. AMPKα1 deletion resulted in increased levels of ROS and HO-1 protein expression in the spinal dorsal horn.

(A) shows representative images of ROS intensity in the spinal dorsal horn of AMPKα1 knockout and AMPKα1 littermate mice. Scale bar = 40 µm. Bar graphs (B) show summaries (mean + S.E.) of ROS density in the spinal dorsal horn obtained from AMPKα1 knockout and AMPKα1 littermate mice. The mean (+ S.E.) relative density of HO-1 protein expressions to β-actin in the spinal dorsal horn, obtained from AMPKα1 knockout and AMPKα1 littermate mice, are shown in (C). Samples of Western blot gels of HO-1 are shown below. *, p < 0.05; ***, p < 0.001.
The generation of ROS is increased when intracellular heme is accumulated, which is degraded by heme oxygenase-1 (HO-1) [70, 71]. HO-1 is typically expressed at undetectable levels under normal conditions and becomes activated with oxidative stress [63, 64]. To explore whether increased levels of ROS in AMPKα1 knockout mice results from a reduction of HO-1, the protein expression of HO-1 was measured by Western blotting. We found that the protein expression of HO-1 was significantly higher (p < 0.05) in AMPKα1 knockout mice (n = 5) than AMPKα1 littermate controls (n = 4) (Figure 3C), suggesting that the increased levels of ROS in AMPKα1 knockout mice are not caused by a reduction in HO-1. Instead, our results demonstrate that increased HO-1 expression acts in a compensatory mechanism to counteract the increased ROS level in AMPKα1 knockout mice.
ROS are the key down-stream signaling molecules mediating the behavioral hypersensitivity induced by deficiency of AMPKα1
We then hypothesized that ROS are master signaling molecules mediating the enhanced nociceptive behaviors in AMPKα1 knockout mice. To test this assumption, we examined whether mechanical allodynia in AMPKα1 knockout mice can be reversed by suppressing ROS. We first used a free radical scavenger (phenyl-N-tert-butylnitrone, PBN) [39, 72]. Previous studies have shown that an intraperitoneal injection of PBN (100 mg/kg) attenuates mechanical allodynia induced by spinal nerve ligation in rats [73]. Mice were separated into four groups: AMPKα1 Knockout + PBN, AMPKα1 Knockout + Vehicle, AMPKα1 Littermate + PBN, and AMPKα1 Littermate + Vehicle. After taking baseline (BL) hind paw mechanical threshold measurements, we intraperitoneally injected PBN (100 mg/kg, in a volume of 0.5 mL) or vehicle (0.5 mL) into the mice. As shown in Figure 4A, in comparison with their baseline readings, mechanical thresholds of hind paw withdrawal responses in AMPKα1 knockout mice receiving PBN was significantly increased at 30 minutes following injection (p < 0.05, n = 8). At the same time point, AMPKα1 knockout mice administered with PBN had significantly (p < 0.05) increased mechanical withdrawal threshold compared to AMPKα1 knockout mice receiving vehicle (n = 6). Meanwhile, neither PBN nor vehicle given to AMPKα1 littermate controls altered the mechanical thresholds of hind paw withdrawal responses in the mice.
Figure 4. ROS mediated the behavioral hypersensitivity induced by deficiency of AMPKα1.

Line plots show measurements (mean + S.E.) of mechanical thresholds collected at baseline (BL) and then 30 min, 1, 2, 4, 6 and 24 hrs after the intraperitoneal administration of the tested agents in different groups.(A) shows that the ROS scavenger (100 mg/kg, PBN) attenuated mechanical allodynia in AMPKα1 knockout mice. Comparisons between data collected at baseline and each time point are indicated with + for the AMPKα1 Knockout + PBN group. Comparisons between the AMPKα1 Knockout + Vehicle group and the AMPKα1 Knockout + PBN group are labeled with #. (B) shows that pharmacological activation of HO-1 with CoPP (5 mg/kg) attenuated mechanical allodynia in AMPKα1 knockout mice. Comparisons between data collected at baseline and each time point are indicated with + for the AMPKα1 Knockout + CoPP group. Comparisons between the AMPKα1 Knockout + Vehicle group and the AMPKα1 Knockout + CoPP group are labeled with #. One symbol, p < 0.05; Two symbol, p < 0.01, Three symbols, p < 0.001.
It is known that activation of HO-1 reduces ROS levels in different cell types [74, 75]. To further validate the role of ROS in the genesis of mechanical allodynia in AMPKα1 knockout mice, we examined the effects of HO-1 activator, Cobalt protoporphyrin IX (CoPP) [76], on AMPKα1 knockout mice and AMPKα1 littermate controls. We grouped mice into four groups: AMPKα1 Knockout + CoPP, AMPKα1 Knockout + Vehicle, AMPKα1 Littermate + CoPP, and AMPKα1 Littermate + Vehicle. CoPP (5 mg/kg, in a volume of 0.5 mL) or vehicle (0.5 mL) was intraperitoneally injected into the mice. Mechanical thresholds of hind paw withdrawal responses were measured before and after the injection. As shown in Figure 4B, mechanical thresholds of hind paw withdrawal responses in AMPKα1 knockout mice (n = 9) receiving CoPP treatment were significantly increased at 30 minutes following the injection in comparison with their own baseline readings (p < 0.01), or in comparison with AMPKα1 knockout mice (p < 0.001, n = 6) treated with vehicle (Fig. 4B). Mechanical thresholds in AMPKα1 littermate controls were not significantly altered by treatment of CoPP (n = 9) or vehicle (n = 9). Together with data collected in Figure 4A, these data strongly indicate that ROS are the culprit in the behavioral hypersensitivity in mice with AMPKα1 knockout. These results further indicate that normal nociception is not regulated by endogenous ROS under normal conditions, which is consistent with previous studies [77]
Levels of ROS in the spinal dorsal horn in AMPKa1 knockout mice were reduced by the ROS scavenger and HO-1 activator
PBN can scavenge both ROS and peroxynitrite [39, 72]. To directly demonstrate the effect of PBN on ROS levels in the spinal dorsal horn of AMPKα1 knockout mice, we directly monitored ROS levels of live spinal slices before, after 15 min of bath-perfusion, and after washout of PBN. The ROS levels were determined by analyzing the fluorescent intensity created by the interaction between ROS and Carboxy-H2DCFDA. After capturing baseline ROS levels in the spinal dorsal horn of AMPKα1 knockout mice, we bath-perfused PBN at 1 mM concentration, a concentration known to suppress long term potentiation at the spinal dorsal horn [78]. Bath perfusion of PBN significantly (p < 0.001) reduced the ROS levels by 13.70 ± 2.44%. Such effects were reversed after PBN was washed out (Figure 5A and B). We also found that bath-perfusion of the HO-1 activator (CoPP) at a concentration of 10 µM significantly (p < 0.001) reduced the ROS levels by 12.77 ± 2.41% in the spinal dorsal horn of AMPKα1 knockout mice (Figure 5C and D). These effects disappeared after washout of CoPP. These findings directly demonstrate that the ROS levels in the spinal dorsal horn of AMPKα1 knockout mice can be effectively suppressed by PBN and CoPP.
Figure 5. Levels of ROS in the spinal dorsal horn in AMPKα1 knockout mice were reduced by the ROS scavenger and HO-1 activator.

Representative images show ROS intensity in the spinal dorsal horn of AMPKα1 knockout mice before (baseline), during and after washout of PBN (A) or CoPP (C) perfusion. Bar graphs show summaries (mean + S.E.) of ROS density normalized to their baseline before, during and after washout of PBN (B) or CoPP (D) perfusion. Scale bar = 40 µm. **, p < 0.01; ***, p < 0.001.
The ROS scavenger and HO-1 activator effectively attenuated the enhanced glutamatergic synaptic activities in the spinal dorsal horn of AMPKα1 knockout mice
Finally, we examined the effects induced by the ROS scavenger and HO-1 activator on glutamatergic synaptic activities in the spinal dorsal horn of AMPKα1 knockout mice. mEPSCs in dorsal horn neurons of AMPKα1 knockout mice were recorded from live spinal slices before and after the bath perfusion of 1 mM PBN. As shown in Figure 6A, B, and C, PBN perfusion significantly reduced mEPSC frequencies from 6.54 ± 0.46 Hz (n = 9) at baseline to 3.69 ± 0.29 Hz (p < 0.001), and amplitudes from 29.04 ± 0.96 pA at baseline to 22.45 ± 0.70 pA (p < 0.001). These PBN induced effects disappeared when PBN was washed out. Similarly, mEPSC frequencies and amplitudes in the spinal dorsal horn neurons of AMPKα1 knockout mice were significantly reduced when 10 µM CoPP was bath-perfused. mEPSC frequencies were reduced from 6.41 ± 0.23 Hz (n = 14) to 3.98 ± 0.13 Hz (p < 0.001) and amplitudes were reduced from 28.93 ± 0.58 pA to 23.51 ± 0.58 (p < 0.001) (Figure 6D, E, and F). Such effects went away upon washout of CoPP. These data provide direct evidence that elevated ROS levels are a causal factor for the enhanced glutamatergic synaptic activation in the spinal dorsal horn neurons and the behavioral hypersensitivity in AMPKα1 knockout mice.
Figure 6. The ROS scavenger and inducer of HO-1 effectively attenuated the enhanced glutamatergic synaptic activities in the spinal dorsal horn of AMPKα1 knockout mice.

Raw data show samples of continuous mEPSC recordings collected from dorsal horn neurons of AMPKα1 knockout mice before, during and after washout of PBN (A) or CoPP (D) perfusion. Bar graphs show the mean (+ S.E.) frequency and amplitude of mEPSCs in AMPKα1 knockout mice before (baseline), during and after washout of PBN (B and C) or CoPP (E and F). *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Discussion
Given that multiple AMPK activators like metformin and resveratrol are FDA approved or currently in clinical trials for a range of conditions such as diabetes, investigating the role of AMPK in the pain signaling system may bring a novel clinical application for this class of drugs. In this study, we for the first time revealed the spinal molecular and synaptic mechanisms by which AMPKα1 regulates nociceptive behaviors. Our study suggests that AMPKα1 may be a new target for the development of analgesics.
Recent studies of the role of AMPK in the pain signaling system are mainly focused on the effects induced by pharmacological activation of AMPK on different animal pain models. These studies in general demonstrated that activation of AMPK produces analgesic effects in rodents with pathological pain but does not alter normal nociception. Activation of AMPK with peritoneal injection of metformin and A769662 reverses pre-existing mechanical allodynia induced by spinal nerve ligation [33]. These analgesic effects are associated with suppression of the translation regulation signaling pathways, eIF4F complex formation, and nascent protein synthesis in injured nerves [33]. Systemic administration of metformin or AICAR suppresses the development of mechanical allodynia and thermal hyperalgesia in diabetic rats induced by streptozotocin, which is accompanied by suppression of the increased expressions of PGC-1α, Sirt-3 and nNOS in the sciatic nerve in the animals [35]. Metformin treatment also prevents the reduction in density of intra-epidermal nerve fibers (IENFs) in the paw that develops as a result of cisplatin [34]. Inflammatory pain and paw edema induced by zymosan A in mice is attenuated by peritoneal administration of metformin or AICAR [79]. These above studies suggest that AMPK activators may produce analgesic effects through acting at the peripheral nerves and tissues. Other studies provide evidence that AMPK activators can suppress pathological pain through their action at the CNS (spinal dorsal horn). For example, peritoneal injection of metformin or AICAR attenuates the formalin-induced phase II nociceptive behaviors [79], which is likely mediated by spinal cord mechanisms [64, 80]. More directly, we demonstrated that spinal intrathecal administration of AICAR attenuates neuropathic pain induced by partial sciatic nerve ligation in rats [9]. Together, these studies demonstrated that AMPK activators produce analgesic effects through both peripheral and central mechanisms.
The critical role of AMPK in the pain signaling system is further supported by recent studies that demonstrated a reduction of endogenous AMPK activity at the peripheral nerves or CNS of animals with behavioral hypersensitivity. Suppression of AMPK activities was found in the sciatic nerve of rats with diabetic painful neuropathy [35]. We recently reported that phosphorylation levels (activities) of AMPK at the spinal dorsal horn are decreased in rats with neuropathic pain induced by partial sciatic nerve ligation [9]. Pharmacological inhibition of AMPK activities in the spinal cord induces thermal hyperalgesia in rats. More importantly, rats with spinal AMPK gene knockdown by siRNA exhibit thermal hyperalgesia. Interestingly, the integrity of AMPK in the hypothalamus is required for acupuncture to produce full analgesic effects in rats [81]. Endogenous AMPKα1 and AMPKα2 appear to play differing roles in nociceptive sensory processing. Animals with complete or selective AMPKα2 knockout in peripheral sensory neurons display normal sensitivities to mechanical and thermal stimuli, but have exaggerated phase II nociceptive behavioral responses in the formalin test and stronger hypersensitivity induced by zymosan A [79]. Our current and previous studies demonstrated that animals with AMPKα1 deletion in the CNS have hypersensitivity to peripheral stimuli, indicating that AMPKα1 is not dispensable for maintaining normal nociception. Furthermore, our current study revealed that allodynia in AMPKα1 knockout mice is ascribed to the increased glutamatergic synaptic activities in the spinal dorsal horn.
It has been well documented that increased neuronal activation in the spinal dorsal horn plays a key role in the genesis of different pathological pain conditions. These include the sciatic nerve-injury induced neuropathic pain [82, 83], diabetic neuropathic pain [84], paclitaxel [54] or vincristine-chemotherapy [85] induced neuropathic pain, and CFA-induced inflammatory pain [86]. Neuronal activation depends on neuronal passive and active membrane properties, and excitatory and inhibitory synaptic activities. Glutamate is the major excitatory neurotransmitter released from the primary afferents [87]. Therefore, activities of excitatory glutamatergic synapses in the spinal dorsal horn neurons predominantly control activation of spinal dorsal horn neuron. Activation of glutamatergic synapses are governed by three factors: the amount of glutamate released from presynaptic terminals, the function and number of postsynaptic glutamate receptors, and the function of glutamate transporters [88]. Our current and previous studies together demonstrated that suppression of AMPK activities results in enhanced glutamatergic synaptic activities via controlling all of these three factors. Our present study shows that spinal dorsal horn neurons in AMPKα1 knockout mice have increased frequencies and amplitudes of mEPSCs, indicating an increase of glutamate release from presynaptic terminals and function of glutamate receptors at the postsynaptic neuron. In other words, normal AMPKα1 activity limits the release of glutamate from presynaptic terminals and activities of postsynaptic glutamate receptors. We have previously demonstrated that the activity of glial glutamate transporters are reduced upon AMPK inhibition but increased on AMPK activation [9]. We recently also reported that deficient glial glutamate transport results in weakening of GABAergic strength in spinal dorsal horn neurons due to insufficient GABA synthesis through the glutamate-glutamine cycle between astrocytes and neurons [89]. Thus, it is conceivable that in addition to enhancing glutamatergic synaptic activities, suppression of AMPK may reduce GABAergic inhibitory synaptic activities in spinal dorsal horn neurons as well, both resulting in enhanced neuronal activation and pain signaling.
Signaling molecular pathways triggered by alterations of AMPK have been extensively studied in forebrain areas and cell lines. AMPK is known to regulate many key molecules important to the development of inflammation [90]. In contrast, there are only two reports about signaling molecules altered by AMPK in the spinal dorsal horn. Peritoneal injection of an AMPK activator (AICAR) increases spinal activities of AMPK (phosphorylation levels of AMPK), and attenuates the formalin-induced activation of p38 and JNK2 MAP kinases in the spinal cord [79]. We recently reported that spinal AMPKα knockdown and AMPKα1 knockout induces activation of astrocytes, over-production of IL-1β, and increased activation of GSK3β in the spinal dorsal horn. Our current study confirm our previous findings and further unveils that AMPKα1 conditional knockout results in activation of p38, ERK, over-production of ROS, and increased protein expression of HO-1 in the spinal dorsal horn. We also demonstrated for the first time that over-production of ROS mediates the increased glutamatergic synaptic activities in the spinal dorsal horn of mice and allodynia in mice with AMPKα1 knockout. This indicates that ROS are the key down-stream signaling molecules mediating the behavioral hypersensitivity in AMPKα1 knockout mice. Activation of glial cells, p38, ERK, increased production of ROS, and pro-inflammatory cytokines are widely reported in many animals with pathological pain [73, 77, 91-93]. Given these facts, our findings provide compelling evidence for the use of AMPK activators as a powerful tool to suppress spinal neuroinflammation and restore normal neuronal activities and pain signaling at the spinal dorsal horn.
Conclusions
In conclusion, we identified the spinal molecular signaling pathways and synaptic mechanisms controlled by AMPKα1. We found that increased spinal ROS levels and glutamatergic synaptic activities mediate the enhanced nociceptive behaviors in mice deficient for AMPKα1. The knockout of AMPKα1 increased ERK and p-38 activities, and the production of IL-1β, ROS, and HO-1 in the spinal dorsal horn. An intraperitoneal injection of a ROS scavenger or inducer of HO-1 attenuated mechanical allodynia in AMPKα1 knockout mice. Furthermore, the bath-perfusion of these pharmacological agents effectively attenuated increased spinal ROS and glutamatergic synaptic activities in the spinal dorsal horn of mice. These results suggest that targeting spinal AMPK may represent an effective approach for the treatment of pathological pain conditions involving increased ROS and neuroinflammation in the spinal dorsal horn.
Highlights.
Pain behavior and glutamatergic synaptic activity increase in AMPKα1 knockout mice.
AMPKα1 knockout elevates levels of spinal p-38, ERK activity, IL-1β, ROS and HO-1.
ROS scavenging and HO-1 activation attenuate AMPKα1 knockout induced allodynia.
ROS inhibition reduces glutamatergic synaptic activity in AMPKα1 knockout mice.
Acknowledgments
This project was supported by the NIH RO1 grant (NS064289) to H.R.W. and in part by the National Natural Science Foundation of China (number 81300662) to X.Y. The authors declare no conflict of interest.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Steinberg GR, Kemp BE. AMPK in health and disease. Science Signaling. 2009;89:1025. doi: 10.1152/physrev.00011.2008. [DOI] [PubMed] [Google Scholar]
- [2].Potter WB, O'Riordan KJ, Barnett D, Osting SM, Wagoner M, Burger C, Roopra A. Metabolic regulation of neuronal plasticity by the energy sensor AMPK. PLoS One. 2010;5:e8996. doi: 10.1371/journal.pone.0008996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Douglas PM, Dillin A. Protein homeostasis and aging in neurodegeneration. J Cell Biol. 2010;190:719–729. doi: 10.1083/jcb.201005144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Salminen A, Kaarniranta K, Haapasalo A, Soininen H, Hiltunen M. AMP-activated protein kinase: a potential player in Alzheimer's disease. J Neurochem. 2011;118:460–474. doi: 10.1111/j.1471-4159.2011.07331.x. [DOI] [PubMed] [Google Scholar]
- [5].Li J, McCullough LD. Effects of AMP-activated protein kinase in cerebral ischemia. J Cereb Blood Flow Metab. 2010;30:480–492. doi: 10.1038/jcbfm.2009.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Price TJ, Dussor G. AMPK: An emerging target for modification of injury-induced pain plasticity. Neuroscience Letters. 2013;557:9–18. doi: 10.1016/j.neulet.2013.06.060. Part A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Taylor A, Westveld AH, Szkudlinska M, Guruguri P, Annabi E, Patwardhan A, Price TJ, Yassine HN. The use of metformin is associated with decreased lumbar radiculopathy pain. Journal of pain research. 2013;6:755. doi: 10.2147/JPR.S52205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Tillu DV, Melemedjian OK, Asiedu MN, Qu N, De Felice M, Dussor G, Price TJ. Resveratrol engages AMPK to attenuate ERK and mTOR signaling in sensory neurons and inhibits incision-induced acute and chronic pain. Molecular Pain. 2012;8:5. doi: 10.1186/1744-8069-8-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Maixner D, Yan X, Gao M, Yadav R, Weng H. Adenosine Monophosphate-activated Protein Kinase Regulates Interleukin-1β Expression and Glial Glutamate Transporter Function in Rodents with Neuropathic Pain. Anesthesiology. 2015 doi: 10.1097/ALN.0000000000000619. DOI. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Woolf CJ. Central sensitization: implications for the diagnosis and treatment of pain. Pain. 2011;152:S2–S15. doi: 10.1016/j.pain.2010.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zhao YL, Chen SR, Chen H, Pan HL. Chronic opioid potentiates presynaptic but impairs postsynaptic N-methyl-D-aspartic acid receptor activity in spinal cords: implications for opioid hyperalgesia and tolerance. The Journal of biological chemistry. 2012;287:25073–25085. doi: 10.1074/jbc.M112.378737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zeng J, Thomson LM, Aicher SA, Terman GW. Primary afferent NMDA receptors increase dorsal horn excitation and mediate opiate tolerance in neonatal rats. Journal of Neuroscience. 2006;26:12033–12042. doi: 10.1523/JNEUROSCI.2530-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Yan X, Jiang E, Gao M, Weng H-R. Endogenous activation of presynaptic NMDA receptors enhances glutamate release from the primary afferents in the spinal dorsal horn in a rat model of neuropathic pain. Journal of Physiology. 2013;591:2001. doi: 10.1113/jphysiol.2012.250522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kawasaki Y, Zhang L, Cheng J-K, Ji R-R. Cytokine mechanisms of central sensitization: distinct and overlapping role of interleukin-1β, interleukin-6, and tumor necrosis factor-α in regulating synaptic and neuronal activity in the superficial spinal cord. The Journal of neuroscience. 2008;28:5189–5194. doi: 10.1523/JNEUROSCI.3338-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Watkins LR, Maier SF. Beyond Neurons: Evidence That Immune and Glial Cells Contribute to Pathological Pain States. Physiological Reviews. 2002;82:981–1011. doi: 10.1152/physrev.00011.2002. [DOI] [PubMed] [Google Scholar]
- [16].Watkins LR, Milligan ED, Maier SF. Glial activation: a driving force for pathological pain. Trends in Neurosciences. 2001;24:450–455. doi: 10.1016/s0166-2236(00)01854-3. [DOI] [PubMed] [Google Scholar]
- [17].Raghavendra V, Tanga FY, DeLeo JA. Complete Freunds adjuvant-induced peripheral inflammation evokes glial activation and proinflammatory cytokine expression in the CNS. European Journal of Neuroscience. 2004;20:467–473. doi: 10.1111/j.1460-9568.2004.03514.x. [DOI] [PubMed] [Google Scholar]
- [18].Sweitzer S, Colburn R, Rutkowski M, DeLeo J. Acute peripheral inflammation induces moderate glial activation and spinal IL-1β expression that correlates with pain behavior in the rat. Brain Research. 1999;829:209–221. doi: 10.1016/s0006-8993(99)01326-8. [DOI] [PubMed] [Google Scholar]
- [19].Tanga FY, Raghavendra V, DeLeo JA. Quantitative real-time RT-PCR assessment of spinal microglial and astrocytic activation markers in a rat model of neuropathic pain. Neurochemistry International. 2004;45:397–407. doi: 10.1016/j.neuint.2003.06.002. [DOI] [PubMed] [Google Scholar]
- [20].Yan X, Weng H-R. Endogenous Interleukin-1β in Neuropathic Rats Enhances Glutamate Release from the Primary Afferents in the Spinal Dorsal Horn through Coupling with Presynaptic N-Methyl-d-aspartic Acid Receptors. Journal of Biological Chemistry. 2013;288:30544–30557. doi: 10.1074/jbc.M113.495465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Gao YJ, Ji RR. Chemokines, neuronal-glial interactions, and central processing of neuropathic pain. Pharmacology & therapeutics. 2010;126:56. doi: 10.1016/j.pharmthera.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ji R-R, Samad TA, Jin S-X, Schmoll R, Woolf CJ. p38 MAPK activation by NGF in primary sensory neurons after inflammation increases TRPV1 levels and maintains heat hyperalgesia. Neuron. 2002;36:57–68. doi: 10.1016/s0896-6273(02)00908-x. [DOI] [PubMed] [Google Scholar]
- [23].Ji R-R, Suter MR. p38 MAPK, microglial signaling, and neuropathic pain. Mol Pain. 2007;3:33. doi: 10.1186/1744-8069-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].White FA, Jung H, Miller RJ. Chemokines and the pathophysiology of neuropathic pain. Proceedings of the National Academy of Sciences. 2007;104:20151–20158. doi: 10.1073/pnas.0709250104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Scholz J, Woolf CJ. The neuropathic pain triad: neurons, immune cells and glia. Nature Neuroscience. 2007;10:1361–1368. doi: 10.1038/nn1992. [DOI] [PubMed] [Google Scholar]
- [26].Ji R-R, Gereau RW, Malcangio M, Strichartz GR. MAP kinase and pain. Brain Research Reviews. 2009;60:135–148. doi: 10.1016/j.brainresrev.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Zhuang Z-Y, Gerner P, Woolf CJ, Ji R-R. ERK is sequentially activated in neurons, microglia, and astrocytes by spinal nerve ligation and contributes to mechanical allodynia in this neuropathic pain model. Pain. 2005;114:149–159. doi: 10.1016/j.pain.2004.12.022. [DOI] [PubMed] [Google Scholar]
- [28].Zhuang Z-Y, Kawasaki Y, Tan P-H, Wen Y-R, Huang J, Ji R-R. Role of the CX3CR1/p38 MAPK pathway in spinal microglia for the development of neuropathic pain following nerve injury-induced cleavage of fractalkine. Brain, behavior, and immunity. 2007;21:642–651. doi: 10.1016/j.bbi.2006.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Zhao X, Zmijewski JW, Lorne E, Liu G, Park YJ, Tsuruta Y, Abraham E. Activation of AMPK attenuates neutrophil proinflammatory activity and decreases the severity of acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2952008:L497–504. doi: 10.1152/ajplung.90210.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Bae HB, Zmijewski JW, Deshane JS, Tadie JM, Chaplin DD, Takashima S, Abraham E. AMP-activated protein kinase enhances the phagocytic ability of macrophages and neutrophils. Faseb J. 2011;25:4358–4368. doi: 10.1096/fj.11-190587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Giri S, Nath N, Smith B, Viollet B, Singh AK, Singh I. 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside inhibits proinflammatory response in glial cells: a possible role of AMP-activated protein kinase. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2004;24:479–487. doi: 10.1523/JNEUROSCI.4288-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Meares GP, Qin H, Liu Y, Holdbrooks AT, Benveniste EN. AMP-Activated Protein Kinase Restricts IFN-gamma Signaling. J Immunol. 2012 doi: 10.4049/jimmunol.1202390. DOI 10.4049/jimmunol.1202390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Melemedjian OK, Asiedu MN, Tillu DV, Sanoja R, Yan J, Lark A, Khoutorsky A, Johnson J, Peebles KA, Lepow T. Targeting adenosine monophosphate-activated protein kinase (AMPK) in preclinical models reveals a potential mechanism for the treatment of neuropathic pain. Molecular Pain. 2011;7:70. doi: 10.1186/1744-8069-7-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Mao-Ying QL, Kavelaars A, Krukowski K, Huo XJ, Zhou W, Price TJ, Cleeland C, Heijnen CJ. The anti-diabetic drug metformin protects against chemotherapy-induced peripheral neuropathy in a mouse model. PloS one. 2014;9:e100701. doi: 10.1371/journal.pone.0100701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Ma J, Yu H, Liu J, Chen Y, Wang Q, Xiang L. Metformin attenuates hyperalgesia and allodynia in rats with painful diabetic neuropathy induced by Streptozotocin. European Journal of Pharmacology. 2015 doi: 10.1016/j.ejphar.2015.06.010. DOI. [DOI] [PubMed] [Google Scholar]
- [36].Finkel T. Oxygen radicals and signaling. Current opinion in cell biology. 1998;10:248–253. doi: 10.1016/s0955-0674(98)80147-6. [DOI] [PubMed] [Google Scholar]
- [37].Rhee SG. Redox signaling: hydrogen peroxide as intracellular messenger. Experimental and Molecular Medicine. 1999;31:53–59. doi: 10.1038/emm.1999.9. [DOI] [PubMed] [Google Scholar]
- [38].Thannickal VJ, Fanburg BL. Reactive oxygen species in cell signaling. American Journal of Physiology-Lung Cellular and Molecular Physiology. 2000;279:L1005–L1028. doi: 10.1152/ajplung.2000.279.6.L1005. [DOI] [PubMed] [Google Scholar]
- [39].Salvemini D, Little JW, Doyle T, Neumann WL. Roles of reactive oxygen and nitrogen species in pain. Free Radical Biology and Medicine. 2011;51:951–966. doi: 10.1016/j.freeradbiomed.2011.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Janes K, Neumann WL, Salvemini D. Anti-superoxide and anti-peroxynitrite strategies in pain suppression. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease. 2012;1822:815–821. doi: 10.1016/j.bbadis.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Park E-S, Gao X, Chung JM, Chung K. Levels of mitochondrial reactive oxygen species increase in rat neuropathic spinal dorsal horn neurons. Neuroscience letters. 2006;391:108–111. doi: 10.1016/j.neulet.2005.08.055. [DOI] [PubMed] [Google Scholar]
- [42].Siniscalco D, Fuccio C, Giordano C, Ferraraccio F, Palazzo E, Luongo L, Rossi F, Roth KA, Maione S, de Novellis V. Role of reactive oxygen species and spinal cord apoptotic genes in the development of neuropathic pain. Pharmacological research. 2007;55:158–166. doi: 10.1016/j.phrs.2006.11.009. [DOI] [PubMed] [Google Scholar]
- [43].Kim HK, Zhang YP, Gwak YS, Abdi S. Phenyl N-tert-butylnitrone, a free radical scavenger, reduces mechanical allodynia in chemotherapy-induced neuropathic pain in rats. The Journal of the American Society of Anesthesiologists. 2010;112:432–439. doi: 10.1097/ALN.0b013e3181ca31bd. [DOI] [PubMed] [Google Scholar]
- [44].Khattab MM. TEMPOL, a membrane-permeable radical scavenger, attenuates peroxynitrite-and superoxide anion-enhanced carrageenan-induced paw edema and hyperalgesia: a key role for superoxide anion. European journal of pharmacology. 2006;548:167–173. doi: 10.1016/j.ejphar.2006.08.007. [DOI] [PubMed] [Google Scholar]
- [45].Doyle T, Bryant L, Muscoli C, Cuzzocrea S, Esposito E, Chen Z, Salvemini D. Spinal NADPH oxidase is a source of superoxide in the development of morphine-induced hyperalgesia and antinociceptive tolerance. Neuroscience letters. 2010;483:85–89. doi: 10.1016/j.neulet.2010.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Doyle T, Esposito E, Bryant L, Cuzzocrea S, Salvemini D. NADPH-oxidase 2 activation promotes opioid-induced antinociceptive tolerance in mice. Neuroscience. 2013;241:1–9. doi: 10.1016/j.neuroscience.2013.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Muscoli C, Cuzzocrea S, Ndengele MM, Mollace V, Porreca F, Fabrizi F, Esposito E, Masini E, Matuschak GM, Salvemini D. Therapeutic manipulation of peroxynitrite attenuates the development of opiate-induced antinociceptive tolerance in mice. The Journal of clinical investigation. 2007;117:3530–3539. doi: 10.1172/JCI32420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Hardie DG. AMP-activated protein kinase—an energy sensor that regulates all aspects of cell function. Genes & development. 2011;25:1895–1908. doi: 10.1101/gad.17420111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Qin S, Rodrigues GA. Differential roles of AMPKα1 and AMPKα2 in regulating 4-HNE-induced RPE cell death and permeability. Experimental eye research. 2010;91:818–824. doi: 10.1016/j.exer.2010.10.007. [DOI] [PubMed] [Google Scholar]
- [50].Ost M, Werner F, Dokas J, Klaus S, Voigt A. Activation of AMPKα2 is not crucial for mitochondrial uncoupling-induced metabolic effects but required to maintain skeletal muscle integrity. PloS one. 2014;9 doi: 10.1371/journal.pone.0094689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Fu X, Zhao J-X, Zhu M-J, Foretz M, Viollet B, Dodson MV, Du M. AMP-activated protein kinase α1 but not α2 catalytic subunit potentiates myogenin expression and myogenesis. Molecular and cellular biology. 2013;33:4517–4525. doi: 10.1128/MCB.01078-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Zhuo L, Theis M, Alvarez-Maya I, Brenner M, Willecke K, Messing A. hGFAP-cre transgenic mice for manipulation of glial and neuronal function in vivo. genesis. 2001;31:85–94. doi: 10.1002/gene.10008. [DOI] [PubMed] [Google Scholar]
- [53].Nakada D, Saunders TL, Morrison SJ. Lkb1 regulates cell cycle and energy metabolism in haematopoietic stem cells. Nature. 2010;468:653–658. doi: 10.1038/nature09571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Yan X, Maixner DW, Yadav R, Gao M, Li P, Bartlett MG, Weng H-R. Paclitaxel induces acute pain via directly activating toll like receptor 4. Molecular Pain. 2015;11:10. doi: 10.1186/s12990-015-0005-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Moore SA, Saito F, Chen J, Michele DE, Henry MD, Messing A, Cohn RD, Ross-Barta SE, Westra S, Williamson RA, Hoshi T, Campbell KP. Deletion of brain dystroglycan recapitulates aspects of congenital muscular dystrophy. Nature. 2002;418:422–425. doi: 10.1038/nature00838. [DOI] [PubMed] [Google Scholar]
- [56].Garrett AM, Weiner JA. Control of CNS synapse development by γ-protocadherin-mediated astrocyte–neuron contact. The Journal of Neuroscience. 2009;29:11723–11731. doi: 10.1523/JNEUROSCI.2818-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Weng H-R, Gao M, Maixner DW. Glycogen synthase kinase 3 beta regulates glial glutamate transporter protein expression in the spinal dorsal horn in rats with neuropathic pain. Experimental neurology. 2014;252:18–27. doi: 10.1016/j.expneurol.2013.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Weng HR, Chen J, Pan Z, Nie H. Glial glutamate transporter 1 regulates the spatial and temporal coding of glutamatergic synaptic transmission in spinal lamina II neurons. Neuroscience. 2007;149:898–907. doi: 10.1016/j.neuroscience.2007.07.063. [DOI] [PubMed] [Google Scholar]
- [59].Zhang J, Malik A, Choi HB, Ko RW, Dissing-Olesen L, MacVicar BA. Microglial CR3 activation triggers long-term synaptic depression in the hippocampus via NADPH oxidase. Neuron. 2014;82:195–207. doi: 10.1016/j.neuron.2014.01.043. [DOI] [PubMed] [Google Scholar]
- [60].Yan X, Yadav R, Gao M, Weng HR. Interleukin-1 beta enhances endocytosis of glial glutamate transporters in the spinal dorsal horn through activating protein kinase C. Glia. 2014 doi: 10.1002/glia.22665. DOI. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Nakatsuka T, Tsuzuki K, Ling JX, Sonobe H, Gu JG. Distinct roles of P2X receptors in modulating glutamate release at different primary sensory synapses in rat spinal cord. Journal of neurophysiology. 2003;89:3243–3252. doi: 10.1152/jn.01172.2002. [DOI] [PubMed] [Google Scholar]
- [62].Weng H, Chen J, Cata J. Inhibition of glutamate uptake in the spinal cord induces hyperalgesia and increased responses of spinal dorsal horn neurons to peripheral afferent stimulation. Neuroscience. 2006;138:1351–1360. doi: 10.1016/j.neuroscience.2005.11.061. [DOI] [PubMed] [Google Scholar]
- [63].Yoshimura M, Jessell T. Primary afferent-evoked synaptic responses and slow potential generation in rat substantia gelatinosa neurons in vitro. J Neurophysiol. 1989;62:96–108. doi: 10.1152/jn.1989.62.1.96. [DOI] [PubMed] [Google Scholar]
- [64].Ji R-R, Baba H, Brenner GJ, Woolf CJ. Nociceptive-specific activation of ERK in spinal neurons contributes to pain hypersensitivity. Nature Neuroscience. 1999;2:1114–1119. doi: 10.1038/16040. [DOI] [PubMed] [Google Scholar]
- [65].Son Y, Cheong Y-K, Kim N-H, Chung H-T, Kang DG, Pae H-O. Mitogen-activated protein kinases and reactive oxygen species: how can ROS activate MAPK pathways? Journal of signal transduction. 2011;2011 doi: 10.1155/2011/792639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Son Y, Kim S, Chung H-T, Pae H-O. Reactive oxygen species in the activation of MAP kinases. Methods In Enzymology. 2012;528:27–48. doi: 10.1016/B978-0-12-405881-1.00002-1. [DOI] [PubMed] [Google Scholar]
- [67].Vereker E, O'Donnell E, Lynch M. The inhibitory effect of interleukin-1β on long-term potentiation is coupled with increased activity of stress-activated protein kinases. The Journal of neuroscience. 2000;20:6811–6819. doi: 10.1523/JNEUROSCI.20-18-06811.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Fekete Á, Vizi ES, Kovács KJ, Lendvai B, Zelles T. Layer-specific differences in reactive oxygen species levels after oxygen–glucose deprivation in acute hippocampal slices. Free Radical Biology and Medicine. 2008;44:1010–1022. doi: 10.1016/j.freeradbiomed.2007.11.022. [DOI] [PubMed] [Google Scholar]
- [69].Halliwell B, Whiteman M. Measuring reactive species and oxidative damage in vivo and in cell culture: how should you do it and what do the results mean? British journal of pharmacology. 2004;142:231–255. doi: 10.1038/sj.bjp.0705776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Al-Huseini LM, Aw Yeang HX, Hamdam JM, Sethu S, Alhumeed N, Wong W, Sathish JG. Heme oxygenase-1 regulates dendritic cell function through modulation of p38MAPK-CREB/ATF1 signalling. J Biol Chem. 2014 doi: 10.1074/jbc.M113.532069. DOI 10.1074/jbc.M113.532069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Kim D, You B, Jo E-K, Han S-K, Simon MI, Lee SJ. NADPH oxidase 2-derived reactive oxygen species in spinal cord microglia contribute to peripheral nerve injury-induced neuropathic pain. Proceedings of the National Academy of Sciences. 2010;107:14851–14856. doi: 10.1073/pnas.1009926107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Dhainaut A, Tizot A, Raimbaud E, Lockhart B, Lestage P, Goldstein S. Synthesis, structure, and neuroprotective properties of novel imidazolyl nitrones. Journal of medicinal chemistry. 2000;43:2165–2175. doi: 10.1021/jm991154w. [DOI] [PubMed] [Google Scholar]
- [73].Kim HK, Park SK, Zhou J-L, Taglialatela G, Chung K, Coggeshall RE, Chung JM. Reactive oxygen species (ROS) play an important role in a rat model of neuropathic pain. Pain. 2004;111:116–124. doi: 10.1016/j.pain.2004.06.008. [DOI] [PubMed] [Google Scholar]
- [74].Chow J-M, Shen S-C, Huan SK, Lin H-Y, Chen Y-C. Quercetin, but not rutin and quercitrin, prevention of H2O2-induced apoptosis via anti-oxidant activity and heme oxygenase 1 gene expression in macrophages. Biochemical pharmacology. 2005;69:1839–1851. doi: 10.1016/j.bcp.2005.03.017. [DOI] [PubMed] [Google Scholar]
- [75].Takahashi T, Morita K, Akagi R, Sassa S. Heme oxygenase-1: a novel therapeutic target in oxidative tissue injuries. Current medicinal chemistry. 2004;11:1545–1561. doi: 10.2174/0929867043365080. [DOI] [PubMed] [Google Scholar]
- [76].Drummond GS, Kappas A. The cytochrome P-450-depleted animal: an experimental model for in vivo studies in chemical biology. Proceedings of the National Academy of Sciences. 1982;79:2384–2388. doi: 10.1073/pnas.79.7.2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Gao X, Kim HK, Chung JM, Chung K. Reactive oxygen species (ROS) are involved in enhancement of NMDA-receptor phosphorylation in animal models of pain. Pain. 2007;131:262–271. doi: 10.1016/j.pain.2007.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Lee KY, Chung K, Chung JM. Involvement of reactive oxygen species in long-term potentiation in the spinal cord dorsal horn. Journal of neurophysiology. 2010;103:382–391. doi: 10.1152/jn.90906.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Russe OQ, Möser CV, Kynast KL, King TS, Stephan H, Geisslinger G, Niederberger E. Activation of the AMP-Activated Protein Kinase Reduces Inflammatory Nociception. The Journal of Pain. 2013;14:1330–1340. doi: 10.1016/j.jpain.2013.05.012. [DOI] [PubMed] [Google Scholar]
- [80].Yamamoto T, Yaksh T. Comparison of the antinociceptive effects of pre-and posttreatment with intrathecal morphine and MK801, an NMDA antagonist, on the formalin test in the rat. Anesthesiology. 1992;77:757–763. doi: 10.1097/00000542-199210000-00021. [DOI] [PubMed] [Google Scholar]
- [81].Kim SK, Sun B, Yoon H, Lee JH, Lee G, Sohn SH, Kim H, Quan FS, Shim I, Ha J, Min BI, Bae H. Expression levels of the hypothalamic AMPK gene determines the responsiveness of the rats to electroacupuncture-induced analgesia. BMC Complement Altern Med. 2014;14:211. doi: 10.1186/1472-6882-14-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Nie H, Weng HR. Impaired glial glutamate uptake induces extrasynaptic glutamate spillover in the spinal sensory synapses of neuropathic rats. Journal of neurophysiology. 2010;103:2570–2580. doi: 10.1152/jn.00013.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Xie W, Strong JA, Zhang JM. Early blockade of injured primary sensory afferents reduces glial cell activation in two rat neuropathic pain models. Neuroscience. 2009;160:847–857. doi: 10.1016/j.neuroscience.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Li JQ, Chen SR, Chen H, Cai YQ, Pan HL. Regulation of increased glutamatergic input to spinal dorsal horn neurons by mGluR5 in diabetic neuropathic pain. Journal of neurochemistry. 2010;112:162–172. doi: 10.1111/j.1471-4159.2009.06437.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Weng HR, Cordella JV, Dougherty PM. Changes in sensory processing in the spinal dorsal horn accompany vincristine-induced hyperalgesia and allodynia. Pain. 2003;103:131–138. doi: 10.1016/s0304-3959(02)00445-1. [DOI] [PubMed] [Google Scholar]
- [86].Ren K, Dubner R. Inflammatory models of pain and hyperalgesia. ILAR Journal. 1999;40:111–118. doi: 10.1093/ilar.40.3.111. [DOI] [PubMed] [Google Scholar]
- [87].De Biasi S, Rustioni A. Glutamate and substance P coexist in primary afferent terminals in the superficial laminae of spinal cord. Proceedings of the National Academy of Sciences. 1988;85:7820–7824. doi: 10.1073/pnas.85.20.7820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Danbolt NC. Glutamate uptake. Progress in Neurobiology. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- [89].Jiang E, Yan X, Weng HR. Glial glutamate transporter and glutamine synthetase regulate GABAergic synaptic strength in the spinal dorsal horn. Journal of neurochemistry. 2012 doi: 10.1111/j.1471-4159.2012.07694.x. DOI. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Salminen A, Hyttinen JM, Kaarniranta K. AMP-activated protein kinase inhibits NF-κB signaling and inflammation: impact on healthspan and lifespan. Journal of molecular medicine. 2011;89:667–676. doi: 10.1007/s00109-011-0748-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Chung JM. The role of reactive oxygen species (ROS) in persistent pain. Molecular interventions. 2004;4:248. doi: 10.1124/mi.4.5.3. [DOI] [PubMed] [Google Scholar]
- [92].Lee DZ, Chung JM, Chung K, Kang M-G. Reactive oxygen species (ROS) modulate AMPA receptor phosphorylation and cell-surface localization in concert with pain-related behavior. PAIN®. 2012;153:1905–1915. doi: 10.1016/j.pain.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Yowtak J, Lee KY, Kim HY, Wang J, Kim HK, Chung K, Chung JM. Reactive oxygen species contribute to neuropathic pain by reducing spinal GABA release. PAIN®. 2011;152:844–852. doi: 10.1016/j.pain.2010.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]